Monitoring the Fate of Orally Administered PLGA Nanoformulation for Local Delivery of Therapeutic Drugs

 , , , ,
, , , ,  and
and
Abstract
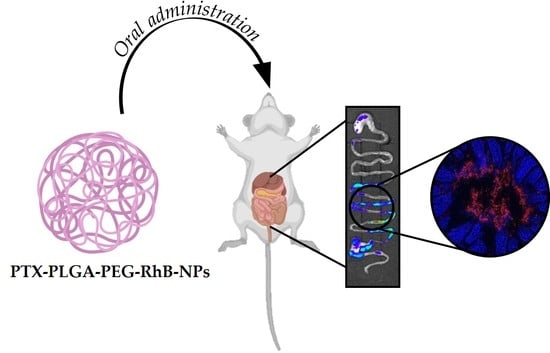
:
1. Introduction
2. Materials and Methods
2.1. Nanoparticles Synthesis
2.1.1. Materials
2.1.2. PLGA-PEG-RhB Synthesis
2.1.3. Synthesis of PLGA-PEG-RhB-NPs and PTX-PLGA-PEG-RhB-NPs
2.2. Nanoparticles Characterization
2.2.1. Dynamic Light Scattering (DLS) and Zeta Potential Measurements
2.2.2. Scanning Transmission Electron Microscopy (STEM) Analysis
2.2.3. In Vitro Release Studies
2.2.4. Nanoparticles Stability in Different Buffers
2.3. Cells
2.4. Animals
2.5. Treatments
2.6. Molecular Imaging and Histology
2.7. NPs Characterization from Homogenates and Biological Fluids
2.8. Pharmacokinetics
2.9. Statistical Analysis
3. Results and Discussion
3.1. NPs Synthesis and Characterization
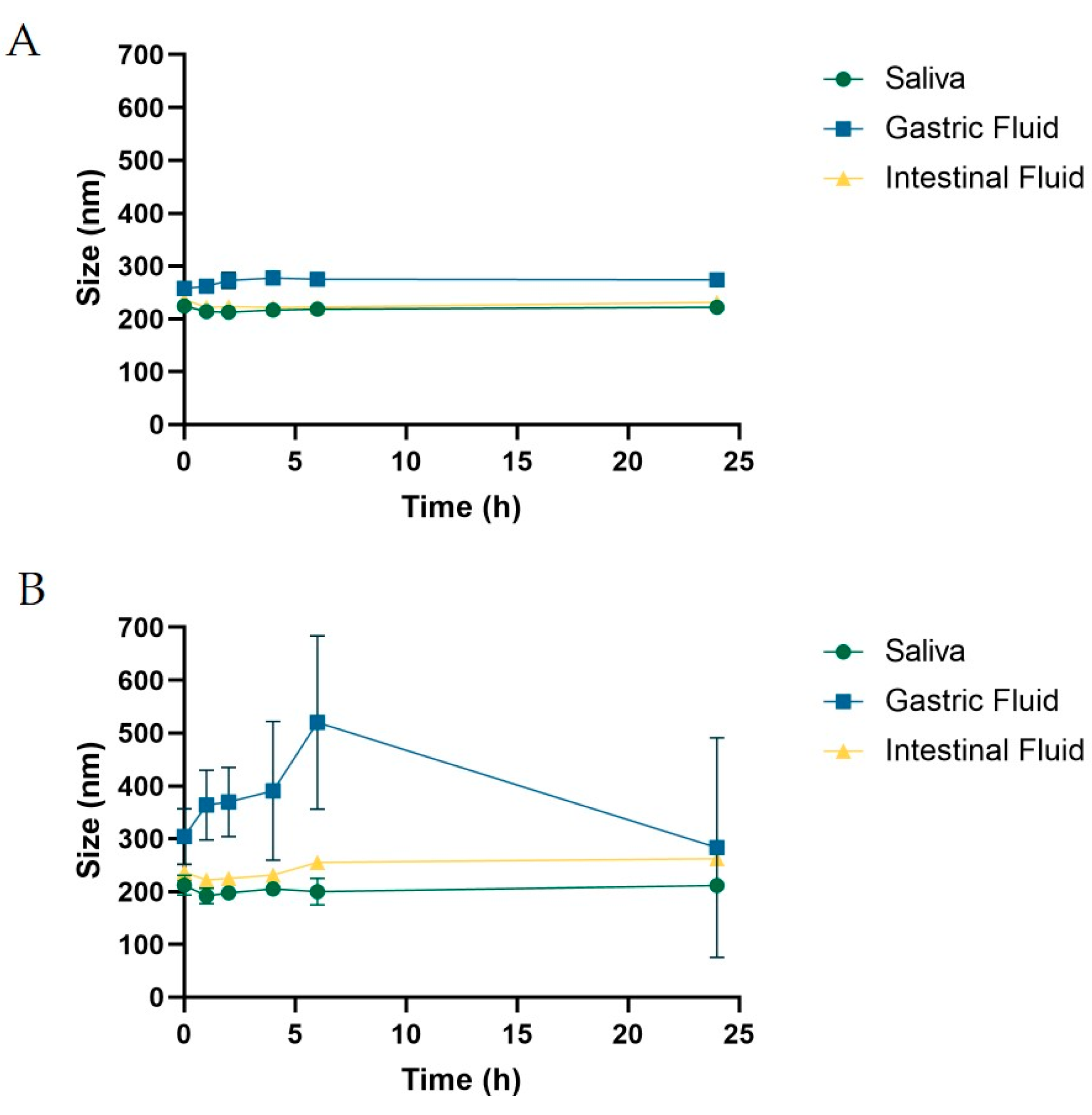
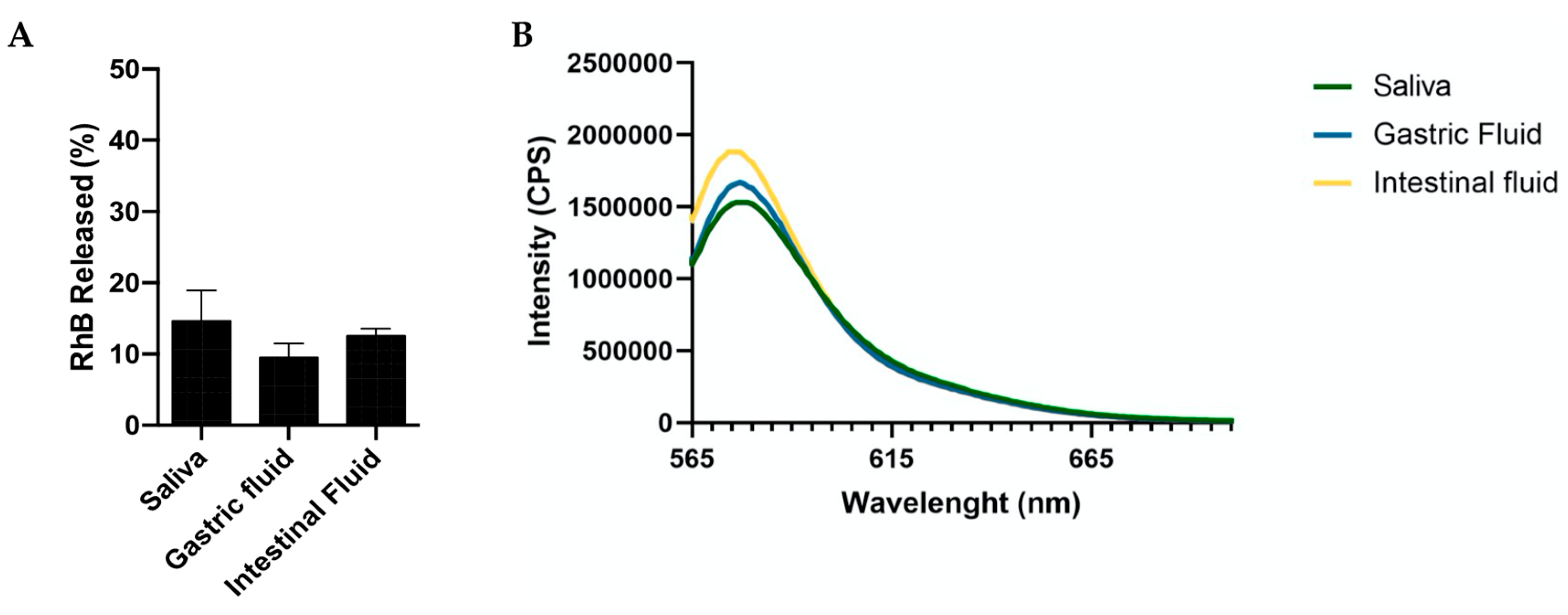
Stability in Mimicking Biological Fluids
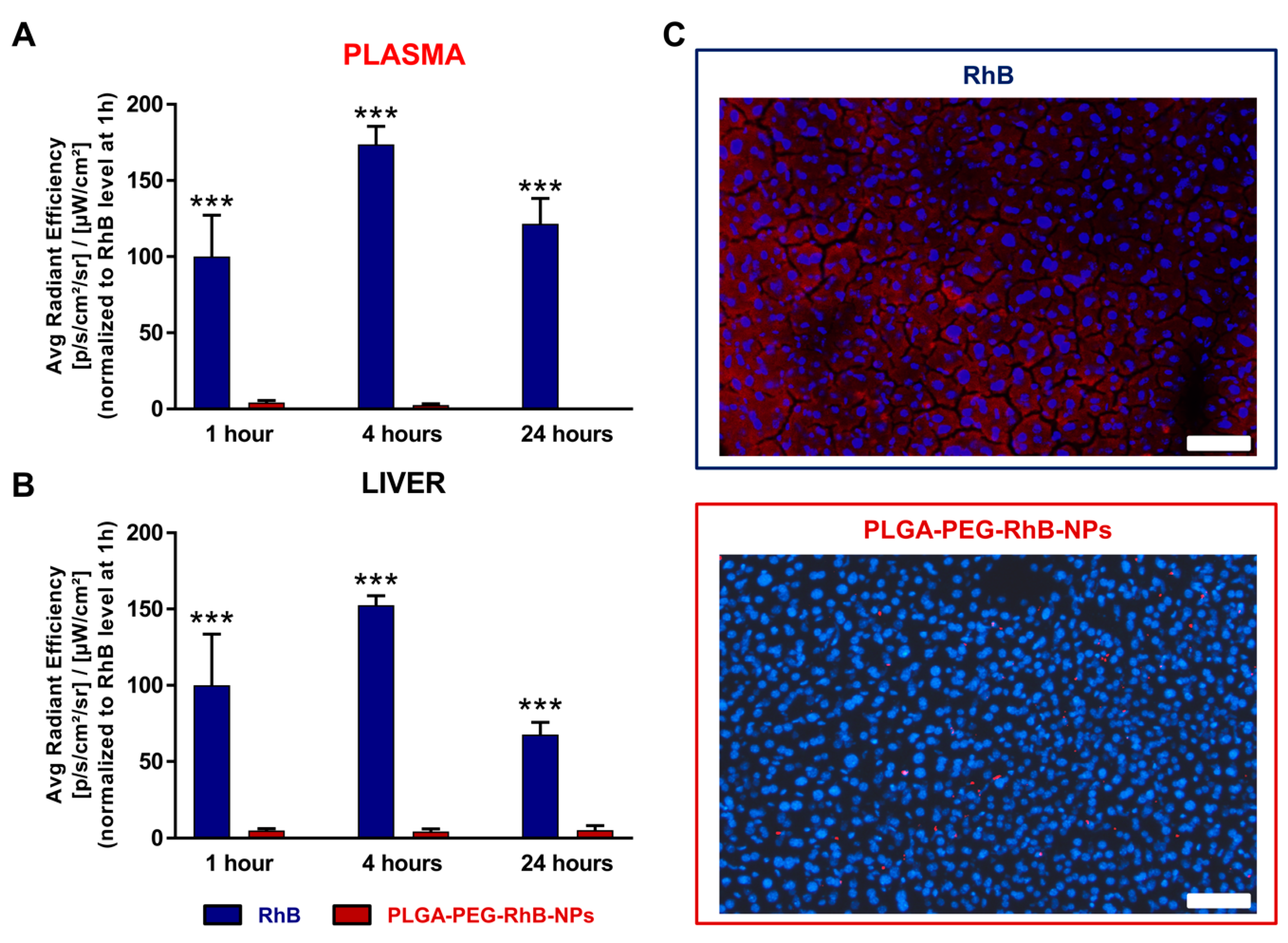
3.2. Pharmacokinetics
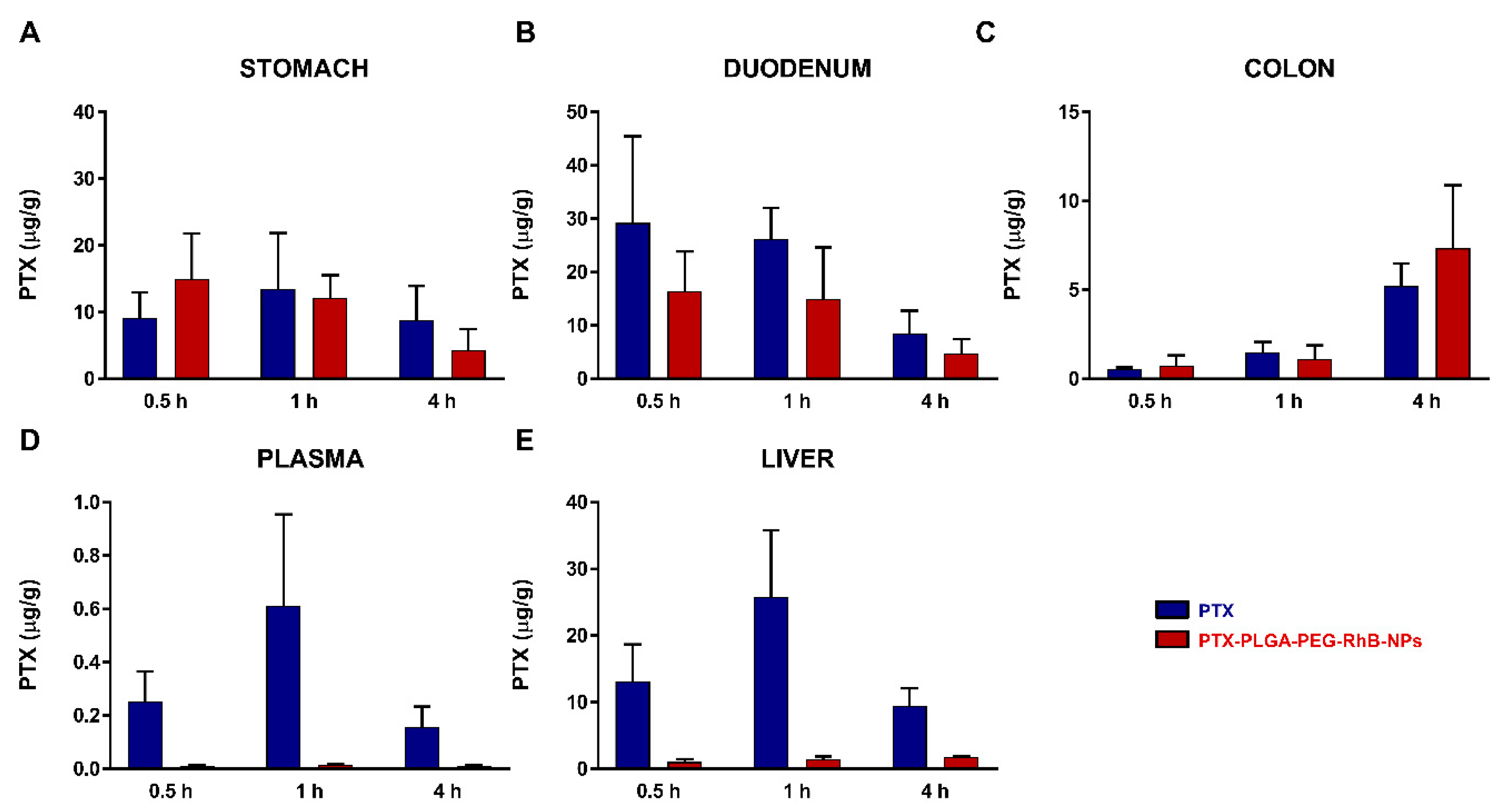
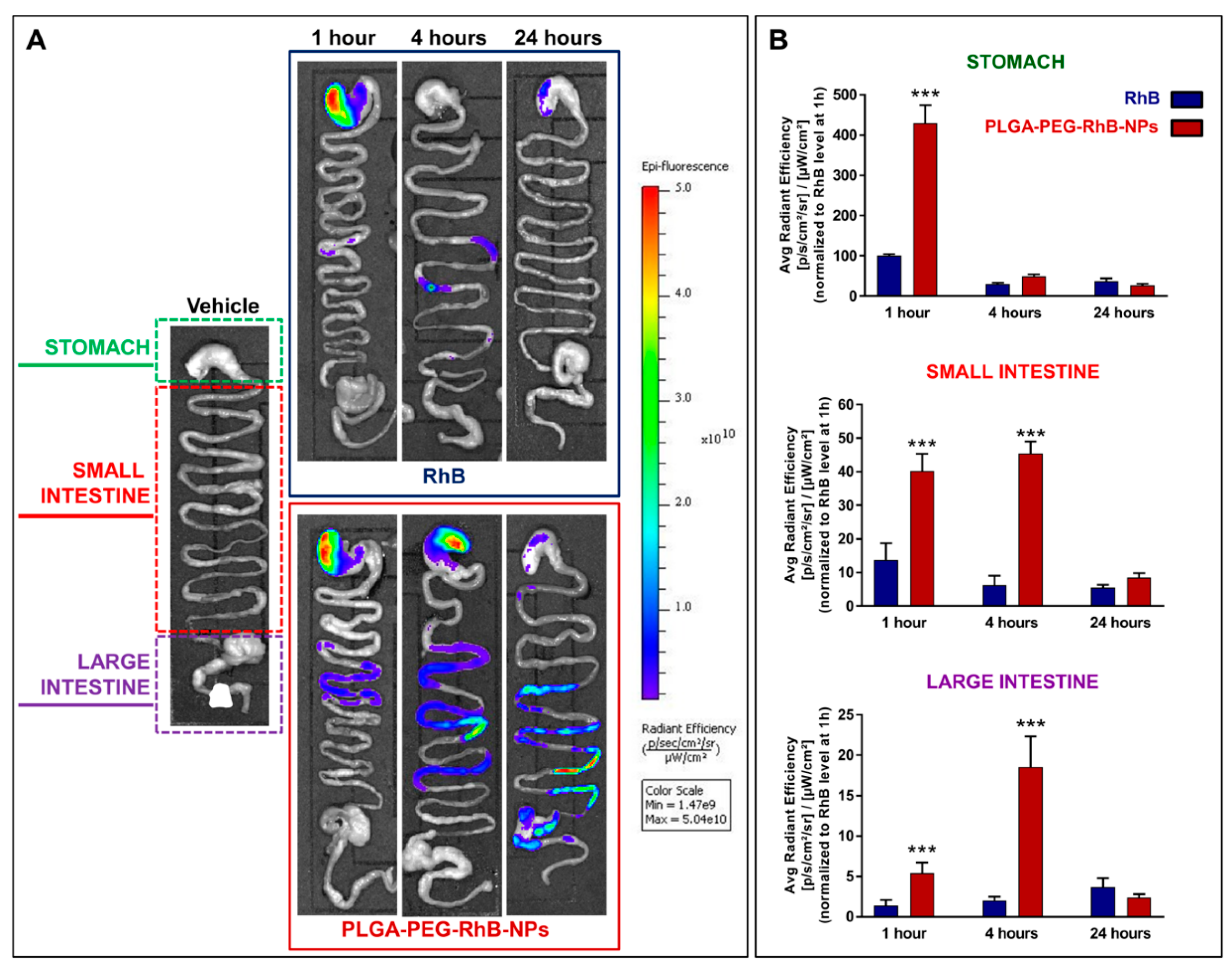
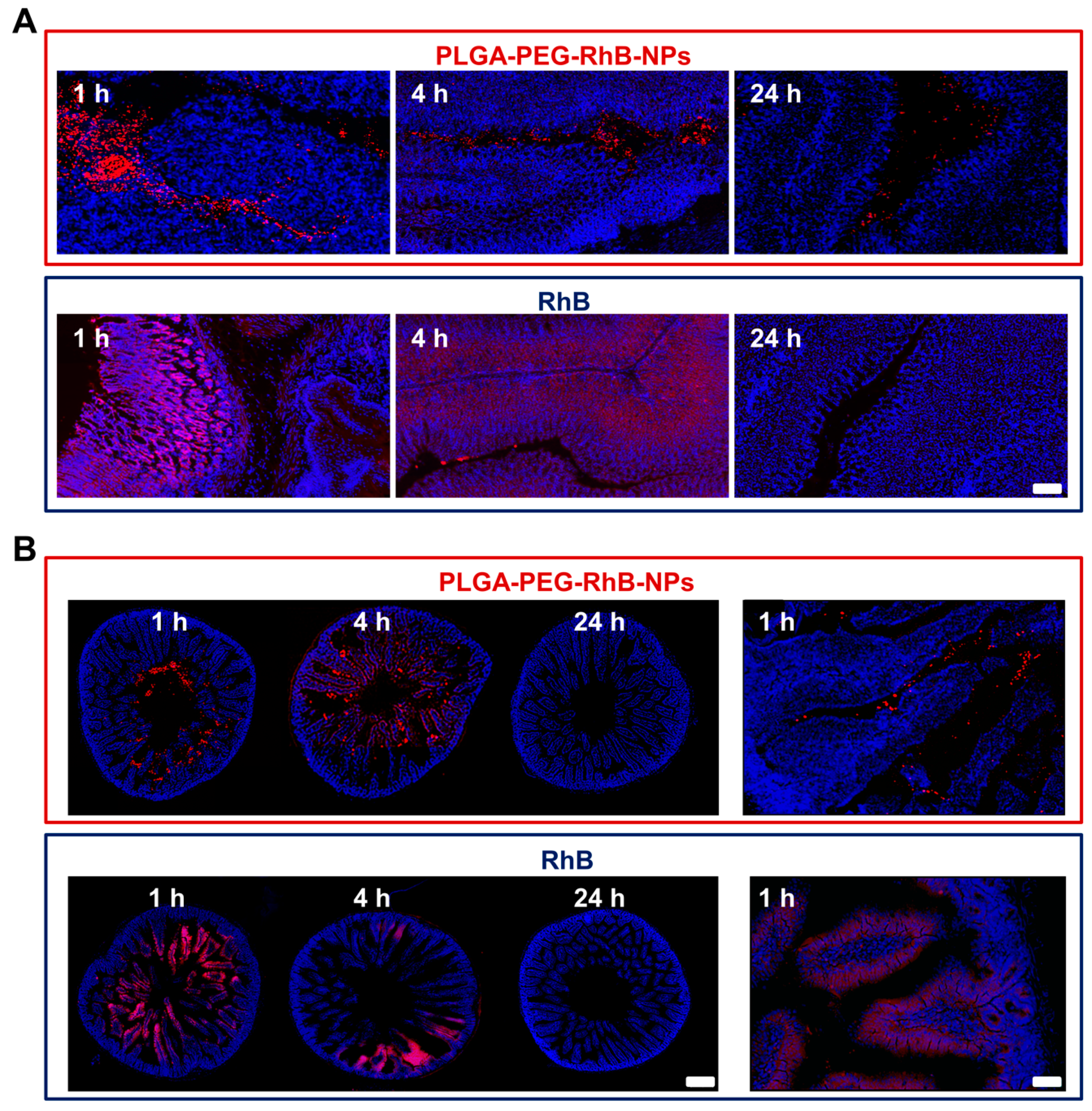
3.3. NPs Biodistribution and Nanosafety
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.R. Nanotechnologies for biomedical science and translational medicine. Proc. Natl. Acad. Sci. USA 2015, 112, 14436–14443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Violatto, M.B.; Casarin, E.; Talamini, L.; Russo, L.; Baldan, S.; Tondello, C.; Messmer, M.; Hintermann, E.; Rossi, A.; Passoni, A.; et al. Dexamethasone Conjugation to Biodegradable Avidin-Nucleic-Acid-Nano-Assemblies Promotes Selective Liver Targeting and Improves Therapeutic Efficacy in an Autoimmune Hepatitis Murine Model. ACS Nano 2019, 13, 4410–4423. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Scheyda, M.K.; Warzecha, K.T.; Rizzo, Y.L.; Hittatiya, K.; Luedde, T.; Storm, G.; Trautwein, C.; Lammers, T.; Tacke, F. Fluorescent cell-traceable dexamethasone-loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials 2015, 37, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef] [Green Version]
- Uziely, B.; Jeffers, S.; Isacson, R.; Kutsch, K.; Wei-Tsao, D.; Yehoshua, Z.; Libson, E.; Muggia, F.M.; Gabizon, A. Liposomal doxorubicin: Antitumor activity and unique toxicities during two complementary phase I studies. J. Clin. Oncol. 1995, 13, 1777–1785. [Google Scholar] [CrossRef]
- Feng, S.; Mu, L.; Win, K.Y. Nanoparticles of Biodegradable Polymers for Clinical Administration of Paclitaxel. Curr. Med. Chem. 2004, 11, 413–424. [Google Scholar] [CrossRef]
- Bhatt, P.; Lalani, R.; Vhora, I.; Patil, S.; Amrutiya, J.; Misra, A.; Mashru, R. Liposomes encapsulating native and cyclodextrin enclosed paclitaxel: Enhanced loading efficiency and its pharmacokinetic evaluation. Int. J. Pharm. 2018, 536, 95–107. [Google Scholar] [CrossRef]
- Venugopal, V.; Krishnan, S.; Palanimuthu, V.R.; Sankarankutty, S.; Kalaimani, J.K.; Karupiah, S.; Kit, N.S.; Hock, T.T. Anti-EGFR anchored paclitaxel loaded PLGA nanoparticles for the treatment of triple negative breast cancer.In-vitro and in-vivo anticancer activities. PLoS ONE 2018, 13, e0206109. [Google Scholar] [CrossRef]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [Green Version]
- Colzani, B.; Pandolfi, L.; Hoti, A.; Iovene, P.A.; Natalello, A.; Avvakumova, S.; Colombo, M.; Prosperi, D. Investigation of antitumor activities of trastuzumab delivered by PLGA nanoparticles. Int. J. Nanomed. 2018, 13, 957–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Fong, P.M.; Lu, J.; Russell, K.S.; Booth, C.J.; Saltzman, W.M.; Fahmy, T.M. PEGylated PLGA nanoparticles for the improved delivery of doxorubicin. Nanomedicine 2009, 5, 410–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramezani, M.; Ebrahimian, M.; Hashemi, M. Current Strategies in the Modification of PLGA-based Gene Delivery System. Curr. Med. Chem. 2017, 24, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sabliov, C.M. PLA/PLGA nanoparticles for delivery of drugs across the blood-brain barrier. Nanotechnol. Rev. 2013, 2, 241–257. [Google Scholar] [CrossRef]
- Gamboa, J.M.; Leong, K.W. In vitro and in vivo models for the study of oral delivery of nanoparticles. Adv. Drug Deliv. Rev. 2013, 65, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Wagner, A.M.; Gran, M.P.; Peppas, N.A. Designing the new generation of intelligent biocompatible carriers for protein and peptide delivery. Acta Pharm. Sin. B 2018, 8, 147–164. [Google Scholar] [CrossRef]
- Santini, B.; Zanoni, I.; Marzi, R.; Cigni, C.; Bedoni, M.; Gramatica, F.; Palugan, L.; Corsi, F.; Granucci, F.; Colombo, M. Cream formulation impact on topical administration of engineered colloidal nanoparticles. PLoS ONE 2015, 10, e0126366. [Google Scholar] [CrossRef]
- Musazzi, U.M.; Santini, B.; Selmin, F.; Marini, V.; Corsi, F.; Allevi, R.; Prosperi, D.; Cilurzo, F.; Colombo, M.; Minghetti, P. Impact of semi-solid formulations on skin penetration of iron oxide nanoparticles. J. Nanobiotechnol. 2017, 15, 14. [Google Scholar] [CrossRef] [Green Version]
- Cunha, S.; Amaral, M.H.; Lobo, J.M.S.; Silva, A.C. Lipid Nanoparticles for Nasal/Intranasal Drug Delivery. Crit Rev. Ther. Drug Carr. Syst. 2017, 34, 257–282. [Google Scholar] [CrossRef]
- Codullo, V.; Cova, E.; Pandolfi, L.; Breda, S.; Morosini, M.; Frangipane, V.; Malatesta, M.; Calderan, L.; Cagnone, M.; Pacini, C.; et al. Imatinib-loaded gold nanoparticles inhibit proliferation of fibroblasts and macrophages from systemic sclerosis patients and ameliorate experimental bleomycin-induced lung fibrosis. J. Control. Release 2019, 310, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Salvioni, L.; Fiandra, L.; Del Curto, M.D.; Mazzucchelli, S.; Allevi, R.; Truffi, M.; Sorrentino, L.; Santini, B.; Cerea, M.; Palugan, L.; et al. Oral delivery of insulin via polyethylene imine-based nanoparticles for colonic release allows glycemic control in diabetic rats. Pharmacol. Res. 2016, 110, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inchaurraga, L.; Martín-Arbella, N.; Zabaleta, V.; Quincoces, G.; Peñuelas, I.; Irache, J.M. In vivo study of the mucus-permeating properties of PEG-coated nanoparticles following oral administration. Eur. J. Pharm. Biopharm. 2015, 97 Pt A, 280–289. [Google Scholar] [CrossRef]
- Aggarwal, S.; Gupta, S.; Pabla, D.; Murty, R.S.R. Gemcitabine-loaded PLGA-PEG immunonanoparticles for targeted chemotherapy of pancreatic cancer. Cancer Nanotechnol. 2013, 4, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Lazzari, S.; Moscatelli, D.; Codari, F.; Salmona, M.; Morbidelli, M.; Diomede, L. Colloidal stability of polymeric nanoparticles in biological fluids. J. Nanopart. Res. 2012, 14, 920. [Google Scholar] [CrossRef] [Green Version]
- Fruscio, R.; Lissoni, A.A.; Frapolli, R.; Corso, S.; Mangioni, C.; D’Incalci, M.; Zucchetti, M. Clindamycin-paclitaxel pharmacokinetic interaction in ovarian cancer patients. Cancer Chemother. Pharmacol. 2006, 58, 319–325. [Google Scholar] [CrossRef]
- Enlow, E.M.; Luft, J.C.; Napier, M.E.; DeSimone, J.M. Potent engineered PLGA nanoparticles by virtue of exceptionally high chemotherapeutic loadings. Nano Lett. 2011, 11, 808–813. [Google Scholar] [CrossRef] [Green Version]
- Abouelmagd, S.A.; Sun, B.; Chang, A.C.; Ku, Y.J.; Yeo, Y. Release Kinetics Study of Poorly Water-Soluble Drugs from Nanoparticles: Are We Doing It Right? Mol. Pharm. 2015, 12, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Gullotti, E.; Yeo, Y. Beyond the imaging: Limitations of cellular uptake study in the evaluation of nanoparticles. J. Control. Release 2012, 164, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Shi, K.; Shi, J.; He, X. Use of the fluorescence of rhodamine B for the pH sensing of a glycine solution. Proc. SPIE 2016, 10155. [Google Scholar] [CrossRef]
- Capasso Palmiero, U.; Morosi, L.; Lupi, M.; Ponzo, M.; Frapolli, R.; Zucchetti, M.; Ubezio, P.; Morbidelli, M.; D’Incalci, M.; Bello, E.; et al. Self-Assembling PCL-Based Nanoparticles as PTX Solubility Enhancer Excipients. Macromol. Biosci. 2018, 18, 1800164. [Google Scholar] [CrossRef]
- Colombo, C.; Morosi, L.; Bello, E.; Ferrari, R.; Licandro, S.A.; Lupi, M.; Ubezio, P.; Morbidelli, M.; Zucchetti, M.; D’Incalci, M.; et al. PEGylated Nanoparticles Obtained through Emulsion Polymerization as Paclitaxel Carriers. Mol. Pharm. 2016, 13, 40–46. [Google Scholar] [CrossRef]
- Tenuta, T.; Monopoli, M.; Kim, J.; Salvati, A.; Dawson, K.; Sandin, P.; Lynch, I. Elution of Labile Fluorescent Dye from Nanoparticles during Biological Use. PLoS ONE 2011, 6, e25556. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Size (nm ± SD) | PDI | Z-Potential (mV ± SD) | Process Yield (%, w/w ± SD) |
|---|---|---|---|---|
| PLGA-PEG-NPs | 190.3 ± 12.7 | 0.078 ± 0.032 | +17.9 ± 5.9 | 53.3 ± 7.7 |
| PLGA-PEG-RhB-NPs | 205.6 ± 18.6 | 0.168 ± 0.030 | −18.2 ± 1.5 | 50.1 ± 13.4 |
| PTX-PLGA-PEG-RhB-NPs | 201.6 ± 26.2 | 0.205 ± 0.032 | −13.3 ± 1.7 | 39.3 ± 7.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morelli, L.; Gimondi, S.; Sevieri, M.; Salvioni, L.; Guizzetti, M.; Colzani, B.; Palugan, L.; Foppoli, A.; Talamini, L.; Morosi, L.; et al. Monitoring the Fate of Orally Administered PLGA Nanoformulation for Local Delivery of Therapeutic Drugs. Pharmaceutics 2019, 11, 658. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11120658
Morelli L, Gimondi S, Sevieri M, Salvioni L, Guizzetti M, Colzani B, Palugan L, Foppoli A, Talamini L, Morosi L, et al. Monitoring the Fate of Orally Administered PLGA Nanoformulation for Local Delivery of Therapeutic Drugs. Pharmaceutics. 2019; 11(12):658. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11120658
Chicago/Turabian StyleMorelli, Lucia, Sara Gimondi, Marta Sevieri, Lucia Salvioni, Maria Guizzetti, Barbara Colzani, Luca Palugan, Anastasia Foppoli, Laura Talamini, Lavinia Morosi, and et al. 2019. "Monitoring the Fate of Orally Administered PLGA Nanoformulation for Local Delivery of Therapeutic Drugs" Pharmaceutics 11, no. 12: 658. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11120658