Bioavailability Enhancement of Poorly Water-Soluble Drugs via Nanocomposites: Formulation–Processing Aspects and Challenges


Abstract
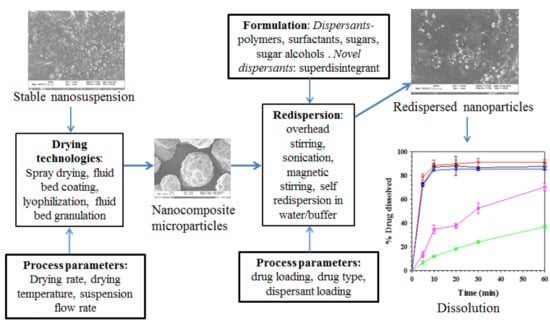
:
1. Introduction
2. Preparation of Drug Nanosuspensions and Their Stabilization
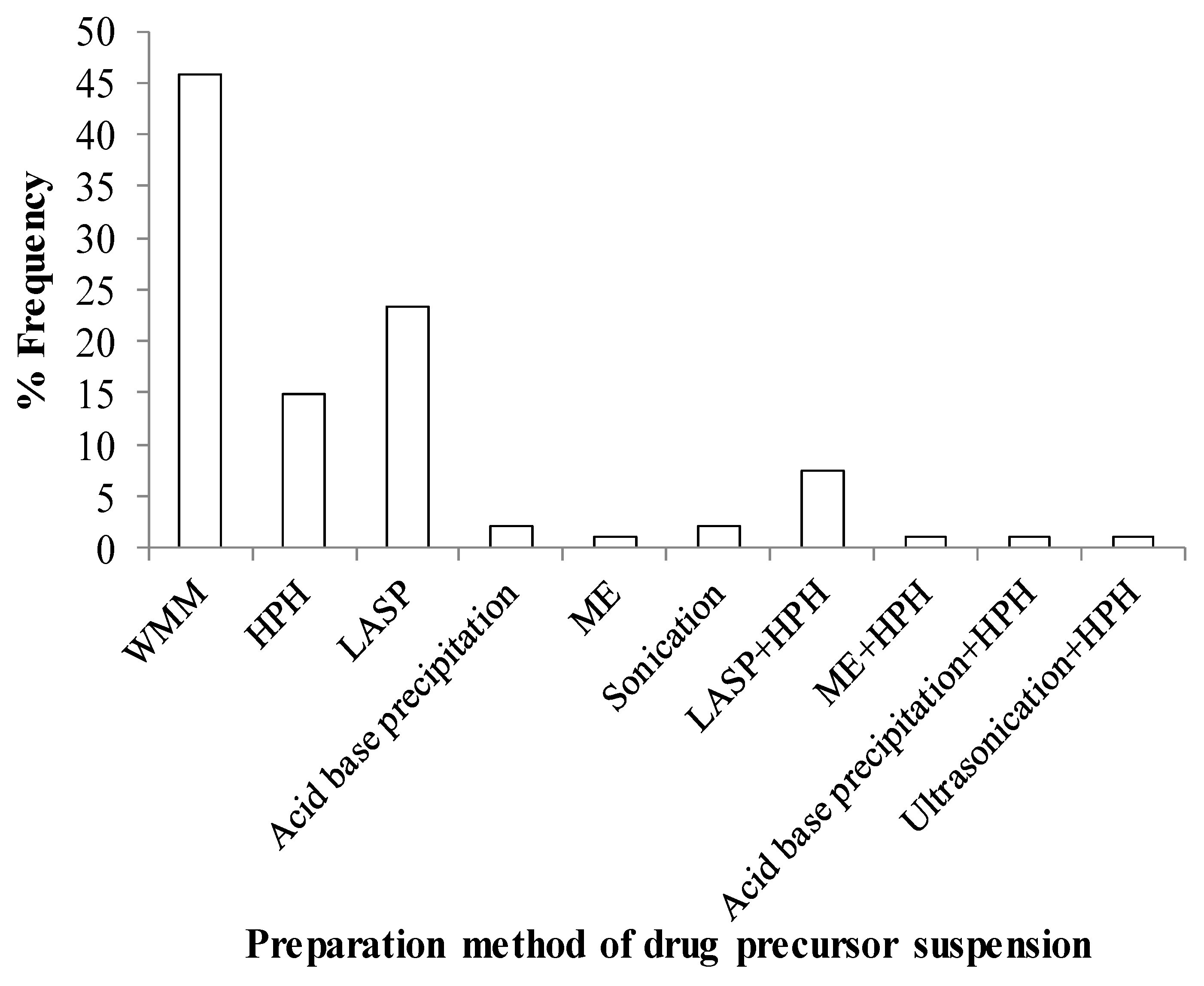
2.1. Preparation Methods
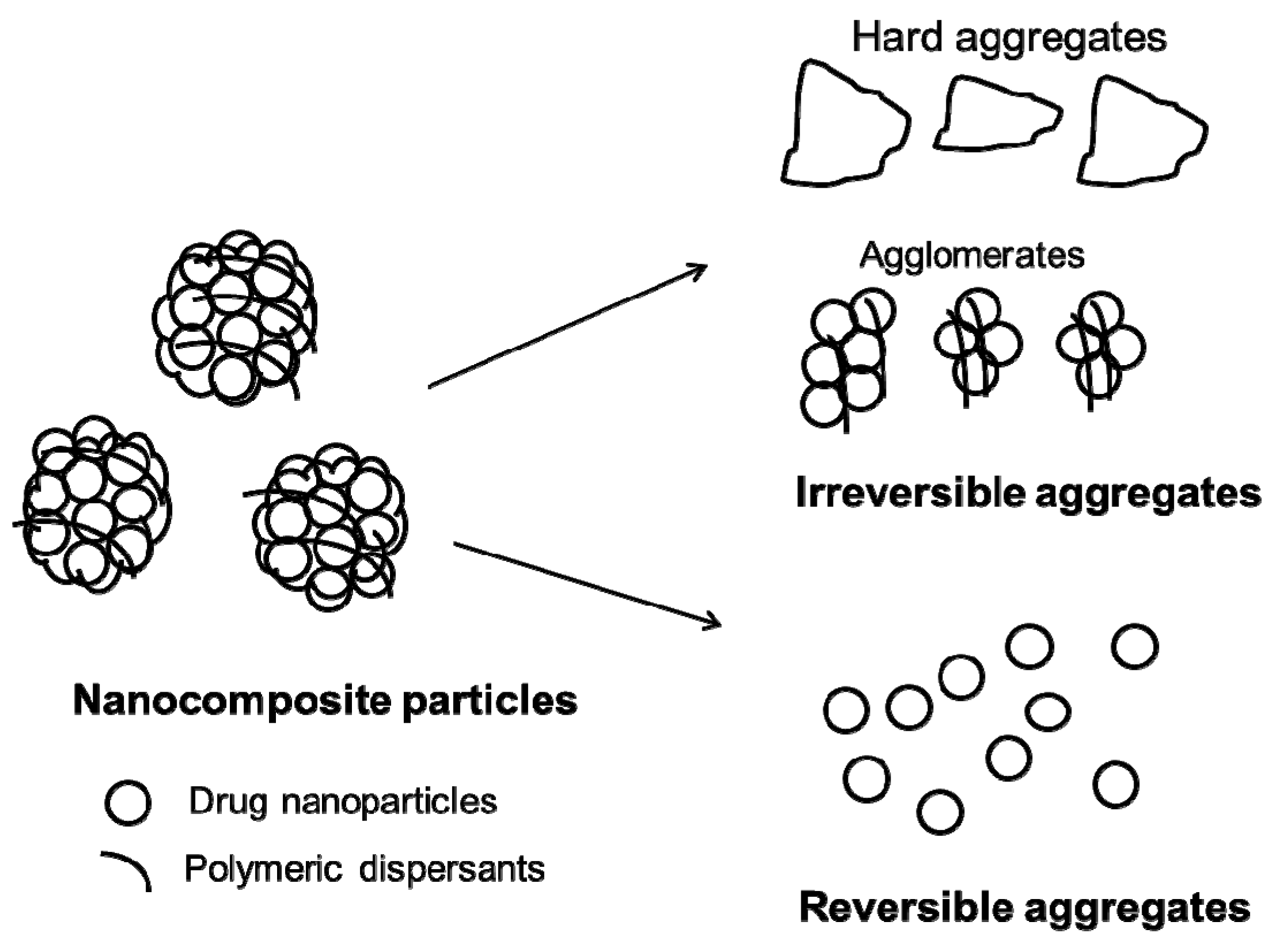
2.2. More on the Stabilization of Drug Nanosuspensions
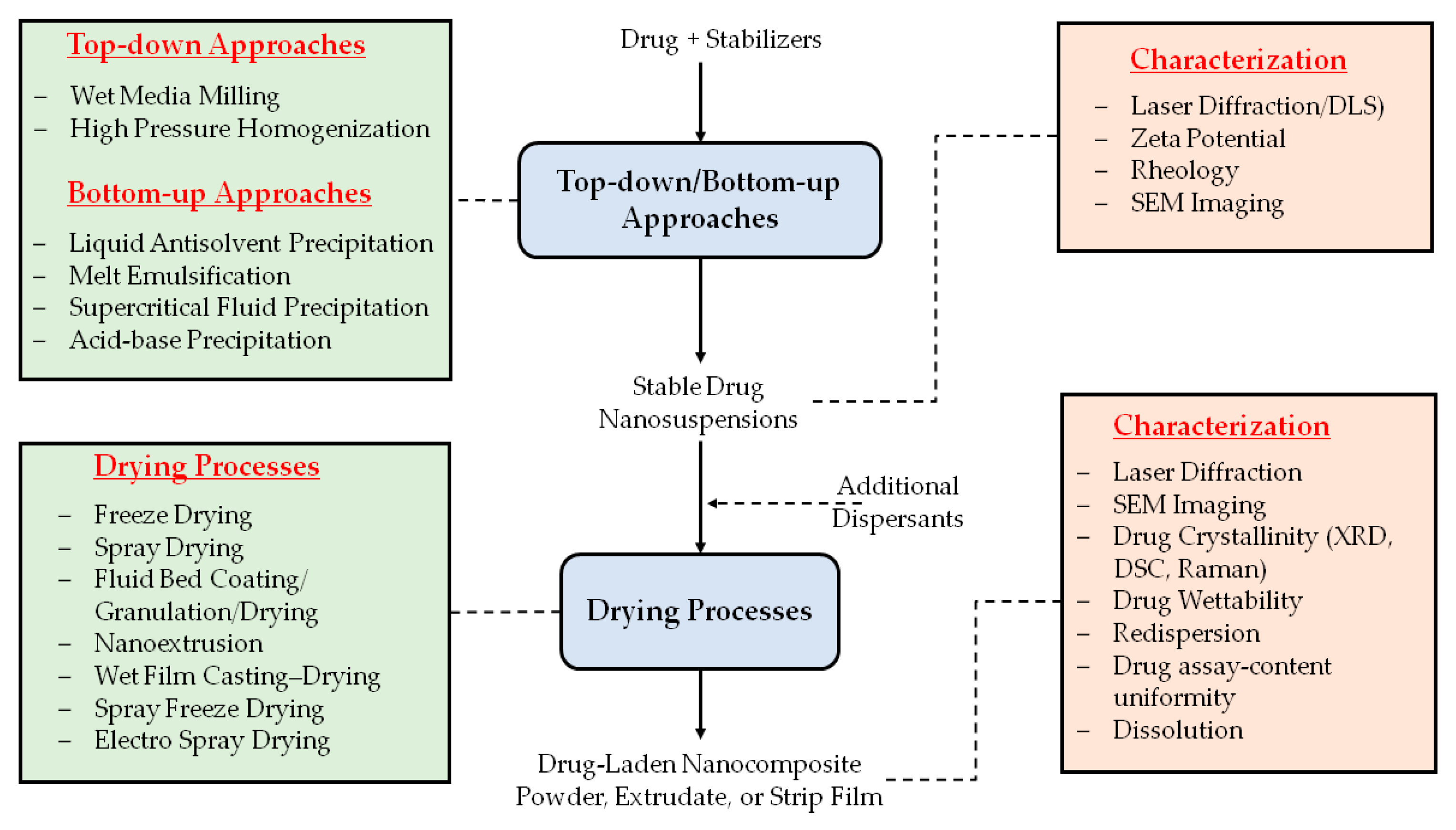
3. Preparation, Characterization, and Formulation of Drug-Laden Nanocomposites
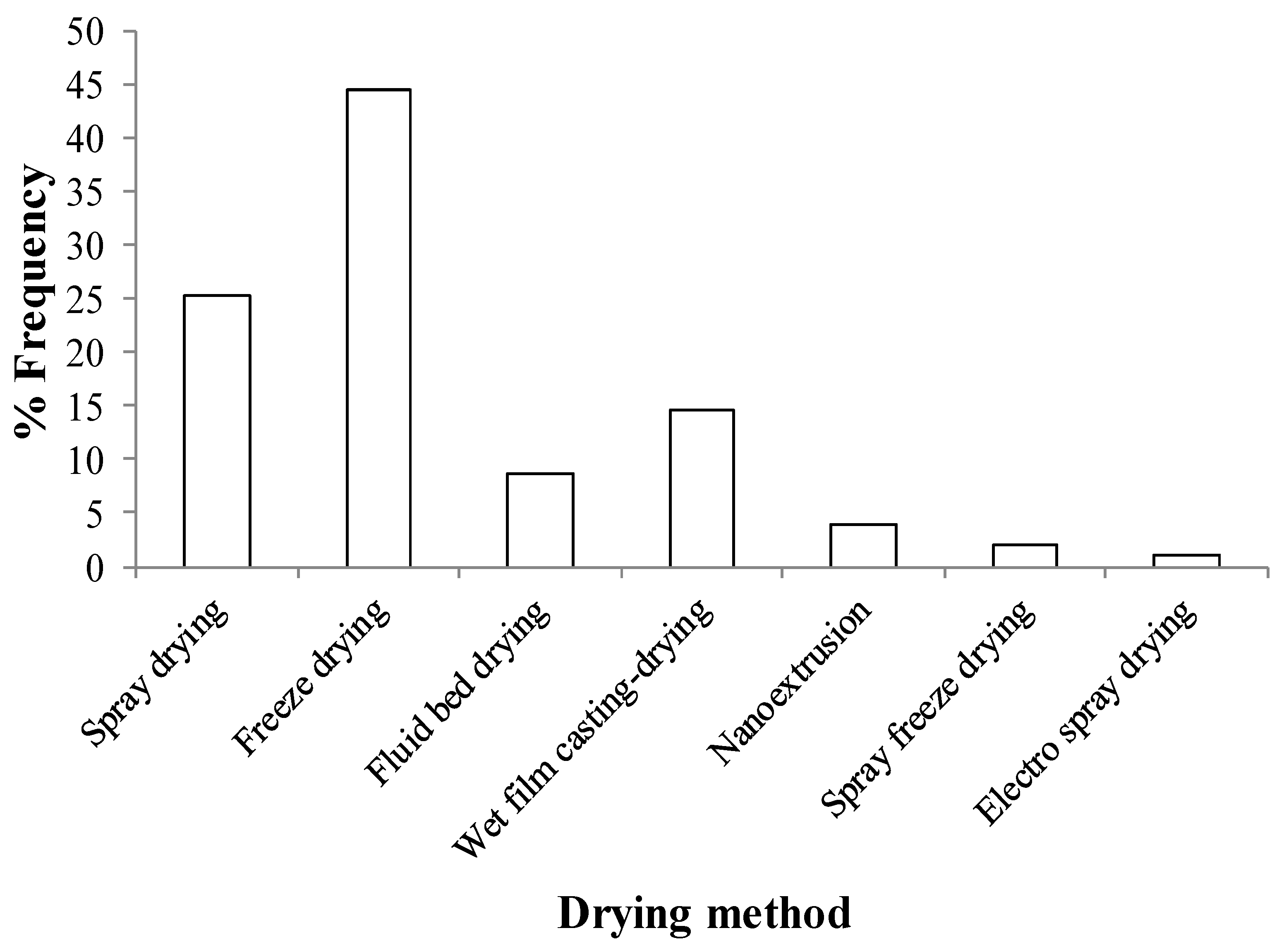
3.1. Drying Processes for the Production of Nanocomposites
3.2. Characterization of the Nanocomposites
3.2.1. Particle Sizing
3.2.2. Scanning Electron Microscope (SEM) Imaging
3.2.3. Drug Crystallinity
3.2.4. Redispersion Methods
3.2.5. Dissolution Testing
3.2.6. Drug Wettability
3.3. Nanocomposite Formulations and Functionalities of Dispersants
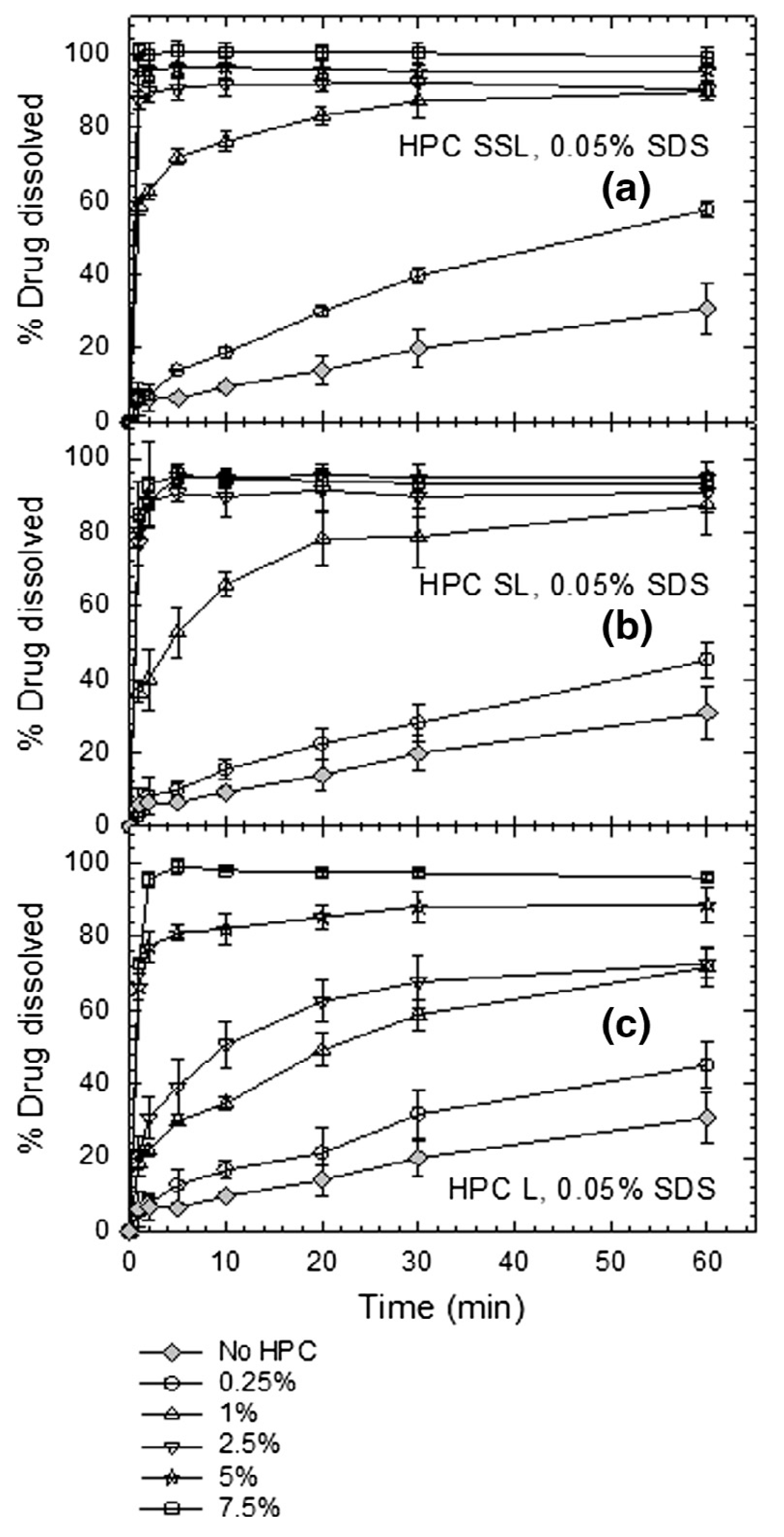
3.3.1. Impact of Soluble Polymers on Redispersion–Drug Release
3.3.2. Impact of Surfactants on Redispersion–Drug Release
3.3.3. Impact of Soluble Polymer–Surfactant on Redispersion–Drug Release
3.3.4. Impact of Other Classes of Dispersants
3.4. Impact of Drying Method
3.5. Incorporating Nanocomposite Intermediate into Final Oral Solid Dosage Forms
4. Some Engineering Considerations for Rational Selection of a Drying Process
5. Additional Insights into Development of Redispersible, Fast-Dissolving Nanocomposites
6. Concluding Remarks and Future Research Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C. Poor aqueous solubility—An industry wide problem in drug discovery. Am. Pharm. Rev. 2002, 5, 82–85. [Google Scholar]
- Keserü, G.M.; Makara, G.M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 2009, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A. Innovative strategies for the oral delivery of drugs and peptides. Trends Biotechnol. 1998, 16, 152–157. [Google Scholar] [CrossRef]
- Müllertz, A.; Ogbonna, A.; Ren, S.; Rades, T. New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J. Pharm. Pharmacol. 2010, 62, 1622–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, T.; Danjo, K. Design of self-dispersible dry nanosuspension through wet milling and spray freeze-drying for poorly water-soluble drugs. Eur. J. Pharm. Sci. 2013, 50, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Bhakay, A.; Azad, M.; Vizzotti, E.; Dave, R.N.; Bilgili, E. Enhanced recovery and dissolution of griseofulvin nanoparticles from surfactant-free nanocomposite microparticles incorporating wet-milled swellable dispersants. Drug Dev. Ind. Pharm. 2014, 40, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- Humberstone, A.J.; Charman, W.N. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv. Drug Deliv. Rev. 1997, 25, 103–128. [Google Scholar] [CrossRef]
- Hauss, D.J.; Fogal, S.E.; Ficorilli, J.V.; Price, C.A.; Roy, T.; Jayaraj, A.A.; Keirns, J.J. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water-soluble LTB4 inhibitor. J. Pharm. Sci. 1998, 87, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Rumondor, A.C.; Dhareshwar, S.S.; Kesisoglou, F. Amorphous solid dispersions or prodrugs: Complementary strategies to increase drug absorption. J. Pharm. Sci. 2016, 105, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Nakagami, H. Solid dispersions of indomethacin and griseofulvin in non-porous fumed silicon dioxide, prepared by melting. Chem. Pharm. Bull. 1991, 39, 2417–2421. [Google Scholar] [CrossRef]
- Serajuddin, A. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Rahman, Z.; Zidan, A.S.; Samy, R.; Sayeed, V.A.; Khan, M.A. Improvement of physicochemical properties of an antiepileptic drug by salt engineering. AAPS PharmSciTech 2012, 13, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.P.; Holm, R.; de Diego, H.L. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, N.; Newman, A. Pharmaceutical cocrystals and their physicochemical properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Shete, A.; Dabke, A.; Kulkarni, P.; Sakhare, S. Co-crystals: A novel approach to modify physicochemical properties of active pharmaceutical ingredients. Indian J. Pharm. Sci. 2009, 71, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Aleem, O.; Kuchekar, B.; Pore, Y.; Late, S. Effect of β-cyclodextrin and hydroxypropyl β-cyclodextrin complexation on physicochemical properties and antimicrobial activity of cefdinir. J. Pharm. Biomed. Anal. 2008, 47, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Srivalli, K.M.R.; Mishra, B. Improved aqueous solubility and antihypercholesterolemic activity of ezetimibe on formulating with hydroxypropyl-β-cyclodextrin and hydrophilic auxiliary substances. AAPS PharmSciTech 2016, 17, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Bhakay, A.; Merwade, M.; Bilgili, E.; Dave, R.N. Novel aspects of wet milling for the production of microsuspensions and nanosuspensions of poorly water-soluble drugs. Drug Dev. Ind. Pharm. 2011, 37, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.; Afolabi, A.; Bilgili, E. Continuous production of drug nanoparticle suspensions via wet stirred media milling: A fresh look at the Rehbinder effect. Drug Dev. Ind. Pharm. 2013, 39, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Azad, M.; Davé, R.; Bilgili, E. Nanomilling of drugs for bioavailability enhancement: A holistic formulation-process perspective. Pharmaceutics 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Benita, S.; Böhm, B.H. Nanosuspensions. In Emulsions and Nanosuspensions for the Formulation of Poorly Soluble Drugs; Benita, S., Böhm, B.H., Eds.; Medpharm Scientific: Stuttgart, Germany, 1998; pp. 149–173. [Google Scholar]
- Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef]
- Zhang, X.; Guan, J.; Ni, R.; Li, L.C.; Mao, S. Preparation and solidification of redispersible nanosuspensions. J. Pharm. Sci. 2014, 103, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.; Arteaga, C.; Abdelmalek, B.; Davé, R.; Bilgili, E. Spray drying of drug-swellable dispersant suspensions for preparation of fast-dissolving, high drug-loaded, surfactant-free nanocomposites. Drug Dev. Ind. Pharm. 2015, 41, 1617–1631. [Google Scholar] [CrossRef] [PubMed]
- Kesisoglou, F.; Panmai, S.; Wu, Y. Nanosizing—Oral formulation development and biopharmaceutical evaluation. Adv. Drug Deliv. Rev. 2007, 59, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Junghanns, J.-U.A.; Müller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–309. [Google Scholar] [Green Version]
- Srivalli, K.M.R.; Mishra, B. Drug nanocrystals: A way toward scale-up. Saudi Pharm. J. 2016, 24, 386–404. [Google Scholar] [CrossRef] [PubMed]
- Malamatari, M.; Taylor, K.M.; Malamataris, S.; Douroumis, D.; Kachrimanis, K. Pharmaceutical nanocrystals: Prodcution by wet media milling and applications. Drug Discov. Today 2018, 23, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.; Bohm, B.; Grau, J. Nanosuspensions: A formulation approach for poorly soluble and poorly bioavailable drugs. Handb. Pharm. Control. Release Technol. 2000, 17, 345–357. [Google Scholar]
- Lee, J. Drug nano-and microparticles processed into solid dosage forms: Physical properties. J. Pharm. Sci. 2003, 92, 2057–2068. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Froyen, L.; Van Humbeeck, J.; Martens, J.A.; Augustijns, P.; Van den Mooter, G. Drying of crystalline drug nanosuspensions—The importance of surface hydrophobicity on dissolution behavior upon redispersion. Eur. J. Pharm. Sci. 2008, 35, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Liversidge, G.G.; Cundy, K.C.; Bishop, J.F.; Czekai, D.A. Surface Modified Drug Nanoparticles. U.S. Patent 5,145,684, 8 September 1992. [Google Scholar]
- Beck, C.; Dalvi, S.V.; Dave, R.N. Controlled liquid antisolvent precipitation using a rapid mixing device. Chem. Eng. Sci. 2010, 65, 5669–5675. [Google Scholar] [CrossRef]
- De Zordi, N.; Moneghini, M.; Kikic, I.; Grassi, M.; Castillo, A.E.D.R.; Solinas, D.; Bolger, M.B. Applications of supercritical fluids to enhance the dissolution behaviors of Furosemide by generation of microparticles and solid dispersions. Eur. J. Pharm. Biopharm. 2012, 81, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, J.C.; Bevilacqua, G.; Rosa, P.d.T.V.e.; Sosnik, A. Production of pure indinavir free base nanoparticles by a supercritical anti-solvent (SAS) method. Drug Dev. Ind. Pharm. 2014, 40, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Knieke, C.; Rawtani, A.; Davé, R.N. Concentrated fenofibrate nanoparticle suspensions from melt emulsification for enhanced drug dissolution. Chem. Eng. Technol. 2014, 37, 157–167. [Google Scholar] [CrossRef]
- Bhakay, A.; Vizzotti, E.; Li, M.; Davé, R.; Bilgili, E. Incorporation of fenofibrate nanoparticles prepared by melt emulsification into polymeric films. J. Pharm. Innov. 2016, 11, 53–63. [Google Scholar] [CrossRef]
- Salazar, J.; Ghanem, A.; Müller, R.H.; Möschwitzer, J.P. Nanocrystals: Comparison of the size reduction effectiveness of a novel combinative method with conventional top-down approaches. Eur. J. Pharm. Biopharm. 2012, 81, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Bruno, J.A.; Doty, B.D.; Gustow, E.; Illig, K.J.; Rajagopalan, N.; Sarpotdar, P. Method of Grinding Pharmaceutical Substances. U.S. Patent No. 5,518,187, 21 May 1996. [Google Scholar]
- Sommer, M.; Stenger, F.; Peukert, W.; Wagner, N. Agglomeration and breakage of nanoparticles in stirred media mills—A comparison of different methods and models. Chem. Eng. Sci. 2006, 61, 135–148. [Google Scholar] [CrossRef]
- Knieke, C.; Sommer, M.; Peukert, W. Identifying the apparent and true grinding limit. Powder Technol. 2009, 195, 25–30. [Google Scholar] [CrossRef]
- Su, J.-C.; Liang, S.Y.; Liu, W.L.; Jan, T.C. Ceramic micro/nanoparticle size evolution in wet grinding in stirred ball mill. J. Manuf. Sci. Eng. 2004, 126, 779–786. [Google Scholar] [CrossRef]
- Peukert, W.; Schwarzer, H.-C.; Stenger, F. Control of aggregation in production and handling of nanoparticles. Chem. Eng. Process. Process Intensif. 2005, 44, 245–252. [Google Scholar] [CrossRef]
- Cerdeira, A.M.; Mazzotti, M.; Gander, B. Miconazole nanosuspensions: Influence of formulation variables on particle size reduction and physical stability. Int. J. Pharm. 2010, 396, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Napper, D.H. Colloid stability. Ind. Eng. Chem. Prod. Res. Dev. 1970, 9, 467–477. [Google Scholar] [CrossRef]
- Muller, R. Zetapotential und Partikelladung—Kurze Theorie, Praktische Meûdurchfu Ehrung, Daten Interpretation; Wissenschaftliche Verlagsgesellschaft: Stuttgart, Germany, 1996. [Google Scholar]
- Riddick, T.M. Control of Colloid Stability Through Zeta Potential; Zeta-Meter Inc. via Livingston Publishing Company: Lynnewood, PA, USA, 1968. [Google Scholar]
- Lakshmi, P.; Kumar, G.A. Nanosuspension technology: A review. Int. J. Pharm. Sci. 2010, 2, 35–40. [Google Scholar]
- Kim, C.-J. Advanced Pharmaceutics: Physicochemical Principles; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Ghosh, I.; Bose, S.; Vippagunta, R.; Harmon, F. Nanosuspension for improving the bioavailability of a poorly soluble drug and screening of stabilizing agents to inhibit crystal growth. Int. J. Pharm. 2011, 409, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kumar, S.; Gokhale, R.; Burgess, D.J. Physical stability of nanosuspensions: Investigation of the role of stabilizers on ostwald ripening. Int. J. Pharm. 2011, 406, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Chin, W.W.L.; Parmentier, J.; Widzinski, M.; Tan, E.H.; Gokhale, R. A brief literature and patent review of nanosuspensions to a final drug product. J. Pharm. Sci. 2014, 103, 2980–2999. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, L.; Hirvonen, J. Pharmaceutical nanocrystals by nanomilling: Critical process parameters, particle fracturing and stabilization methods. J. Pharm. Pharmacol. 2010, 62, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, J.; Watanabe, W. Physical and chemical stability of drug nanoparticles. Adv. Drug Deliv. Rev. 2011, 63, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Tewa-Tagne, P.; Briançon, S.; Fessi, H. Preparation of redispersible dry nanocapsules by means of spray-drying: Development and characterisation. Eur. J. Pharm. Sci. 2007, 30, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Froyen, L.; Van Humbeeck, J.; Martens, J.A.; Augustijns, P.; Van Den Mooter, G. Alternative matrix formers for nanosuspension solidification: Dissolution performance and X-ray microanalysis as an evaluation tool for powder dispersion. Eur. J. Pharm. Sci. 2008, 35, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Vertzoni, M.; Kunath, K.; Reppas, C.; Dressman, J.B. Cogrinding enhances the oral bioavailability of EMD 57033, a poorly water soluble drug, in dogs. Eur. J. Pharm. Biopharm. 2008, 68, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Kho, K.; Cheow, W.S.; Lie, R.H.; Hadinoto, K. Aqueous re-dispersibility of spray-dried antibiotic-loaded polycaprolactone nanoparticle aggregates for inhaled anti-biofilm therapy. Powder Technol. 2010, 203, 432–439. [Google Scholar] [CrossRef]
- Hu, J.; Ng, W.K.; Dong, Y.; Shen, S.; Tan, R.B. Continuous and scalable process for water-redispersible nanoformulation of poorly aqueous soluble APIs by antisolvent precipitation and spray-drying. Int. J. Pharm. 2011, 404, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Ryde, N.P.; Ruddy, S.B. Solid dose Nanoparticulate Compositions Comprising a Synergistic Combination of a Polymeric Surface Stabilizer and Dioctyl Sodium Sulfosuccinate. U.S. Patent 6,375,986, 23 April 2002. [Google Scholar]
- Möschwitzer, J.; Müller, R.H. Spray coated pellets as carrier system for mucoadhesive drug nanocrystals. Eur. J. Pharm. Biopharm. 2006, 62, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Basa, S.; Muniyappan, T.; Karatgi, P.; Prabhu, R.; Pillai, R. Production and in vitro characterization of solid dosage form incorporating drug nanoparticles. Drug Dev. Ind. Pharm. 2008, 34, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Bhakay, A.; Davé, R.; Bilgili, E. Recovery of BCS Class II drugs during aqueous redispersion of core-shell type nanocomposite particles produced via fluidized bed coating. Powder Technol. 2013, 236, 221–234. [Google Scholar] [CrossRef]
- Cheow, W.S.; Ng, M.L.L.; Kho, K.; Hadinoto, K. Spray freeze-drying production of thermally sensitive polymeric nanoparticle aggregates for inhaled drug delivery: Effect of freeze-drying adjuvants. Int. J. Pharm. 2011, 404, 289–300. [Google Scholar] [CrossRef] [PubMed]
- De Waard, H.; Hinrichs, W.; Frijlink, H. A novel bottom-up process to produce drug nanocrystals: Controlled crystallization during freeze-drying. J. Control. Release 2008, 128, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Ng, W.K.; Shen, S.; Kim, S.; Tan, R.B. Controlled antisolvent precipitation of spironolactone nanoparticles by impingement mixing. Int. J. Pharm. 2011, 410, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, Y.; Tian, Y.; Xu, X.; Chen, Y.; Mu, L.; Zhang, Y.; Fang, L. Preparation and in vitro/in vivo evaluation of revaprazan hydrochloride nanosuspension. Int. J. Pharm. 2011, 408, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Beirowski, J.; Inghelbrecht, S.; Arien, A.; Gieseler, H. Freeze-drying of nanosuspensions, part 3: Investigation of factors compromising storage stability of highly concentrated drug nanosuspensions. J. Pharm. Sci. 2012, 101, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Chung, N.-O.; Lee, M.K.; Lee, J. Mechanism of freeze-drying drug nanosuspensions. Int. J. Pharm. 2012, 437, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-Y.; Park, C.H.; Lee, J. Effect of polymer molecular weight on nanocomminution of poorly soluble drug. Drug Deliv. 2008, 15, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J. Effective polymeric dispersants for vacuum, convection and freeze drying of drug nanosuspensions. Int. J. Pharm. 2010, 397, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Khinast, J.; Baumgartner, R.; Roblegg, E. Nano-extrusion: A one-step process for manufacturing of solid nanoparticle formulations directly from the liquid phase. AAPS PharmSciTech 2013, 14, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, R.; Eitzlmayr, A.; Matsko, N.; Tetyczka, C.; Khinast, J.; Roblegg, E. Nano-extrusion: A promising tool for continuous manufacturing of solid nano-formulations. Int. J. Pharm. 2014, 477, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Patil, H.; Feng, X.; Tiwari, R.V.; Lu, J.; Gryczke, A.; Kolter, K.; Langley, N.; Majumdar, S.; Neupane, D. Conjugation of hot-melt extrusion with high-pressure homogenization: A novel method of continuously preparing nanocrystal solid dispersions. AAPS PharmSciTech 2016, 17, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ioannidis, N.; Gogos, C.; Bilgili, E. A comparative assessment of nanocomposites vs. amorphous solid dispersions prepared via nanoextrusion for drug dissolution enhancement. Eur. J. Pharm. Biopharm. 2017, 119, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Sievens-Figueroa, L.; Bhakay, A.; Jerez-Rozo, J.I.; Pandya, N.; Romañach, R.J.; Michniak-Kohn, B.; Iqbal, Z.; Bilgili, E.; Davé, R.N. Preparation and characterization of hydroxypropyl methyl cellulose films containing stable BCS Class II drug nanoparticles for pharmaceutical applications. Int. J. Pharm. 2012, 423, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Susarla, R.; Afolabi, A.; Patel, D.; Bilgili, E.; Davé, R.N. Novel use of superdisintegrants as viscosity enhancing agents in biocompatible polymer films containing griseofulvin nanoparticles. Powder Technol. 2015, 285, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Krull, S.M.; Patel, H.V.; Li, M.; Bilgili, E.; Davé, R.N. Critical material attributes (CMAs) of strip films loaded with poorly water-soluble drug nanoparticles: I. Impact of plasticizer on film properties and dissolution. Eur. J. Pharm. Sci. 2016, 92, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krull, S.M.; Ammirata, J.; Bawa, S.; Li, M.; Bilgili, E.; Davé, R.N. Critical material attributes of strip films loaded with poorly water-soluble drug nanoparticles: II. Impact of polymer molecular weight. J. Pharm. Sci. 2017, 106, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Krull, S.M.; Moreno, J.; Li, M.; Bilgili, E.; Davé, R.N. Critical material attributes (CMAs) of strip films loaded with poorly water-soluble drug nanoparticles: III. Impact of drug nanoparticle loading. Int. J. Pharm. 2017, 523, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhakay, A. Improved Recovery and Dissolution from Dried Stabilized Aqueous Nanomilled Suspensions of Poorly Water-Soluble Drugs. Ph.D. Thesis, New Jersey Institute of Technology, Newark, NJ, USA, 2013. [Google Scholar]
- Bhakay, A.; Azad, M.; Bilgili, E.; Dave, R. Redispersible fast dissolving nanocomposite microparticles of poorly water-soluble drugs. Int. J. Pharm. 2014, 461, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lopez, N.; Bilgili, E. A study of the impact of polymer–surfactant in drug nanoparticle coated pharmatose composites on dissolution performance. Adv. Powder Technol. 2016, 27, 1625–1636. [Google Scholar] [CrossRef]
- Li, M. Assessment of Nanocomposites Versus Amorphous Solid Dispersions for Dissolution Enhancement of BCS Class II Drugs. Ph.D. Thesis, New Jersey Institute of Technology, Newark, NJ, USA, 2017. [Google Scholar]
- Wang, B.; Zhang, W.; Zhang, W.; Mujumdar, A.S.; Huang, L. Progress in drying technology for nanomaterials. Dry. Technol. 2005, 23, 7–32. [Google Scholar] [CrossRef]
- Vehring, R. Pharmaceutical particle engineering via spray drying. Pharm. Res. 2008, 25, 999–1022. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Kim, M.Y.; Kim, S.; Lee, J. Cryoprotectants for freeze drying of drug nano-suspensions: Effect of freezing rate. J. Pharm. Sci. 2009, 98, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.; Moreno, J.; Bilgili, E.; Davé, R. Fast dissolution of poorly water soluble drugs from fluidized bed coated nanocomposites: Impact of carrier size. Int. J. Pharm. 2016, 513, 319–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhakay, A.; Davé, R.; Bilgili, E. Quiescent and agitated redispersion as a tool for evaluating dispersant effectiveness in dissolution enhancement of drug-laden nanocomposites. AAPS PharmSciTech 2018, 19, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Brough, C.; Williams, R.O. Amorphous solid dispersions and nano-crystal technologies for poorly water-soluble drug delivery. Int. J. Pharm. 2013, 453, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Chogale, M.M.; Ghodake, V.N.; Patravale, V.B. Performance parameters and characterizations of nanocrystals: A brief review. Pharmaceutics 2016, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, L.; Hirvonen, J. Drug nanocrystals—Versatile option for formulation of poorly soluble materials. Int. J. Pharm. 2018, 537, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.; Jacobs, C.; Kayser, O. Nanosuspensions as particulate drug formulations in therapy: Rationale for development and what we can expect for the future. Adv. Drug Deliv. Rev. 2001, 47, 3–19. [Google Scholar] [CrossRef]
- Kwade, A. Wet comminution in stirred media mills—Research and its practical application. Powder Technol. 1999, 105, 14–20. [Google Scholar] [CrossRef]
- Merisko-Liversidge, E.; Liversidge, G.G.; Cooper, E.R. Nanosizing: A formulation approach for poorly-water-soluble compounds. Eur. J. Pharm. Sci. 2003, 18, 113–120. [Google Scholar] [CrossRef]
- Bilgili, E.; Afolabi, A. A combined microhydrodynamics-polymer adsorption analysis for elucidation of the roles of stabilizers in wet stirred media milling. Int. J. Pharm. 2012, 439, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Knieke, C.; Azad, M.; Davé, R.; Bilgili, E. A study of the physical stability of wet media-milled fenofibrate suspensions using dynamic equilibrium curves. Chem. Eng. Res. Des. 2013, 91, 1245–1258. [Google Scholar] [CrossRef]
- Bilgili, E.; Li, M.; Afolabi, A. Is the combination of cellulosic polymers and anionic surfactants a good strategy for ensuring physical stability of BCS Class II drug nanosuspensions? Pharm. Dev. Technol. 2016, 21, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Afolabi, A.; Akinlabi, O.; Bilgili, E. Impact of process parameters on the breakage kinetics of poorly water-soluble drugs during wet stirred media milling: A microhydrodynamic view. Eur. J. Pharm. Sci. 2014, 51, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yaragudi, N.; Afolabi, A.; Dave, R.; Bilgili, E. Sub-100 nm drug particle suspensions prepared via wet milling with low bead contamination through novel process intensification. Chem. Eng. Sci. 2015, 130, 207–220. [Google Scholar] [CrossRef]
- Li, M.; Alvarez, P.; Bilgili, E. A microhydrodynamic rationale for selection of bead size in preparation of drug nanosuspensions via wet stirred media milling. Int. J. Pharm. 2017, 524, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Möschwitzer, J.P. Drug nanocrystals in the commercial pharmaceutical development process. Int. J. Pharm. 2013, 453, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.D.; Shen, C.Y.; Yuan, X.D.; Bai, J.X.; Lv, Q.Y.; Xu, H.; Dai, L.; Yu, C.; Han, J.; Yuan, H.L. Development and characterization of an orodispersible film containing drug nanoparticles. Eur. J. Pharm. Biopharm. 2013, 85, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Ni, R.; Zhang, X.; Li, L.C.; Mao, S. Spray drying of a poorly water-soluble drug nanosuspension for tablet preparation: Formulation and process optimization with bioavailability evaluation. Drug Dev. Ind. Pharm. 2015, 41, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, H.; Tian, B.; Yuan, K.; Pan, H.; Ma, S.; Yang, X.; Pan, W. Fabrication of carvedilol nanosuspensions through the anti-solvent precipitation–ultrasonication method for the improvement of dissolution rate and oral bioavailability. AAPS PharmSciTech 2012, 13, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Sievens-Figueroa, L.; Gärtner, K.; Jerez-Rozo, J.I.; Romañach, R.J.; Bilgili, E.; Davé, R.N. Effects of stabilizers on particle redispersion and dissolution from polymer strip films containing liquid antisolvent precipitated griseofulvin particles. Powder Technol. 2013, 236, 37–51. [Google Scholar] [CrossRef]
- Freag, M.S.; Elnaggar, Y.S.; Abdallah, O.Y. Development of novel polymer-stabilized diosmin nanosuspensions: In vitro appraisal and ex vivo permeation. Int. J. Pharm. 2013, 454, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Elham, G.; Mahsa, P.; Vahid, R. Spray drying of nanoparticles to form fast dissolving glipizide. Asian J. Pharm. 2015, 213–218. [Google Scholar]
- Kumar, V.; Prud’homme, R.K. Nanoparticle stability: Processing pathways for solvent removal. Chem. Eng. Sci. 2009, 64, 1358–1361. [Google Scholar] [CrossRef]
- Fu, Q.; Ma, M.; Li, M.; Wang, G.; Guo, M.; Li, J.; Hou, Y.; Fang, M. Improvement of oral bioavailability for nisoldipine using nanocrystals. Powder Technol. 2017, 305, 757–763. [Google Scholar] [CrossRef]
- Tuomela, A.; Hirvonen, J.; Peltonen, L. Stabilizing agents for drug nanocrystals: Effect on bioavailability. Pharmaceutics 2016, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Van den Mooter, G.; Augustijns, P. Top-down production of drug nanocrystals: Nanosuspension stabilization, miniaturization and transformation into solid products. Int. J. Pharm. 2008, 364, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Merisko-Liversidge, E.; Liversidge, G.G. Nanosizing for oral and parenteral drug delivery: A perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv. Drug Deliv. Rev. 2011, 63, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.; Jacobs, C. Buparvaquone mucoadhesive nanosuspension: Preparation, optimisation and long-term stability. Int. J. Pharm. 2002, 237, 151–161. [Google Scholar] [CrossRef]
- Mishra, P.R.; Al Shaal, L.; Müller, R.H.; Keck, C.M. Production and characterization of hesperetin nanosuspensions for dermal delivery. Int. J. Pharm. 2009, 371, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Vermant, J.; Martens, J.A.; Froyen, L.; Van Humbeeck, J.; Augustijns, P.; Van den Mooter, G. A screening study of surface stabilization during the production of drug nanocrystals. J. Pharm. Sci. 2009, 98, 2091–2103. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.; Lee, J. Redispersible drug nanoparticles prepared without dispersant by electro-spray drying. Drug Dev. Ind. Pharm. 2012, 38, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Cerdeira, A.M.; Mazzotti, M.; Gander, B. Formulation and drying of miconazole and itraconazole nanosuspensions. Int. J. Pharm. 2013, 443, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Alvarez, P.; Orbe, P.; Bilgili, E. Multi-faceted characterization of wet-milled griseofulvin nanosuspensions for elucidation of aggregation state and stabilization mechanisms. AAPS PharmSciTech 2018, 19, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, A.; Laaksonen, T.; Laru, J.; Antikainen, O.; Kiesvaara, J.; Ilkka, J.; Oksala, O.; Rönkkö, S.; Järvinen, K.; Hirvonen, J. Solid formulations by a nanocrystal approach: Critical process parameters regarding scale-ability of nanocrystals for tableting applications. Int. J. Pharm. 2015, 485, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, S.-J.; Choi, J.-Y.; Yoo, J.Y.; Ahn, C.-H. Amphiphilic amino acid copolymers as stabilizers for the preparation of nanocrystal dispersion. Eur. J. Pharm. Sci. 2005, 24, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, J.-Y.; Park, C. Characteristics of polymers enabling nano-comminution of water-insoluble drugs. Int. J. Pharm. 2008, 355, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-Y.; Yoo, J.Y.; Kwak, H.-S.; Nam, B.U.; Lee, J. Role of polymeric stabilizers for drug nanocrystal dispersions. Curr. Appl. Phys. 2005, 5, 472–474. [Google Scholar] [CrossRef]
- George, M.; Ghosh, I. Identifying the correlation between drug/stabilizer properties and critical quality attributes (CQAs) of nanosuspension formulation prepared by wet media milling technology. Eur. J. Pharm. Sci. 2013, 48, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.B.; Kompella, U.B. Nanoparticle Technology for Drug Delivery; Taylor & Francis: New York, NY, USA, 2006. [Google Scholar]
- Zhu, W.; Romanski, F.S.; Dalvi, S.V.; Dave, R.N.; Tomassone, M.S. Atomistic simulations of aqueous griseofulvin crystals in the presence of individual and multiple additives. Chem. Eng. Sci. 2012, 73, 218–230. [Google Scholar] [CrossRef]
- Bose, S.; Schenck, D.; Ghosh, I.; Hollywood, A.; Maulit, E.; Ruegger, C. Application of spray granulation for conversion of a nanosuspension into a dry powder form. Eur. J. Pharm. Sci. 2012, 47, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Susarla, R.; Sievens-Figueroa, L.; Bhakay, A.; Shen, Y.; Jerez-Rozo, J.I.; Engen, W.; Khusid, B.; Bilgili, E.; Romanach, R.J.; Morris, K.R. Fast drying of biocompatible polymer films loaded with poorly water-soluble drug nano-particles via low temperature forced convection. Int. J. Pharm. 2013, 455, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.K.; Yeung, C.F.; Ho, S.W.; Chow, S.F.; Chow, A.H.; Baum, L. Highly stabilized curcumin nanoparticles tested in an in vitro blood–brain barrier model and in alzheimer’s disease Tg2576 mice. AAPS J. 2013, 15, 324–336. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Lu, Y.; Qi, J.; Chen, L.; Yin, L.; Wu, W. Formulating food protein-stabilized indomethacin nanosuspensions into pellets by fluid-bed coating technology: Physical characterization, redispersibility, and dissolution. Int. J. Nanomed. 2013, 8, 3119–3128. [Google Scholar]
- Fu, Q.; Sun, J.; Zhang, D.; Li, M.; Wang, Y.; Ling, G.; Liu, X.; Sun, Y.; Sui, X.; Luo, C. Nimodipine nanocrystals for oral bioavailability improvement: Preparation, characterization and pharmacokinetic studies. Colloids Surf. B Biointerfaces 2013, 109, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Ige, P.P.; Baria, R.K.; Gattani, S.G. Fabrication of fenofibrate nanocrystals by probe sonication method for enhancement of dissolution rate and oral bioavailability. Colloids Surf. B Biointerfaces 2013, 108, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Zuo, B.; Sun, Y.; Li, H.; Liu, X.; Zhai, Y.; Sun, J.; He, Z. Preparation and in vitro/in vivo evaluation of fenofibrate nanocrystals. Int. J. Pharm. 2013, 455, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.-L.; Han, Y.-R.; Quan, L.-H.; Liu, C.-Y.; Liao, Y.-H. Oily nanosuspension for long-acting intramuscular delivery of curcumin didecanoate prodrug: Preparation, characterization and in vivo evaluation. Eur. J. Pharm. Sci. 2013, 49, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Gokhale, R.; Burgess, D.J. Sugars as bulking agents to prevent nano-crystal aggregation during spray or freeze-drying. Int. J. Pharm. 2014, 471, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Xu, X.; Gokhale, R.; Burgess, D.J. Formulation parameters of crystalline nanosuspensions on spray drying processing: A DoE approach. Int. J. Pharm. 2014, 464, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Amsa, P.; Tamizharasi, S.; Jagadeeswaran, M.; Sivakumar, T. Preparation and solid state characterization of simvastatin nanosuspensions for enhanced solubility and dissolution. Int. J. Pharm. Pharm. Sci. 2014, 6, 265–269. [Google Scholar]
- Dong, Y.; Ng, W.K.; Hu, J.; Shen, S.; Tan, R.B. Continuous production of redispersible and rapidly-dissolved fenofibrate nanoformulation by combination of microfluidics and spray drying. Powder Technol. 2014, 268, 424–428. [Google Scholar] [CrossRef]
- Patel, G.V.; Patel, V.B.; Pathak, A.; Rajput, S.J. Nanosuspension of efavirenz for improved oral bioavailability: Formulation optimization, in vitro, in situ and in vivo evaluation. Drug Dev. Ind. Pharm. 2014, 40, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Obeidat, W.M.; Sallam, A.-S.A. Evaluation of tadalafil nanosuspensions and their PEG solid dispersion matrices for enhancing its dissolution properties. AAPS PharmSciTech 2014, 15, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Papdiwal, A.P.; Pande, V.V.; Aher, S. Investigation of effect of different stabilizers on formulation of zaltoprofen nanosuspension. Int. J. Pharm. Sci. Rev. Res. 2014, 27, 244–249. [Google Scholar]
- Homayouni, A.; Sadeghi, F.; Varshosaz, J.; Garekani, H.A.; Nokhodchi, A. Promising dissolution enhancement effect of soluplus on crystallized celecoxib obtained through antisolvent precipitation and high pressure homogenization techniques. Colloids Surf. B Biointerfaces 2014, 122, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, I.; Abdelbary, A.A.; Elshafeey, A.H. Nanosizing of a poorly soluble drug: Technique optimization, factorial analysis, and pharmacokinetic study in healthy human volunteers. Int. J. Nanomed. 2014, 9, 2943–2953. [Google Scholar]
- Yao, Q.; Tao, X.; Tian, B.; Tang, Y.; Shao, Y.; Kou, L.; Gou, J.; Li, X.; Yin, T.; Tang, X. Improved oral bioavailability of core–shell structured beads by redispersion of the shell-forming nanoparticles: Preparation, characterization and in vivo studies. Colloids Surf. B Biointerfaces 2014, 113, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Dhingani, A.; Garala, K.; Raval, M.; Sheth, N. Design and development of solid nanoparticulate dosage forms of telmisartan for bioavailability enhancement by integration of experimental design and principal component analysis. Powder Technol. 2014, 258, 331–343. [Google Scholar] [CrossRef]
- Homayouni, A.; Sadeghi, F.; Varshosaz, J.; Garekani, H.A.; Nokhodchi, A. Comparing various techniques to produce micro/nanoparticles for enhancing the dissolution of celecoxib containing PVP. Eur. J. Pharm. Biopharm. 2014, 88, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Bhalekar, M.R.; Upadhaya, P.G.; Reddy, S.; Kshirsagar, S.J.; Madgulkar, A.R. Formulation and evaluation of acyclovir nanosuspension for enhancement of oral bioavailability. Asian J. Pharm. 2014, 8, 110–118. [Google Scholar] [CrossRef]
- Kumar, S.; Jog, R.; Shen, J.; Zolnik, B.; Sadrieh, N.; Burgess, D.J. In vitro and in vivo performance of different sized spray-dried crystalline itraconazole. J. Pharm. Sci. 2015, 104, 3018–3028. [Google Scholar] [CrossRef] [PubMed]
- Knieke, C.; Azad, M.A.; To, D.; Bilgili, E.; Davé, R.N. Sub-100 micron fast dissolving nanocomposite drug powders. Powder Technol. 2015, 271, 49–60. [Google Scholar] [CrossRef]
- Krull, S.M.; Susarla, R.; Afolabi, A.; Li, M.; Ying, Y.; Iqbal, Z.; Bilgili, E.; Davé, R.N. Polymer strip films as a robust, surfactant-free platform for delivery of BCS Class II drug nanoparticles. Int. J. Pharm. 2015, 489, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Shen, J.; Zolnik, B.; Sadrieh, N.; Burgess, D.J. Optimization and dissolution performance of spray-dried naproxen nano-crystals. Int. J. Pharm. 2015, 486, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Wang, C.; Dan, J.; Liu, W.; Wu, Z.; Yang, M. The importance of solidification stress on the redispersibility of solid nanocrystals loaded with harmine. Int. J. Pharm. 2015, 480, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.-Q.; Du, X.-Y.; Huang, X.-N.; Qiao, B. Enhanced oral bioavailability of ursolic acid nanoparticles via antisolvent precipitation with TPGS1000 as a stabilizer. J. Drug Deliv. Sci. Technol. 2015, 29, 210–217. [Google Scholar] [CrossRef]
- Malamatari, M.; Somavarapu, S.; Bloxham, M.; Buckton, G. Nanoparticle agglomerates of indomethacin: The role of poloxamers and matrix former on their dissolution and aerosolisation efficiency. Int. J. Pharm. 2015, 495, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Sahoo, J.; Dixit, P.K. Formulation and process optimization of naproxen nanosuspensions stabilized by hydroxypropyl methylcellulose. Carbohydr. Polym. 2015, 127, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Franceschini, I.; Corrias, F.; Sala, M.C.; Cilurzo, F.; Sinico, C.; Pini, E. Maltodextrin fast dissolving films for quercetin nanocrystal delivery. A feasibility study. Carbohydr. Polym. 2015, 121, 217–223. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Yang, H.; Zhang, R.; Li, Y.; Duan, L. Preparation and in vitro–in vivo evaluation of teniposide nanosuspensions. Int. J. Pharm. 2015, 478, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Cui, B.; Zeng, Z.; Feng, L.; Liu, G.; Cui, H.; Pan, H. Lambda-cyhalothrin nanosuspension prepared by the melt emulsification-high pressure homogenization method. J. Nanomater. 2015, 16, 263–270. [Google Scholar] [CrossRef]
- Krull, S.M.; Ma, Z.; Li, M.; Davé, R.N.; Bilgili, E. Preparation and characterization of fast dissolving pullulan films containing BCS class II drug nanoparticles for bioavailability enhancement. Drug Dev. Ind. Pharm. 2016, 42, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Bonda, A.F.; Rinaldi, M.; Segale, L.; Palugan, L.; Cerea, M.; Vecchio, C.; Pattarino, F. Nanonized itraconazole powders for extemporary oral suspensions: Role of formulation components studied by a mixture design. Eur. J. Pharm. Sci. 2016, 83, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Paredes, A.J.; Llabot, J.M.; Sánchez Bruni, S.; Allemandi, D.; Palma, S.D. Self-dispersible nanocrystals of albendazole produced by high-pressure homogenization and spray-drying. Drug Dev. Ind. Pharm. 2016, 42, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-Q.; Zhang, Z.-Z.; Li, G.; Zhang, J.; Xiao, H.-Y.; Li, X.-F. Solidification drug nanosuspensions into nanocrystals by freeze-drying: A case study with ursodeoxycholic acid. Pharm. Dev. Technol. 2016, 21, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, X.; Zhang, C.; Pan, W.; Liang, Y.; Chen, Y.; Chen, W.; Liu, L.; Wang, X. Nanosuspensions as delivery system for gambogenic acid: Characterization and in vitro/in vivo evaluation. Drug Deliv. 2016, 23, 2772–2779. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.; Ma, Y.; Yue, P.; Xie, Y.; Zheng, Q.; Hu, P.; Zhu, W.; Yang, M. Microcrystalline cellulose-carboxymethyl cellulose sodium as an effective dispersant for drug nanocrystals: A case study. Carbohydr. Polym. 2016, 136, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Ma, L.; Yu, X.; Li, Z.; Guo, Y.; Wang, X. A nanoparticulate drug-delivery system for 20 (S)-protopanaxadiol: Formulation, characterization, increased oral bioavailability and anti-tumor efficacy. Drug Deliv. 2016, 23, 2410–2418. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.A.; Patel, M.; Murdande, S.B.; Dave, R.H. Influence of spray drying and dispersing agent on surface and dissolution properties of griseofulvin micro and nanocrystals. Drug Dev. Ind. Pharm. 2016, 42, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Alaei, S.; Ghasemian, E.; Vatanara, A. Spray drying of cefixime nanosuspension to form stabilized and fast dissolving powder. Powder Technol. 2016, 288, 241–248. [Google Scholar] [CrossRef]
- Steiner, D.; Finke, J.H.; Kwade, A. Efficient production of nanoparticle-loaded orodispersible films by process integration in a stirred media mill. Int. J. Pharm. 2016, 511, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Taneja, S.; Shilpi, S.; Khatri, K. Formulation and optimization of efavirenz nanosuspensions using the precipitation-ultrasonication technique for solubility enhancement. Artif. Cells Nanomed. Biotechnol. 2016, 44, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Cheng, L.; Wang, L.Q.; Zhang, L.H.; Shen, B.D.; Liao, W.B.; Li, J.J.; Zheng, J.; Xu, R.; Yuan, H.L. Formulation of dried lignans nanosuspension with high redispersibility to enhance stability, dissolution, and oral bioavailability. Chin. J. Nat. Med. 2016, 14, 757–768. [Google Scholar] [CrossRef]
- Xie, Y.; Ma, Y.; Xu, J.; Dan, J.; Yue, P.; Wu, Z.; Yang, M.; Zheng, Q. Roles of cryo/thermal strength for redispersibility of drug nanocrystals: A representative study with andrographolide. Arch. Pharm. Res. 2016, 39, 1404–1417. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Sahoo, J.; Dixit, P.K. Enhanced bioavailability of cinnarizine nanosuspensions by particle size engineering: Optimization and physicochemical investigations. Mater. Sci. Eng. C 2016, 63, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Karakucuk, A.; Celebi, N.; Teksin, Z.S. Preparation of ritonavir nanosuspensions by microfluidization using polymeric stabilizers: I. A design of experiment approach. Eur. J. Pharm. Sci. 2016, 95, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.J.; Boeck, G. Development of a nanosuspension for iv administration: From miniscale screening to a freeze dried formulation. Eur. J. Pharm. Sci. 2016, 87, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Chonkar, A.D.; Rao, J.V.; Managuli, R.S.; Mutalik, S.; Dengale, S.; Jain, P.; Udupa, N. Development of fast dissolving oral films containing lercanidipine HCl nanoparticles in semicrystalline polymeric matrix for enhanced dissolution and ex vivo permeation. Eur. J. Pharm. Biopharm. 2016, 103, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.; Li, M.; Guo, M.; Yang, W.; Wang, Y.; Li, J.; Fu, Q.; He, Z. Spironolactone nanocrystals for oral administration: Different pharmacokinetic performances induced by stabilizers. Colloids Surf. B Biointerfaces 2016, 147, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Toziopoulou, F.; Malamatari, M.; Nikolakakis, I.; Kachrimanis, K. Production of aprepitant nanocrystals by wet media milling and subsequent solidification. Int. J. Pharm. 2017, 533, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, W.; Zhao, Z.; Dong, Q.; Yin, L.; Zhou, J.; Ding, Y. Lyophilized nanosuspensions for oral bioavailability improvement of insoluble drugs: Preparation, characterization, and pharmacokinetic studies. J. Pharm. Innov. 2017, 12, 271–280. [Google Scholar] [CrossRef]
- Colombo, M.; Orthmann, S.; Bellini, M.; Staufenbiel, S.; Bodmeier, R. Influence of drug brittleness, nanomilling time, and freeze-drying on the crystallinity of poorly water-soluble drugs and its implications for solubility enhancement. AAPS PharmSciTech 2017, 18, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.N.; Clasen, C.; Van den Mooter, G. Encapsulating darunavir nanocrystals within Eudragit L100 using coaxial electrospraying. Eur. J. Pharm. Biopharm. 2017, 113, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Sartori, G.J.; Prado, L.D.; Rocha, H.V.A. Efavirenz dissolution enhancement IV—Antisolvent nanocrystallization by sonication, physical stability, and dissolution. AAPS PharmSciTech 2017, 18, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Shao, J.; Fu, Q.; Li, J.; Sun, J.; He, Z. Spray-dried nanocrystals for a highly hydrophobic drug: Increased drug loading, enhanced redispersity, and improved oral bioavailability. Int. J. Pharm. 2017, 516, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.; Xu, J.; Xie, Y.; Zheng, Q.; Yue, P.; Yang, M. A natural triterpenoid saponin as multifunctional stabilizer for drug nanosuspension powder. AAPS PharmSciTech 2017, 18, 2744–2753. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.; Finke, J.H.; Kwade, A. Redispersion of nanoparticle-loaded orodispersible films: Preservation of particle fineness. Chem. Ing. Tech. 2017, 89, 1034–1040. [Google Scholar] [CrossRef]
- Xu, J.; Ma, Y.; Xie, Y.; Chen, Y.; Liu, Y.; Yue, P.; Yang, M. Design and evaluation of novel solid self-nanodispersion delivery system for andrographolide. AAPS PharmSciTech 2017, 18, 1572–1584. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hong, J.; Di, J.; Guo, Y.; Han, M.; Liu, M.; Wang, X. 10-Hydroxycamptothecin (HCPT) nanosuspensions stabilized by mPEG1000-HCPT conjugate: High stabilizing efficiency and improved antitumor efficacy. Int. J. Nanomed. 2017, 12, 3681–3695. [Google Scholar] [CrossRef] [PubMed]
- Iurian, S.; Bogdan, C.; Tomuță, I.; Szabó-Révész, P.; Chvatal, A.; Leucuța, S.E.; Moldovan, M.; Ambrus, R. Development of oral lyophilisates containing meloxicam nanocrystals using QbD approach. Eur. J. Pharm. Sci. 2017, 104, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Chang, D.; Zhang, X.; Sui, H.; Kong, Y.; Zhu, R.; Wang, W. Oral fast-dissolving films containing lutein nanocrystals for improved bioavailability: Formulation development, in vitro and in vivo evaluation. AAPS PharmSciTech 2017, 18, 2957–2964. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.S. Development and characterization of oral disintegrating tablet containing nanosuspension of lurasidone hydrochloride antipsychotic drug. Asian J. Pharm. 2017, 11, 102–111. [Google Scholar]
- Konnerth, C.; Braig, V.; Ito, A.; Schmidt, J.; Lee, G.; Peukert, W. Formation of mefenamic acid nanocrystals with improved dissolution characteristics. Chem. Ing. Tech. 2017, 89, 1060–1071. [Google Scholar] [CrossRef]
- Geng, T.; Banerjee, P.; Lu, Z.; Zoghbi, A.; Li, T.; Wang, B. Comparative study on stabilizing ability of food protein, non-ionic surfactant and anionic surfactant on BCS type II drug carvedilol loaded nanosuspension: Physicochemical and pharmacokinetic investigation. Eur. J. Pharm. Sci. 2017, 109, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Lyophilization and development of solid protein pharmaceuticals. Int. J. Pharm. 2000, 203, 1–60. [Google Scholar] [CrossRef]
- Tang, X.C.; Pikal, M.J. Design of freeze-drying processes for pharmaceuticals: Practical advice. Pharm. Res. 2004, 21, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahed, W.; Degobert, G.; Fessi, H. Investigation of nanocapsules stabilization by amorphous excipients during freeze-drying and storage. Eur. J. Pharm. Biopharm. 2006, 63, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Masters, K. Spray Drying Handbook; Halsted Press: New York, NY, USA, 1985. [Google Scholar]
- Davis, M. Recent strategies in spray drying for the enhanced bioavailability of poorly water-soluble drugs. J. Control. Release 2018, 269, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.B.; Patel, J.K.; Chakraborty, S.; Shukla, D. Revealing facts behind spray dried solid dispersion technology used for solubility enhancement. Saudi Pharm. J. 2015, 23, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Azad, M. Dissolution Enhanced Nanocomposites from Stabilized Suspensions of Poorly Water-Soluble Drugs. Ph.D. Thesis, New Jersey Institute of Technology, Newark, NJ, USA, 2013. [Google Scholar]
- Dixit, R.; Puthli, S. Oral strip technology: Overview and future potential. J. Control. Release 2009, 139, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Krull, S.M. Formulation and Dissolution of Polymer Strip Films for the Delivery of Poorly Water-Soluble Drug Nanoparticles. Ph.D. Thesis, New Jersey Institute of Technology, Newark, NJ, USA, 2017. [Google Scholar]
- Maury, M.; Murphy, K.; Kumar, S.; Shi, L.; Lee, G. Effects of process variables on the powder yield of spray-dried trehalose on a laboratory spray-dryer. Eur. J. Pharm. Biopharm. 2005, 59, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Bilgili, E.; Rosen, L.A.; Ko, J.S.; Chen, A.; Smith, E.J.; Fliszar, K.; Wong, G. Experimental study of fluidized bed co-granulation of two active pharmaceutical ingredients: An industrial scale-up perspective. Part. Sci. Technol. 2011, 29, 285–309. [Google Scholar] [CrossRef]
- El-Meliegy, E.; Mabrouk, M.; Kamal, G.M.; Awad, S.M.; El-Tohamy, A.M.; El Gohary, M.I. Anticancer drug carriers using dicalcium phosphate/dextran/CMCnanocomposite scaffolds. J. Drug Deliv. Sci. Technol. 2018, 45, 315–322. [Google Scholar] [CrossRef]
- Grigorov, P.I.; Glasser, B.J.; Muzzio, F.J. Cross-sectional analysis of impregnated excipient particles by energy dispersive X-ray spectroscopy. Powder Technol. 2018, 332, 197–209. [Google Scholar] [CrossRef]
- Parmentier, J.; Tan, E.H.; Low, A.; Möschwitzer, J.P. Downstream drug product processing of itraconazole nanosuspension: Factors influencing drug particle size and dissolution from nanosuspension-layered beads. Int. J. Pharm. 2017, 524, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Nekkanti, V.; Pillai, R.; Venkateshwarlu, V.; Harisudhan, T. Development and characterization of solid oral dosage form incorporating candesartan nanoparticles. Pharm. Dev. Technol. 2009, 14, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Pinal, R.; Carvajal, M.T. Process induced disorder in crystalline materials: Differentiating defective crystals from the amorphous form of griseofulvin. J. Pharm. Sci. 2008, 97, 3207–3221. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Xu, S.; Li, S. Understanding a relaxation behavior in a nanoparticle suspension for drug delivery. Int. J. Pharm. 2008, 351, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.; Afolabi, A.; Bhakay, A.; Leonardi, J.; Davé, R.; Bilgili, E. Enhanced physical stabilization of fenofibrate nanosuspensions via wet co-milling with a superdisintegrant and an adsorbing polymer. Eur. J. Pharm. Biopharm. 2015, 94, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Sofie, V.; Martens, J.A.; Jan, V.; Ludo, F.; Jan, V.H.; Van Den Mooter, G.; Patrick, A. Microcrystalline cellulose, a useful alternative for sucrose as a matrix former during freeze-drying of drug nanosuspensions—A case study with itraconazole. Eur. J. Pharm. Biopharm. 2008, 70, 590–596. [Google Scholar] [PubMed]
- Pu, X.; Sun, J.; Wang, Y.; Wang, Y.; Liu, X.; Zhang, P.; Tang, X.; Pan, W.; Han, J.; He, Z. Development of a chemically stable 10-hydroxycamptothecin nanosuspensions. Int. J. Pharm. 2009, 379, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Liu, X.; Guo, Y.; Wang, Y.; Wang, X. Preparation, characterization, biodistribution and antitumor efficacy of hydroxycamptothecin nanosuspensions. Int. J. Pharm. 2013, 455, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C.; Mehnert, W. Freeze-drying of drug-free and drug-loaded solid lipid nanoparticles (SLN). Int. J. Pharm. 1997, 157, 171–179. [Google Scholar] [CrossRef]
- Konan, Y.N.; Gurny, R.; Allémann, E. Preparation and characterization of sterile and freeze-dried sub-200 nm nanoparticles. Int. J. Pharm. 2002, 233, 239–252. [Google Scholar] [CrossRef]
- Stamm, A.; Seth, P. Fenofibrate Pharmaceutical Composition Having High Bioavailability and Method for Preparing It. U.S. Patent 6,074,670, 13 June 2000. [Google Scholar]
- Biradar, S.V.; Patil, A.R.; Sudarsan, G.V.; Pokharkar, V.B. A comparative study of approaches used to improve solubility of roxithromycin. Powder Technol. 2006, 169, 22–32. [Google Scholar] [CrossRef]
- Li, C.; Li, C.; Le, Y.; Chen, J.-F. Formation of bicalutamide nanodispersion for dissolution rate enhancement. Int. J. Pharm. 2011, 404, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Froyen, L.; Martens, J.; Blaton, N.; Augustijns, P.; Brewster, M.; Van den Mooter, G. Characterization of physico-chemical properties and pharmaceutical performance of sucrose co-freeze–dried solid nanoparticulate powders of the anti-HIV agent loviride prepared by media milling. Int. J. Pharm. 2007, 338, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Chaubal, M.V.; Popescu, C. Conversion of nanosuspensions into dry powders by spray drying: A case study. Pharm. Res. 2008, 25, 2302–2308. [Google Scholar] [CrossRef] [PubMed]
- Layre, A.-M.; Couvreur, P.; Richard, J.; Requier, D.; Eddine Ghermani, N.; Gref, R. Freeze-drying of composite core-shell nanoparticles. Drug Dev. Ind. Pharm. 2006, 32, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; De Wulf, O.; Laru, J.; Heikkilä, T.; van Veen, B.; Kiesvaara, J.; Hirvonen, J.; Peltonen, L.; Laaksonen, T. Dissolution studies of poorly soluble drug nanosuspensions in non-sink conditions. AAPS PharmSciTech 2013, 14, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Staufenbiel, S.; Hao, S.; Wang, B.; Dashevskiy, A.; Bodmeier, R. Development of a discriminative biphasic in vitro dissolution test and correlation with in vivo pharmacokinetic studies for differently formulated racecadotril granules. J. Control. Release 2017, 255, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Talukder, R.; Reed, C.; Dürig, T.; Hussain, M. Dissolution and solid-state characterization of poorly water-soluble drugs in the presence of a hydrophilic carrier. AAPS PharmSciTech 2011, 12, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Washburn, E.W. The dynamics of capillary flow. Phys. Rev. 1921, 17, 273–283. [Google Scholar] [CrossRef]
- Hołownia, D.; Kwiatkowska, I.; Hupka, J. An investigation on wetting of porous materials. Physicochem. Prob. Miner. Process. 2008, 42, 251–262. [Google Scholar]
- Yalkowsky, S.H.; Roseman, T.J. Techniques of Solubilization of Drugs; M. Dekker: New York, NY, USA, 1981. [Google Scholar]
- Liversidge, G.G.; Cundy, K.C. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int. J. Pharm. 1995, 125, 91–97. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharm. 2013, 2013, 848043. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Machida, M.; Adachi, K.; Otabe, K.; Sugimoto, T.; Hayashi, M.; Awazu, S. Histopathological study of the effects of a single intratracheal instillation of surface active agents on lung in rats. J. Toxicol. Sci. 2000, 25, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Brugger, A.; Khare, A.; Chaubal, M.; Papadopoulos, P.; Rabinow, B.; Kipp, J.; Ning, J. Suspensions for intravenous (IV) injection: A review of development, preclinical and clinical aspects. Adv. Drug Deliv. Rev. 2008, 60, 939–954. [Google Scholar] [CrossRef] [PubMed]
- Lebhardt, T.; Roesler, S.; Uusitalo, H.P.; Kissel, T. Surfactant-free redispersible nanoparticles in fast-dissolving composite microcarriers for dry-powder inhalation. Eur. J. Pharm. Biopharm. 2011, 78, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; Royal Pharmaceutical Society: London, UK, 2009. [Google Scholar]
- U.S. Food and Drug Administration (FDA). Database for Inactive Ingredient Search for Approv-ed Drug Products. 2018. Available online: https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm?event=BasicSearch.page (accessed on 28 June 2018).
- U.S. Food and Drug Administration (FDA). Guidance for Industry: Nonclinical Studies for the Safety Evaluation of Pharmaceutical Excipients; Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER): Rockville, MD, USA, 2005. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM079250.pdf (accessed on 28 June 2018).
- Leung, D.H.; Lamberto, D.J.; Liu, L.; Kwong, E.; Nelson, T.; Rhodes, T.; Bak, A. A new and improved method for the preparation of drug nanosuspension formulations using acoustic mixing technology. Int. J. Pharm. 2014, 473, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, L.; Davé, R.N.; Bilgili, E. An intensified vibratory milling process for enhancing the breakage kinetics during the preparation of drug nanosuspensions. AAPS PharmSciTech 2016, 17, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahed, W.; Degobert, G.; Stainmesse, S.; Fessi, H. Freeze-drying of nanoparticles: Formulation, process and storage considerations. Adv. Drug Deliv. Rev. 2006, 58, 1688–1713. [Google Scholar] [CrossRef] [PubMed]
- Bilgili, E.; Dave, R.; Bhakay, A.; Azad, M. Systems and Methods for Superdisintegrant-Based Composite Particles for Dispersion and Dissolution of Agents. U.S. Patent 9,452,107B2, 27 September 2016. [Google Scholar]
- Li, M.; Dave, R.; Bilgili, E. High drug-loaded surfactant-free nanocomposite microparticles for enhanced dissolution of poorly soluble drugs. In Proceedings of the 8th World Congress on Particle Technology, Orlando, FL, USA, 22–26 April 2018. [Google Scholar]
- Dolenc, A.; Kristl, J.; Baumgartner, S.; Planinšek, O. Advantages of celecoxib nanosuspension formulation and transformation into tablets. Int. J. Pharm. 2009, 376, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Mauludin, R.; Müller, R.H.; Keck, C.M. Development of an oral rutin nanocrystal formulation. Int. J. Pharm. 2009, 370, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Cal, K.; Sollohub, K. Spray drying technique. I: Hardware and process parameters. J. Pharm. Sci. 2010, 99, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Walters, R.H.; Bhatnagar, B.; Tchessalov, S.; Izutsu, K.-I.; Tsumoto, K.; Ohtake, S. Next generation drying technologies for pharmaceutical applications. J. Pharm. Sci. 2014, 103, 2673–2695. [Google Scholar] [CrossRef] [PubMed]
- Mujumdar, A.; Alterman, D. Drying in the pharmaceutical and biotechnology fields. In Handbook of Downstream Processing; Springer: Dordrecht, The Netherlands, 1997; pp. 235–260. [Google Scholar]
- Möschwitzer, J.P.; Müller, R.H. Factors influencing the release kinetics of drug nanocrystal-loaded pellet formulations. Drug Dev. Ind. Pharm. 2013, 39, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Schwartzbach, H. The Possibilities and Challenges of Spray Drying; Pharmaceutical Technology Europe: London, UK, 2010. [Google Scholar]
- Teunou, E.; Poncelet, D. Batch and continuous fluid bed coating–review and state of the art. J. Food Eng. 2002, 53, 325–340. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Mitra, A. Crystalline nanosuspensions as potential toxicology and clinical oral formulations for BCS II/IV compounds. AAPS J. 2012, 14, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.M.H.; Ullah, F.; Khan, S.; Shah, S.M.M.; de Matas, M.; Hussain, Z.; Minhas, M.U.; AbdEl-Salam, N.M.; Assi, K.H.; Isreb, M. Smart nanocrystals of artemether: Fabrication, characterization, and comparative in vitro and in vivo antimalarial evaluation. Drug Des. Dev. Ther. 2016, 10, 3837–3850. [Google Scholar] [CrossRef] [PubMed]
- Rahim, H.; Sadiq, A.; Khan, S.; Khan, M.A.; Shah, S.M.H.; Hussain, Z.; Ullah, R.; Shahat, A.A.; Ibrahim, K. Aceclofenac nanocrystals with enhanced in vitro, in vivo performance: Formulation optimization, characterization, analgesic and acute toxicity studies. Drug Des. Dev. Ther. 2017, 11, 2443–2452. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name/Company | Drug | Nanoparticle Preparation Method a | Final Dosage | Year Approved |
|---|---|---|---|---|
| Avinza®/King Pharma | Morphine sulfate | WMM | Capsule | 2002 |
| Azopt®/Alcon | Brinzolmid | WMM | Suspension | 1998 |
| Cesamet®/Lilly | Nabilon | Precipitation | Capsule | 2005 |
| Emend®/Merck | Aprepitant | WMM | Capsule | 2003 |
| Focalin XR®/Novartis | Dexmethylphenidate HCl | WMM | Capsule | 2001 |
| Gris-Peg®/Novartis | Griseofulvin | Precipitation | Tablet | 1982 |
| Herbesser®/Mitsubishi | Diltiazem | WMM | Tablet | 2002 |
| Invega Sustenna®/Johnson & Johnson | Paliperidone palmitate | WMM | Suspension | 2009 |
| Megace ES®/Par Pharmaceutical | Megestrol acetate | WMM | Suspension | 2005 |
| Neprelan®/Wyeth | Naproxen sodium | WMM | Tablet | 2006 |
| Rapamune®/Wyeth | Sirolimus (rapamycin) | WMM | Suspension, Tablet | 2000 |
| Ritalin LA®/Novartis | Methylphenidate HCl | WMM | Capsule | 2002 |
| Theodur®/Mitsubishi Tanabe Pharma | Theophylline | WMM | Tablet, Capsule | 2008 |
| Tricor®/Abbott | Fenofibrate | WMM | Tablet | 2004 |
| Triglide®/SkyePharma | Fenofibrate | HPH | Tablet | 2005 |
| Verelan PM®/Schwarz Pharma | Verapamil HCl | WMM | Capsule | 1998 |
| Zanaflex®/Acorda | Tizanidine HCl | WMM | Capsule | 2002 |
| Method for Drug Nanosuspension Preparation | Drying Method | Drug and Its Assay in Nanocomposites (% w/w) | Dispersants in Nanocomposites a,b | Redispersion Method (If Used) | d50c, dvm d, Cumulant e Size Before and After Redispersion (µm) | Reference | |
|---|---|---|---|---|---|---|---|
| Before | After | ||||||
| LASP–Ultrasonication | Freeze drying | Carvedilol (–) | (Alpha tocopherol succinate, SDS, Maltose) | _ | 0.212 | _f | Liu et al., (2012) [108] |
| WMM | Freeze drying | Naproxen (–) | (HPC, PEG Carrageenan), Sucrose | Dried powders were dispersed in 150 mL water and sonicated for 1 min | 0.148 d | 0.150 d,g | Chung et al., (2012) [72] |
| WMM | Freeze drying | Model drug (–) | (Poloxamer 338, PVP K15), Cremophor EL Sucrose, Trehalose | An aqueous solution of 5 mg/mL poloxamer 338 was used as a medium | _ | 0.165 g | Beirowski et al., (2012) [71] |
| WMM | Wet film casting–drying | Griseofulvin (3.8 mg/cm2) Naproxen (3.3 mg/cm2) Fenofibrate (4.8 mg/cm2) | (HPMC E15LV, SDS, Glycerin) | 0.71 cm2 circular films were put in 15 mL water and stirred for 10 min via magnetic stirrer | 0.163 0.144 0.207 | 0.175 g 0.145 g 0.256 f | Sievens et al., (2012) [79] |
| WMM | Electrospray drying | Naproxen (–) | (HPC) | Dried powders were placed in 150 mL water and sonicated for 4 min | ~0.110 d | 0.100 d,g | Ho and Lee (2012) [120] |
| WMM | Fluid bed granulation/drying | Compound A (9.19%) | (Vitamin E TPGS, HPMC 3, Mannitol DC), Lactose monohydrate | _ | ~0.220 | _ f | Bose et al., (2012) [130] |
| WMM | Fluid bed coating/drying | Griseofulvin (12.4%) | (HPC SL, SDS), Mannitol, Pharmatose (core) | 1 g dried sample was dispersed in 30 mL water for 2 min using paddle stirring (200 rpm), pipette stirring, magnetic stirring (100 rpm) and sonication | 0.145 | 0.150 f | Bhakay et al., (2013) [66] |
| LASP–Ultrasonication | Wet film casting–drying | Griseofulvin (3.95%) | (HPMC E15LV, HPMC E4M, Pluronic F127, Glycerin) | Dried films were dispersed in water | 0.580 | ~2.000 f | Beck et al., (2013) [109] |
| WMM | Spray drying | Miconazole (45%) Itraconazole (44%) | (HPC, SDS, Mannitol), MCC | Dried samples were dispersed in water and shaken manually | 0.157 d 0.144 d | ~0.200 d,f ~0.150 d,f | Cerdeira et al., (2013) [121] |
| Freeze drying | Miconazole (47%) Itraconazole (42%) | (HPC, SDS, Mannitol), MCC | Dried samples were dispersed in water and shaken manually | 0.182 d 0.192 d | ~0.198 d,f ~0.200 d | ||
| WMM | Wet film casting–drying | Griseofulvin (1.87 mg/cm2) | (SDS, HPMC E15, Glycerin) | 0.715 cm2 circular films were put in 15 mL water and stirred for 10 min using magnetic stirrer | 0.163 | 0.164 f | Susarla et al., (2013) [131] |
| WMM | Spray-freeze drying | Phenytoin (–) | (PVP, SLS) | Powders equivalent to 2 mg phenytoin were dispersed in 10 mL of dissolution media (pH 1.2 and 6.8) and stirred up to 30 min via magnetic stirrer | 0.170–0.180 | ~0.400 f | Niwa et al., (2013) [7] |
| LASP | Freeze drying | Curcumin (37.6%) | (PEG-PLA, PVP BP, HPBCD) | Dried samples were dispersed in DI-water | 0.055 e | 0.076 e,g | Cheng et al., (2013) [132] |
| LASP–Ultrasonication | Fluid bed coating/drying | Indomethacin (–) | (β-lactoglobulin, PVP K30, Trehalose), Nonpareil (core), Soybean Protein Isolate, Whey protein isolate | 100 mg dried product was dispersed in 10 mL DI-water via manual shaking for 1 min | 0.243 e | 0.289 e,f | He et al., (2013) [133] |
| Acid-base neutralization | Spray drying | Diosmin (–) | (HPMC, Mannitol), MC | Dried powders were dispersed in distilled water | 0.336 e | 0.316 e,f | Freag et al., (2013) [110] |
| LASP–HPH | Freeze drying | Nimodipine (–) | (Poloxamer 407, Sodium deoxycholate, HPMC E5, Mannitol, Maltose) | Beckmann Coulter LS 230 | 0.159 d | 0.148 d,f | Fu et al., (2013) [134] |
| Ultrasonication | Freeze drying | Fenofibrate (–) | (Poloxamer 188, Mannitol), PVP K25, Poloxamer 407, SDS, Tween 80 | _ | 0.460 e | _ f | Ige et al., (2013) [135] |
| WMM | Spray drying | Fenofibrate (–) | (HPMC E5, SDS, Mannitol), Sucrose, Glucose, Maltose, Lactose | 20 mg dry powder was added in 5 mL of DI water and shaken manually | 0.452 | 0.499 f | Zuo et al., (2013) [136] |
| Ultrasonication | Nanoextrusion | TiO2 (–) | (Citric acid monohydrate, SDS, Soluplus), Tween60, Cremophor EL, Cremophor RH 40 | _ | ~0.182 e | _ g | Khinast et al., (2013) [75] |
| HPH | Wet film casting–drying | Herpetrione (10 mg/4 cm2) | (PVP K30, SDS, L-HPC, HPMC E50, MCC, PEG 400, Mannitol) | 2 × 2 cm2 film was placed into distilled water and manually shaken for 30 s | 0.260 e | 0.280 e,f | Shen et al., (2013) [106] |
| WMM | Freeze drying | Curcumin didecanoate (–) | (Poloxamer 188) | 2 mg powder was suspended in peanut oil and sonicated for 1 min | ~0.500 e | 0.517 e,g | Wei et al., (2013) [137] |
| WMM | Spray drying | Naproxen (–) Indomethacin (–) | (HPMC E15), Dowfax 2A 1(Dowfax 2A1), HPMC E15 | Powders were suspended in saturated and filtered solution of the drug in 30% glycerin solution | 0.309 e 0.223 e | 0.400 e,f 0.351 e,f | Kumar et al., (2014a) [138] |
| WMM | Spray drying | Indomethacin (–) | (Dowfax 2A1, Maltose), Trehalose, Lactose, Mannitol, Ficoll PM70, Maltodextrin | Powders were suspended in saturated and filtered solution of indomethacin in 30% glycerin solution | 0.200–0.300 e | 0.179 e,g | Kumar et al., (2014b) [139] |
| Freeze drying | Indomethacin (–) | (Dowfax 2A1, Sucrose), Trehalose, Lactose, Mannitol, Ficoll PM70, Maltodextrin | 0.197 e | 0.208 e,g | |||
| HPH | Freeze drying | Simvastatin (–) | (Soya Lecithin, Mannitol) | _ | 0.316 e | _ f | Asma et al., (2014) [140] |
| WMM | Nanoextrusion | Phenytoin (–) | (Tween 80, Soluplus), Tween 20, Kolliphor P188, Kollicoat IR | _ | 0.335 e | _ f | Baumgartner et al., (2014) [76] |
| LASP | Spray drying | Fenofibrate (–) | (PVP 10, MMT), Lactose | _ | <1.00 e | _ f | Dong et al., (2014) [141] |
| WMM | Freeze drying | Efavirenz (–) | (PVP K30, SLS, Trehalose), Mannitol, Poloxamer 188 and 407 | _ | 0.320 e | _ f | Patel et al., (2014) [142] |
| HPH | Spray drying | Lovastatin (–) | (PVP K17, SDS), HPMC 2910, Polyvinyl alcohol, PVP K30 and K12, Poloxamer 188 and 407 | Dried powders were dispersed in distilled water and shaken manually for 10 s RDI = mean redispersion size/nanosuspesion size ×100 | 0.380 e | 110% f | Zhang et al., (2014) [26] |
| LASP–Ultrasonication | Freeze drying | Tadalafil (–) | (Tween 80, Span80), SLS PEG 4000, PVP K30, Pluronic F-127, Methocel E50 and E5, Span 20 and 60 | _ | 0.193 e | _ f | Obeidat et al., (2014) [143] |
| LASP–Ultrasonication | Freeze drying | Zaltoprofen (–) | (Poloxamer 188, SLS), Poloxamer 407 | Dried samples were diluted with 20 mL distilled water to observe redispersibility | 0.179 e | _ f | Papdiwal et al., (2014) [144] |
| Freeze drying | Quercetin dehydrate (–) | (Tween 80) | _ | ~0.430 e | _ f | ||
| LASP | Freeze drying | Celecoxib (–) | (Soluplus) | _ | 0.293 e | _ f | Homayouni et al., (2014) [145] |
| LASP–HPH | Freeze drying | Celecoxib (–) | (Soluplus) | _ | 0.577 e | _ f | |
| LASP–Ultrasonication–HPH | Freeze drying | Diacerein (–) | (SDS, Mannitol), PVA, Sodium deoxycholate, Sucrose | Malvern Zetasizer® Nano ZS90 | 0.374 e | 0.374 e,f | Elsayed et al., (2014) [146] |
| WMM | Fluid bed coating/drying | Bifendate (–) | (HPC SL, SLS, Mannitol), MCC (core), HPMC E5, PVP K30, Poloxamer 407 and 188 | 5 g dried products were dispersed in 100 mL purified water and paddle stirred at 100 rpm for 5 min | 0.139 d | 0.360 d,f | Yao et al., (2014) [147] |
| WMM | Freeze drying | Telmisartan (–) | (Poloxamer 188, Trehalose), Poloxamer 407, PVA, PVP K30, SLS, Tween 80, HPMC E5, HPC, Glucose, Lactose, Mannitol, Sucrose | 50 mg dried product was dispersed in 5 mL distilled water and sonicated for 15–30 s | ~0.335 e | ~0.337 e,f | Patel et al., (2014) [148] |
| LASP | Spray drying | Celecoxib (–) | (PVP K30) | – | 0.321 e | _ f | Homayouni et al., (2014) [149] |
| Freeze drying | Celecoxib (–) | (PVP K30) | _ | 0.321 e | _ f | ||
| LASP–HPH | Freeze drying | Celecoxib (–) | (PVP K30) | _ | 0.450 e | _ f | |
| LASP–Ultrasonication | Freeze drying | Acyclovir (–) | (Poloxamer 188, PVP K30, Sucrose), TPGS, Tween 80, Mannitol, MCC | Malvern Mastersizer | 0.274 e | 0.353 e,f | Bhalekar et al., (2014) [150] |
| WMM | Fluid bed coating/drying | Griseofulvin (48.8%) Azodicarbonamide (40.4%) | (HPC SL, SDS), Pharmatose (core) | Dried product equivalent to 5 wt.% drug (w.r.t. suspension) dispersed in 20 mL water stirred for 2 min using impeller at 200 rpm | 0.160 0.250 | ~0.160 f ~0.250 f | Bhakay et. al., (2014) [85] |
| Spray drying | Griseofulvin (76.9%) Azodicarbonamide (74%) | (HPC SL, SDS), Mannitol | 0.160 0.250 | ~0.160 f ~0.250 f | |||
| WMM | Spray drying | Itraconazole (–) | (PVP40, SLS), Methocel E3, Methylcellulose A15, HPMC E5, HPMC E15, HPC, PVA, Kollidon 30, PVP40, Poloxamer 188 and 407 | Powders were suspended in saturated and filtered solution of indomethacin in 30% glycerin solution | 0.283 e | 0.310 e,f | Kumar et al., (2015) [151] |
| WMM | Fluid bed coating/drying | Fenofibrate (14.1%) | (HPMC E3, SDS, Mannitol), Pharmatose (core), GranuLac (core), potato starch (core) | 100 mg dried powders were dispersed in 8 mL DI water and shaken manually for 30 s | 0.160 | 0.265 f | Knieke et al., (2015) [152] |
| WMM | Spray drying | Griseofulvin (75.2%) | (HPC SL, SDS), CCS, SSG, Mannitol | Qualitative assessment: 10 mg dried powder was dispersed in 4 mL DI water and observed the cloudiness | 0.161 | Cloudy supernatant f | Azad et al., (2015) [27] |
| WMM | Wet film casting–drying | Fenofibrate (17%) Griseofulvin (14.5%) Naproxen (14.2%) Phenylbutazone (15.1%) Azodicarbonamide (17.7%) | (SDS, HPMC E15 LV, Glycerin) | ~0.71 cm2 circular films were dispersed in 3 mL DI water and vortex mixed for 3–5 min | 0.178 0.16 10.136 0.176 0.278 | 0.283 f 0.164 f 0.134 f 0.184 f 0.352 f | Krull et al., (2015) [153] |
| WMM | Spray drying | Naproxen (–) | (HPMC E15, Trehalose), Tween 80, Lactose | Powders were suspended in saturated and filtered solution of naproxen in 30% glycerin solution | 0.243 e | 0.282 e,f | Kumar et al., (2015) [154] |
| HPH | Freeze drying | Harmine (–) | (HPMC E15, Sorbitol), TPGS, Tween 80, CMS-Na, RH40, Sucrose, Glucose, Trehalose, Mannitol | Malvern Mastersizer RDI = mean redispersion size/nanosuspension size ×100 | 0.500–0.700 | ~100% g | Yue et al., (2015) [155] |
| Spray drying | Harmine (–) | (CMS-Na), HPMC E15, TPGS, Tween 80, CMS-Na, RH40 | 0.500–0.700 | ~100% g | |||
| WMM | Wet film casting–drying | Griseofulvin (15.8%) | (HPMC E15, SDS, Glycerin, CCS/HPMC E4), SSG, CP, GG, XG, Pectin | ~0.71 cm2 circular films were dispersed in 10 mL DI water and vortex mixed for 1 min | 0.160 | 0.160 f | Susarla et al., (2015) [80] |
| LASP | Freeze drying | Ursolic acid (–) | (TPGS 1000, Trehalose), Maltose, Glucose, Sucrose, PEG2000 | Dried powders were dispersed in water and shaken manually | 0.127 e | 0.239 e,f | Ge et al., (2015) [156] |
| WMM | Spray drying | Indomethacin (36.3%) | (Poloxamers 188, Mannitol, L-leucine), Poloxamers 407 and184, TPGS 1000 | Malvern Nano ZS | 0.263 e | 0.417 e,f | Malamatari et al., (2015) [157] |
| HPH | Spray drying | Itraconazole (–) | (Poloxamers 407, SLS, Mannitol) | Malvern Zetasizer | ~0.316 e | ~0.320 e,f | Sun et al., (2015) [107] |
| LASP–Ultrasonication | Freeze drying | Naproxen (9.42%) | (HPMC) | _ | 0.530 e | _ f | Mishra et al., (2015) [158] |
| HPH | Wet film casting–drying | Quercetin (10 mg/6 cm2) | (Maltodextrins, Glycerin, Tween 80, Span 80) | Malvern Zetasizer® Nano ZS | 0.753 e | 0.781 e,f | Lai et al., (2015) [159] |
| Freeze drying | Quercetin (–) | (Tween 80) | 0.753 e | ~0.921 e,f | |||
| LASP–Ultrasonication | Freeze drying | Teniposide (–) | (PVP K30), Poloxamer 188, HPMC | Powders were suspended with 5% glucose and vortexed for 5 s | 0.151 e | 0.151 e,f | He et al., (2015) [160] |
| Acid-base neutralization–Ultrasonication | Spray drying | Glipizide (–) | (SLS, MCC), Poloxamer, PVP, HPMC, Tween 80, Mannitol, Sorbitol | _ | 0.262 | _ f | Elham et al., (2015) [111] |
| ME–HPH | Freeze drying | Lambda-cyhalothrin (–) | (MRES, SDS), PVP K30, PVP K90, SL, Tween 80, PEGNPE, HPMC, Poloxamer 188, Span 80, Mannitol | _ | 0.016 e | _ g | Pan et al., (2015) [161] |
| WMM | Wet film casting–drying | Griseofulvin (8.75%) | (Pullulan, SDS, XG, Glycerin) | 10 mg of the dry film was dispersed in 20 mL DI water and vortexed for 2 min at 1500 rpm | 0.168 | 0.379 | Krull et al., (2016) [162] |
| WMM | Wet film casting–drying | Griseofulvin (15.4%) | (HPMC E15LV, SDS, PEG 400), Triacetin, Glycerin | ~0.7 cm2 circular dried film was dispersed in 3 mL DI water and vortexed at 1500 rpm for 3–5 min | 0.159 | 0.169 f | Krull et al., (2016) [81] |
| ME | Wet film casting–drying | Fenofibrate (5.03%) | (Pluronic F68, HPMC E15 LV, Glycerin) | 2.54 cm diameter film was dispersed in 10 mL DI water and stirred for 5 min at 475 rpm using a magnetic stirrer | ~0.500 | ~0.800 f | Bhakay et al., (2016) [40] |
| WMM | Fluid bed coating/drying | Griseofulvin (12.0%) | (HPC L, SDS), Pharmatose (core), HPC SSL, HPC SL | 1 g of the dried powders was dispersed in 30 mL DI water and paddle stirred for 2 min at 200 rpm | ~0.250 | ~0.250 f | Li et al., (2016) [86] |
| HPH | Spray drying | Itraconazole (–) | (Tween 20, Methocel E5) | Powder corresponding to 0.2 g drug was dispersed in 25 mL water and shaken manually for 30 s | 0.313 e | 0.425 e | Bonda et al., (2016) [163] |
| HPH | Spray drying | Albendazole (–) | (Poloxamer 188), SDS, Poloxamer 407 | 20 mg powders were dispersed in 5 mL DI water and manually shaken for 1 min | 0.450 e | 0.550 e,f | Parades et al., (2016) [164] |
| HPH | Nanoextrusion | Efavirenz (1.02%) | (SLS, Kollidon® 30, Soluplus) | Extrudates were dispersed in water and vortexed for 30 s | 0.320 e | ~0.030 f | Ye et al., (2016) [77] |
| LASP–HPH | Freeze drying | Ursodeoxycholic (–) | (PVP K30, Sucrose/Glucose), TPGS, Poloxamer 188, Lactose Poloxamer 407, RH40, HPMC, SDS, Tween 80, PEG4000, Trehalose, CMS-Na, MCCS, Mannitol, Sorbitol | Malvern Mastersizer RDI = mean redispersion size/nanosuspension size ×100 | 0.600–0.900 | ~100% g | Ma et al., (2016) [165] |
| LASP | Freeze drying | Gambogenic acid (29.7%) | (PVP K30, PEG2000, Trehalose, Mannitol) | Malvern Zetasizer | ~0.184 e | ~0.198 e,f | Yuan et al., (2016) [166] |
| HPH | Spray drying | Model drug (–) | (HPC L, MCCS), HPMC, MCC, CCS, Trehalose, Lactose | Malvern Mastersizer | 0.461 | 0.478 f | Dan et al., (2016) [167] |
| LASP–Ultrasonication | Freeze drying | 20(S)-protopanaxadiol (50%) | (Bovine serum) | _ | 0.222 e | _ f | Han et al., (2016) [168] |
| WMM | Spray drying | Griseofulvin (–) | (HPC SL, Docusate sodium, Mannitol) | Dried powders were dispersed in water and simulated gastric fluid (pH 1.2) using USP II | 0.205 e | 0.200–0.300 e,f | Shah et al., (2016) [169] |
| LASP–Ultrasonication | Spray drying | Cefixime trihydrate (–) | (PVP K30, Lactose monohydrate), Mannitol, Sorbitol | Excess amount of dried powder was dispersed in 2 mL distilled water and sonicated for short time | 0.266 e | ~0.300 e,f | Alaei et al., (2016) [170] |
| WMM | Wet film casting–drying | Naproxen (–) Anthraquinone (–) | (HPMC, Glycerol, PVP VA64, SDS) | 4 cm2 film was dispersed in 0.9 mL liquid (DI water, tap water and saliva substitute) for 1–15 min and further diluted with DI water | 0.270 0.273 | 0.280–0.315 g 0.340–0.420 g | Steiner et al., (2016) [171] |
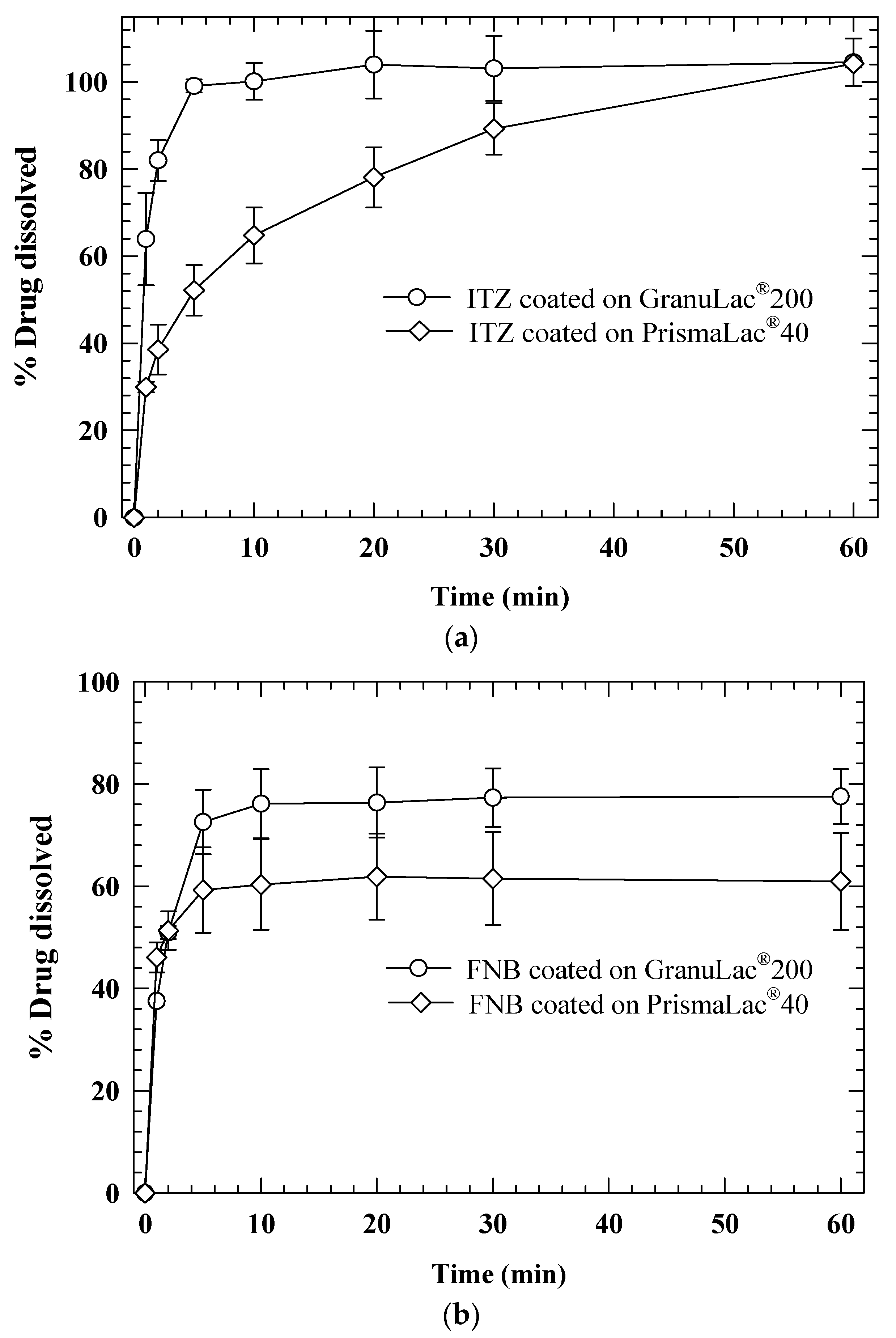
| WMM | Fluid bed coating/drying | Itraconazole (14.8%) Fenofibrate (13.6%) | (HPMC, SDS), Hydrophilic silica), GranuLac® 200 (core), PrismaLac® 40 (core) | 1 g of dried products were dispersed in 30 mL of 7.2 and 2.88 mg/mL SDS and stirred at 110 rpm for 2 min | 0.172 0.171 | 0.490 f 0.290 f | Azad et al., (2016) [91] |
| LASP–Ultrasonication | Freeze drying | Efavirenz (–) | (Poloxamer 407, Soya lecithin, Mannitol) | _ | ~0.184 e | _ f | Taneja et al., (2016) [172] |
| Acid-base neutralization–HPH | Freeze drying | Herpetospermum caudigerum lignans (–) | (SDS, PVP K30, Mannitol), Poloxamer 188, Tween 80, HPMC, PVA, Lecithin | Dried products were dispersed in DI water and shaken for 1 min | 0.243 e | 0.286 e,f | Gang et al., (2016) [173] |
| HPH | Spray drying | Andrographolide (–) | (HPMC E15/MCCS, Lactose), RH 40, TPGS, PVP K30, Tween 80, Sucrose, Trehalose, Mannitol, Sorbitol | Malvern Mastersizer RDI = mean redispersion size/nanosuspension size ×100 | 0.500–0.900 | ~100% g | Xie et al., (2016) [174] |
| Freeze drying | Andrographolide (–) | (HPMC E15, Tween 80, Sucrose/Trehalose/Sorbitol), RH 40, TPGS, MCCS, PVP K30, Mannitol, Lactose | 0.500–0.900 | ~100% g | |||
| LASP–Ultrasonication | Freeze drying | Cinnarizine (9.53%) | (PVA) | _ | 0.621 e | _ f | Mishra et al., (2016) [175] |
| HPH | Freeze drying | Ritonavir (–) | (HPMC 3, SDS, Mannitol), PVP K30 | _ | 0.562 e | _ f | Karakucuk et al., (2016) [176] |
| WMM | Freeze drying | BI XX (–) | (Mannitol, Arginine), Polysorbate 80, PEG 400, Proline, Benzalkonium chloride | 2 mL DI water was added to the dried product and shaken manually | 0.192 | ~0.250 g | Frank and Boeck (2016) [177] |
| LASP–Ultrasonication | Wet film casting–drying | Lercanidipine HCl hemihydrate (3.06 mg) | (TPGS 1000, Hypromellose E15), Hypromellose E5, PVA, PEG 400, Sodium alginate, MC, HPC, HPMC E5 | 4 cm2 films were placed in 10 mL DI water and stirred for 10 min using magnetic stirrer | ~0.277 e | ~0.240 e,f | Chonkar et al., (2016) [178] |
| WMM | Freeze drying | Spironolactone (–) | (HPMC E5, Sorbitol), Sodium deoxycholate, Poloxamer 407, Poloxamer 188, Mannitol | Dried powders were dispersed in water by gentle shaking | 0.374 e | 0.399 e,f | Mu et al., (2016) [179] |
| WMM | Nanoextrusion | Griseofulvin (24.1%) | (HPC SL, SDS), Soluplus | _ | 0.154 | _ f | Li et al., (2017) [78] |
| WMM | Spray drying | Aprepitant (–) | (Pharmacoat 603, SDS, sucrose), HPMC E15, HPC SSL, PVP K30, TPGS 1000, Poloxamer P188, Mannitol | Dried powders were dispersed in few milliliters of distilled water and shaken manually for 30 s | 0.395 e | 0.420 e,g | Toziopoulou et al., (2017) [180] |
| Freeze drying | Aprepitant (–) | (Pharmacoat 603, SDS, Mannitol) | 0.395 e | 0.395 e,g | |||
| LASP–HPH | Freeze drying | P2X7 receptor antagonist (–) | (Poloxamer 188, Mannitol), HPMC, SDS | _ | 0.245 e | _ f | Zhang et al., (2017) [181] |
| WMM | Freeze drying | Nisoldipine (–) | (PVP K30, SDS, Trehalose), HPMC E5 | _ | 0.240 e | _ f | Fu et al., (2017) [113] |
| WMM | Freeze drying | Dexamethasone (–) Tacrolimus (–) | (Poloxamer 407) (Poloxamer 407) | Malvern Zetasizer® Nano ZS90 | 0.403 e0.511 e | 0.300–0.700 e,g 0.300–0.800 e,g | Colombo et al., (2017) [182] |
| WMM | Electrospraying–Freeze drying | Darunavir (–) | (Tween 20, SLS, Eudragit L100, Mannitol), Poloxamer 338, 188, HPMC, Vitamin E TPGS 400 and 1000, Tween 80 | _ | 0.295 e | _ f | Nguyen et al., (2017) [183] |
| LASP–Ultrasonication | Freeze drying | Efavirenz (–) | (HPMC E5, SLS), HPC ELF, Poloxamer 188 and 407, PVP K30 | _ | 0.252 e | _ f | Sartori et al., (2017) [184] |
| WMM | Spray drying | Allisartan Isoproxil (61.7%) | (PVP K30, Mannitol, SDS), Pluronic F127 and F68, Tween 80, HPMC E5, HPMC E50, HPC, PEG 4000 and 6000, Glucose, Sucrose, Maltose, Sorbitol | Dried powders were dispersed in water at a drug concentration of 2.5% (w/v) and diluted to 900-fold with water | ~0.300 e | ~0.304 e,f | Hou et al., (2017) [185] |
| HPH | Freeze drying | Andrographolide (–) | (Glycyrrhizin, Trehalose), Poloxamer 188, Tween 80, TPGS, Sucrose, Lactose | Malvern Mastersizer | 0.487 | 0.491 f | Chen et al., (2017) [186] |
| WMM | Wet film casting–drying | Griseofulvin (50%) | (HPMC E15LV, SDS, Glycerin), HPMC 4M | ~0.7 cm2 circular films were dispersed in DI water and vortexed at 1500 rpm for 3–5 min | 0.150 | <0.200 f | Krull et al., (2017) [83] |
| WMM | Wet film casting–drying | Anthraquinone (–) | (HPMC, Glycerol), HPC, Kollidon VA64, SDS | 4 cm2 film dispersed in 0.9 mL distilled water for 5 min | 0.302 | 0.337 g | Steiner et al., (2017) [187] |
| HPH | Spray drying | Andrographolide (–) | (TPGS, PVP K30, MCC, Lactose), PVPP | Malvern Mastersizer RDI = mean redispersion size/nanosuspension size ×100 | ~0.514 | 101.5% f | Xu et al., (2017) [188] |
| LASP–HPH | Freeze drying | 10-hydroxycamptothecin (94.9%) Camptothecin (91.2%) 7-ethyl-10-hydroxycamptothecin (90.1%) | (mPEG1000-HCPT) | Dried products were reconstituted with water | ~0.093 e ~0.121 e 0.133 e | ~0.094 e,f 0.137 e,g 0.133 e,g | Yang et al., (2017) [189] |
| Ultrasonication–HPH | Freeze drying | Meloxicam (–) | (Poloxamer 188, Mannitol), PVP K25, PEG 4000 | Dried products corresponding 0.5 mL suspension were diluted to 15 mL with purified water and sonicated for 60 s | ~0.463 e | ~0.501 e,f | Iurian et al., (2017) [190] |
| LASP–Ultrasonication | Wet film casting–drying | Lutein (0.23 mg/cm2) | (SPC, SDS, HPMC, PEG 400, Cremophor EL), TPGS, PVP K30, Poloxamer 188, PVA | A piece of film was dispersed in 5 mL water and sonicated for 3 min | 0.220 e | ~0.378 e,f | Liu et al., (2017) [191] |
| WMM | Fluid bed granulation/drying | Lurasidone hydrochloride (–) | (HPMC E3, Polysorbate 80, Mannitol, MCC), HPMC E5, Poloxamer 188, SLS, Span 20, Labrasol | _ | 0.245 e | _ f | Kumar et al., (2017) [192] |
| WMM | Spray drying | Mefenamic acid I (–) Mefenamic acid II (–) | (HPC SSL, Lactose) | _ | 0.188 0.189 | _ f_ f | Konnerth et al., (2017) [193] |
| LASP–Ultrasonication | Freeze drying | Carvedilol (–) | (SDS), Whey protein isolate, Poloxamer 188 | 1 mg sample was dispersed in 3 mL of water and shaken manually for 1 min | ~0.225 e | 0.227 e,f | Geng et al., (2017) [194] |
| Class | Examples | Possible Mechanisms of Action | References |
|---|---|---|---|
| Soluble polymers | HPC, HPMC, PVP, Soluplus, PEG, PVA, Sodium CMC, methyl cellulose, PEG-PLA, Sodium alginate | Steric stabilizer in nanosuspension, enhanced drug wettability in nanocomposites, primary matrix/film former that prevents aggregation during drying and facilitates erosion/disintegration of nanocomposites via dissolution. | Lee et al., 2003 [33], Li et al., 2016 [23], Chin et al. [55] |
| Surfactants | Alpha tocopherol succinate, SDS, TPGS, Poloxamer 338, Pluronic F127, Pluronic F68, Span 20/60, Tween 20/60, Dowfax 2A1, Soy lecithin, Docussate sodium, Cremophor, Sodium deoxycholate | Steric or electrostatic stabilizer depending on its charge, wetting agent, enhanced drug wettability in nanocomposites | Bonda et al., 2016 [163], Li et al., 2016 [23], Chin et al., [55] |
| Other water-soluble dispersants (WSD) | Sucrose, Lactose, Trehalose, Glucose, Maltose, Maltodextrin, Ficoll PM70, HPβ-cyclodextrin, L-Leucine, Mannitol, Arginine, sodium carboxymethyl starch (CMS-Na) | Secondary matrix former that prevents aggregation during drying, facilitates erosion/disintegration of nanocomposite particles via dissolution, and act as cryoprotectant in freeze drying | Abdelwahed et al., 2006 [197], Kesisoglou et al. [28], Chin et al. [55] |
| Water-insoluble dispersants (WID) | Microcrystalline cellulose, anhydrous dicalcium phosphate, colloidal fumed silica, montmorillonite | Secondary matrix former that prevents aggregation during drying and facilitates erosion/disintegration of nanocomposites | Eerdenbrugh et al., 2008 [59] |
| Crosslinked polymers (CLP) | SSG, CCS, CP | Swellable dispersant that facilitates erosion/disintegration of nanocomposite particles via swelling-induced breakage/erosion of nanocomposite matrix | Bhakay et al., 2014 [8], Azad et al., 2015 [27] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhakay, A.; Rahman, M.; Dave, R.N.; Bilgili, E. Bioavailability Enhancement of Poorly Water-Soluble Drugs via Nanocomposites: Formulation–Processing Aspects and Challenges. Pharmaceutics 2018, 10, 86. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics10030086
Bhakay A, Rahman M, Dave RN, Bilgili E. Bioavailability Enhancement of Poorly Water-Soluble Drugs via Nanocomposites: Formulation–Processing Aspects and Challenges. Pharmaceutics. 2018; 10(3):86. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics10030086
Chicago/Turabian StyleBhakay, Anagha, Mahbubur Rahman, Rajesh N. Dave, and Ecevit Bilgili. 2018. "Bioavailability Enhancement of Poorly Water-Soluble Drugs via Nanocomposites: Formulation–Processing Aspects and Challenges" Pharmaceutics 10, no. 3: 86. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics10030086