Sodium Hyaluronate Nanocomposite Respirable Microparticles to Tackle Antibiotic Resistance with Potential Application in Treatment of Mycobacterial Pulmonary Infections

 , , , , and
, , , , and
Abstract
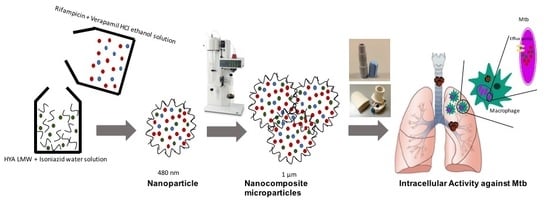
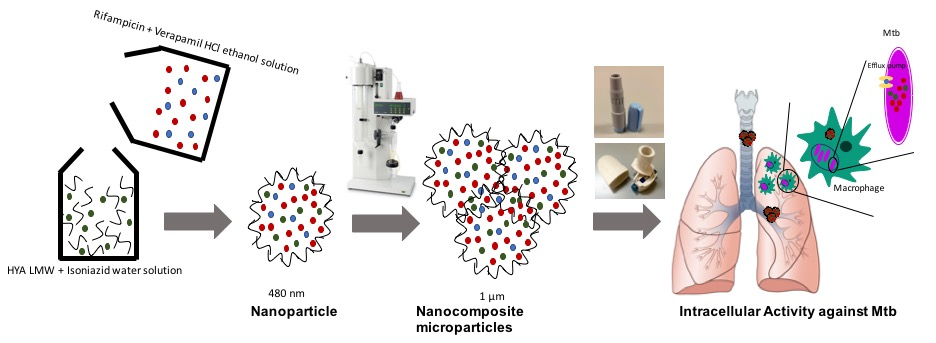
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
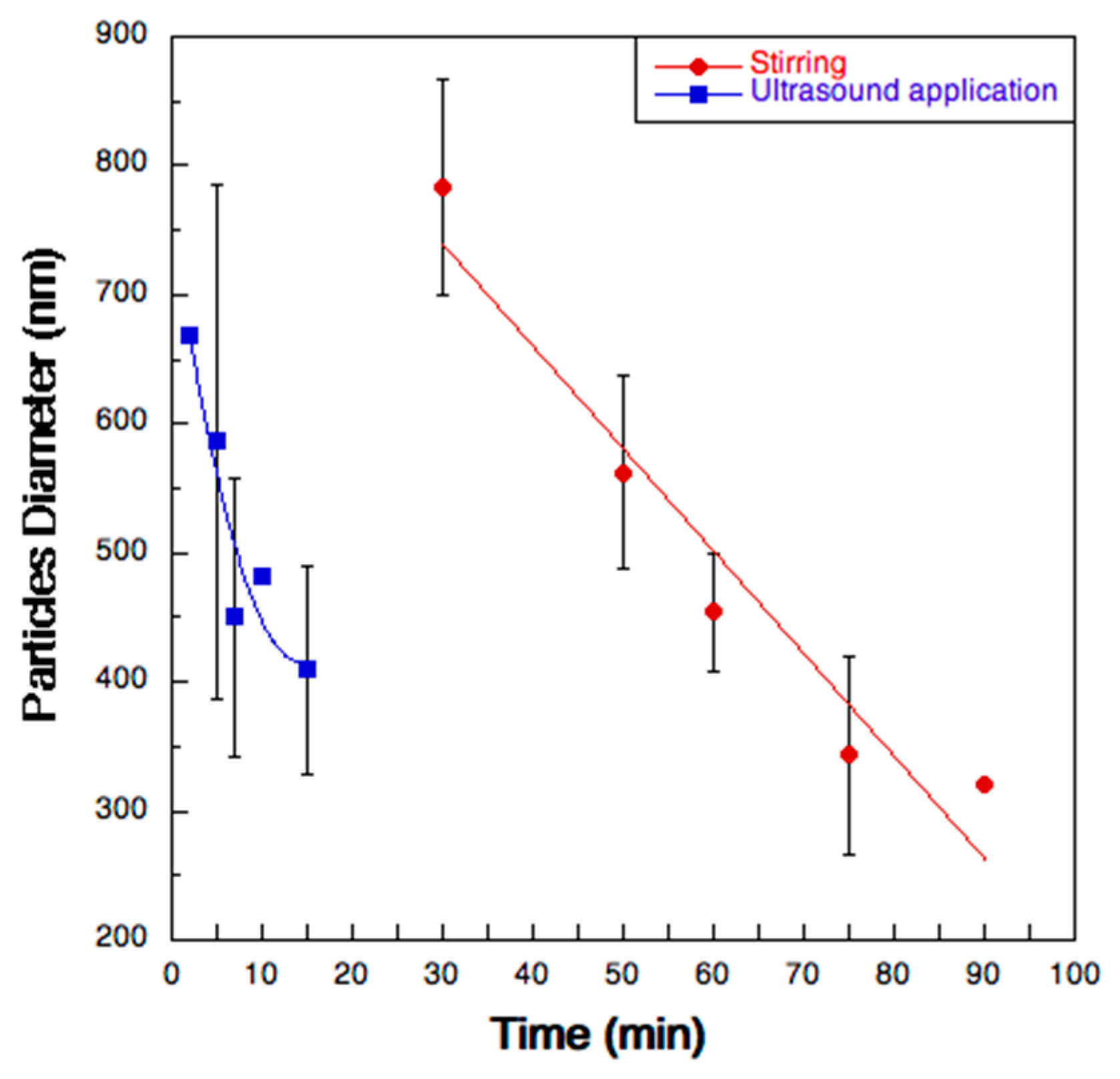
2.2.1. Turbidimetry
2.2.2. Triple Drug Combination HA Nanoparticles-Containing Respirable Powder Produced by Spray Drying
2.2.3. Spray Dried Powder Drug Loading Efficiency and Drug Content
2.2.4. HPLC Analysis
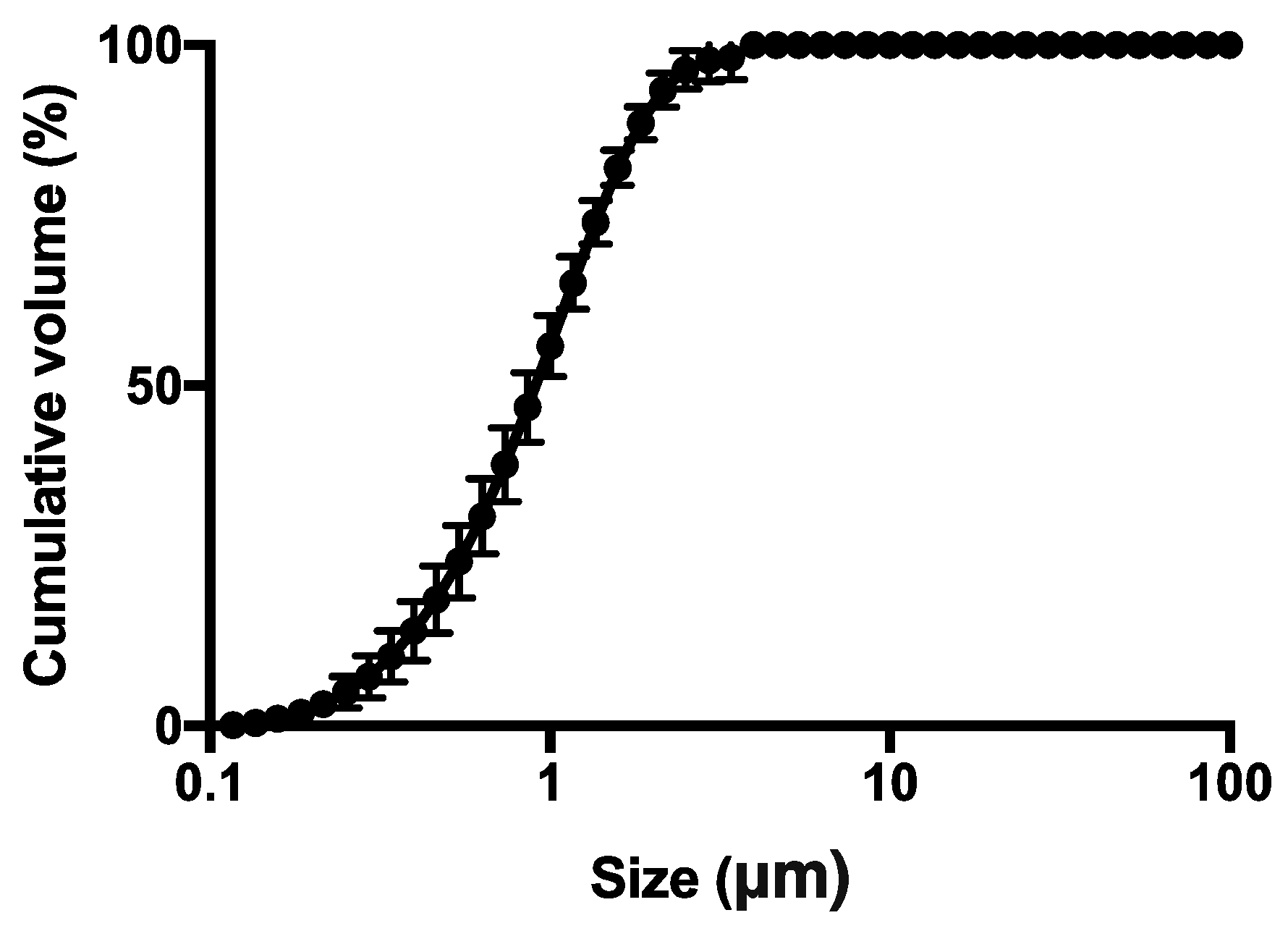
2.2.5. Particle Size Distribution
2.2.6. Thermal Analysis and Solid-State Characterization
2.2.7. Microscopy
2.2.8. Aerodynamic Performance
2.2.9. Preliminary Stability Assessment
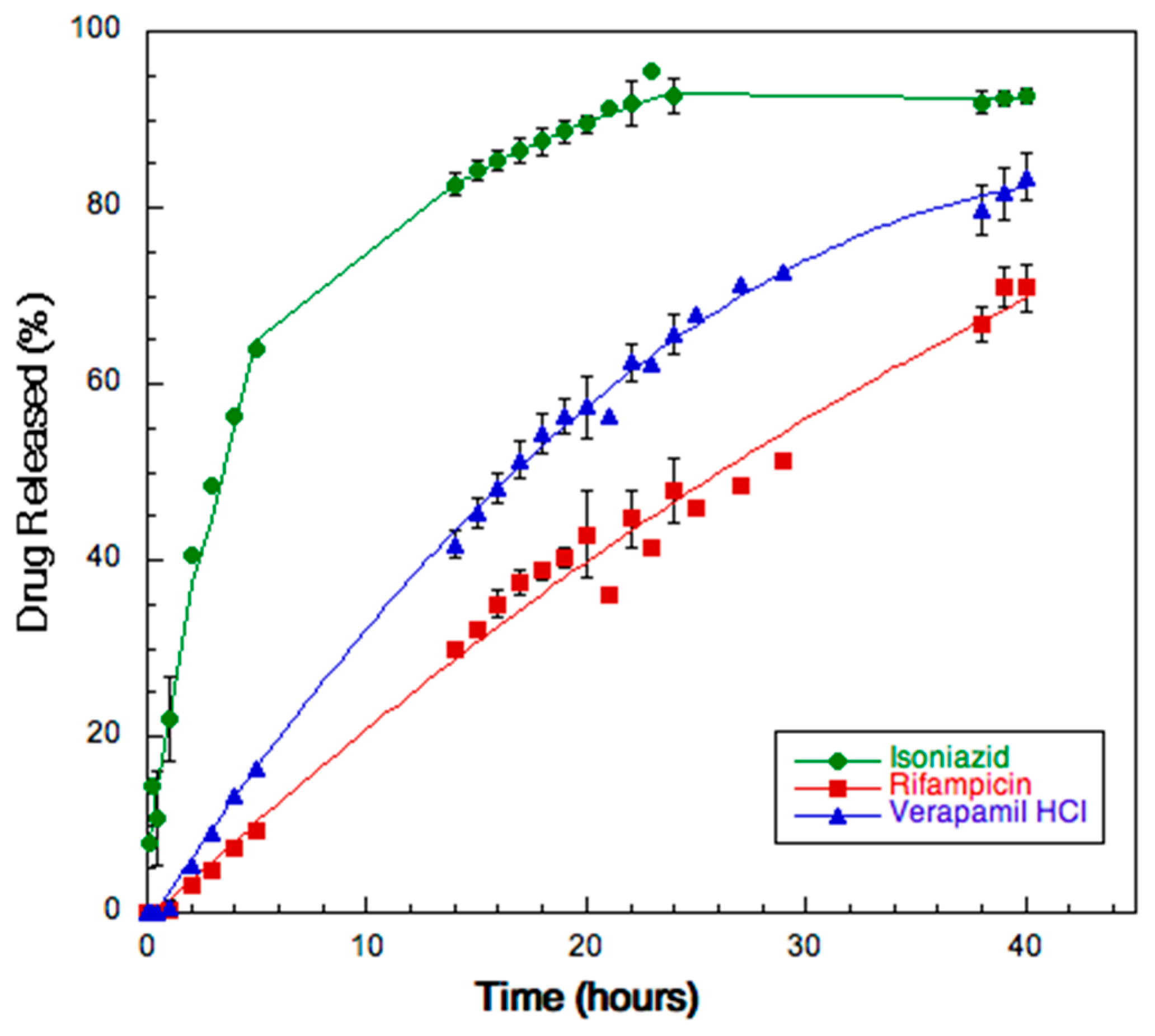
2.2.10. Dissolution
2.2.11. Minimum Inhibitory Concentrations of the Compounds against M. tuberculosis Strains
2.2.12. Evaluation of the Intracellular Activity against M. tuberculosis Strains
Isolation of Human Monocyte-Derived Macrophages
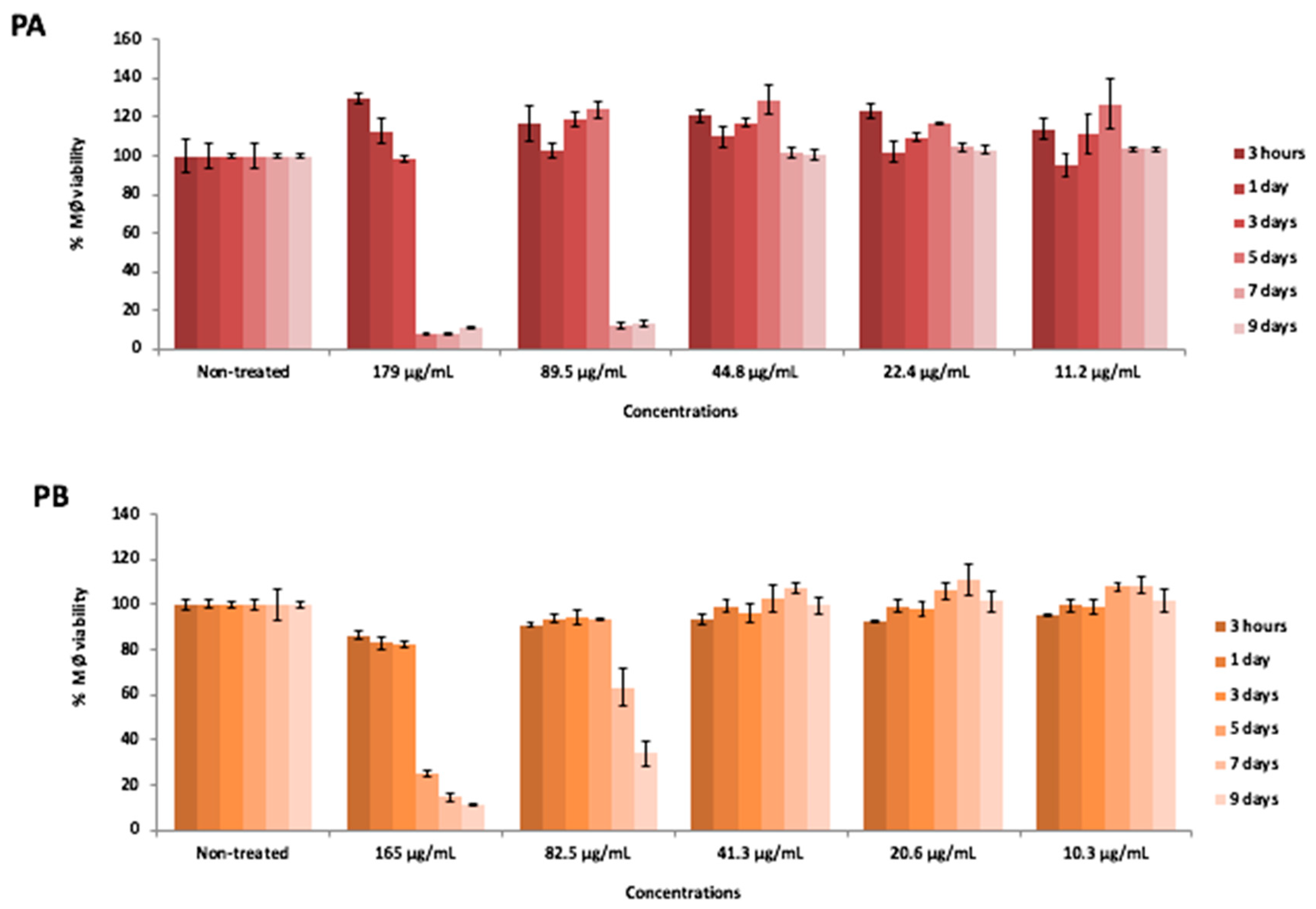
Cytotoxicity Analysis
Ex Vivo Assays
- SD powder A (nanoparticles formulation, NPs)—concentration 89.5 µg/mL, with verapamil-rifampicin 31.3 µg/mL; isoniazid 16.1 µg/mL; verapamil 10.7 µg/mL (as baseline); low molecular weight HA 31.4 µg/mL;
- SD powder B (nanoparticles formulation NPs)—concentration 82.5 µg/mL, without verapamil-rifampicin 31.4 µg/mL (as baseline); isoniazid 19.8 µg/mL; low molecular weight HA 31.4 µg/mL;
- solution A (free solution mixtures, Non-NPs)—rifampicin, 30 µg/mL; isoniazid, 15 µg/mL; verapamil, 10 µg/mL (as baseline);
- solution B (free solution mixtures, Non-NPs)—rifampicin, 30 µg/mL (as baseline); isoniazid 17.6 µg/mL.
3. Results
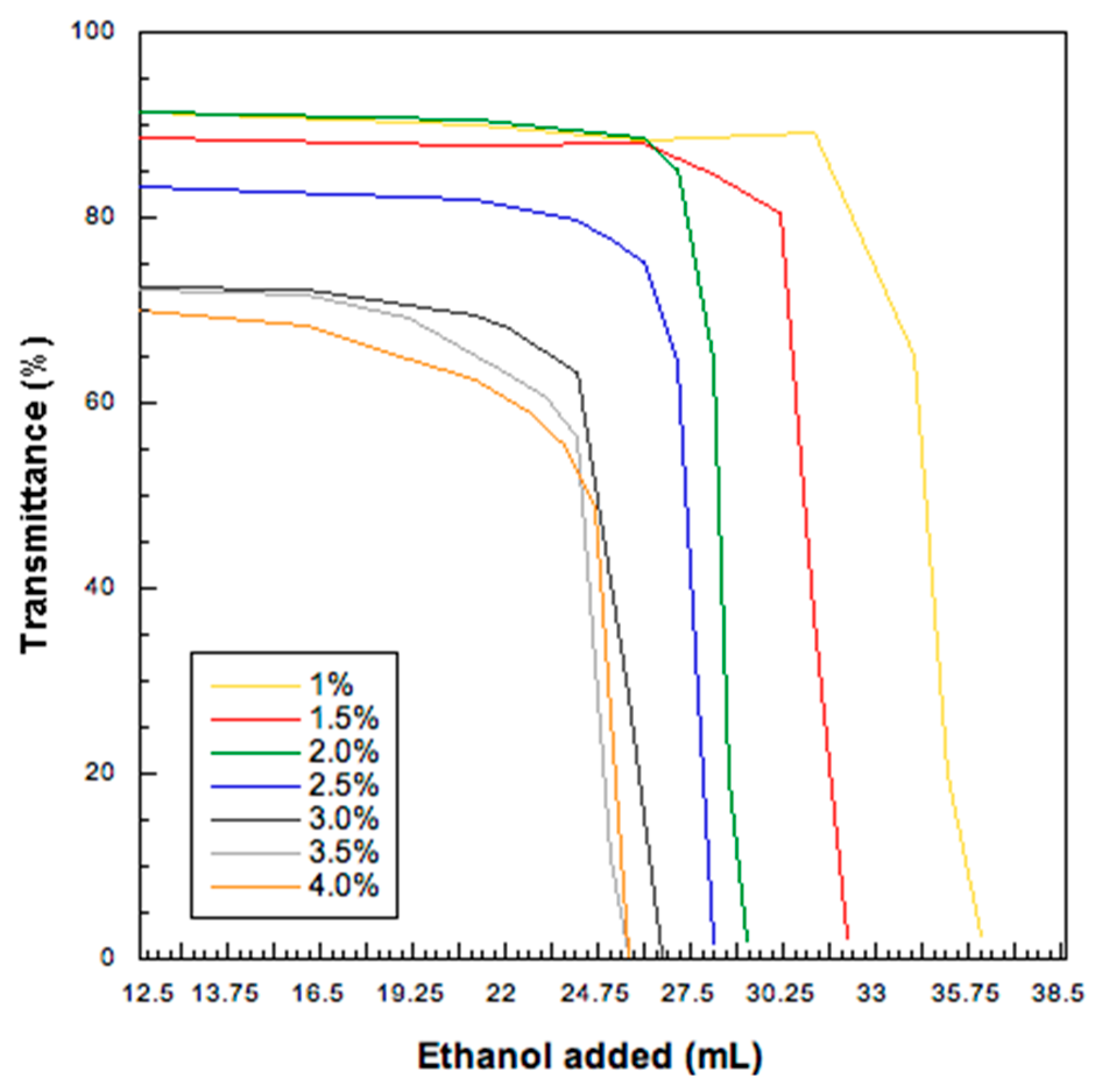
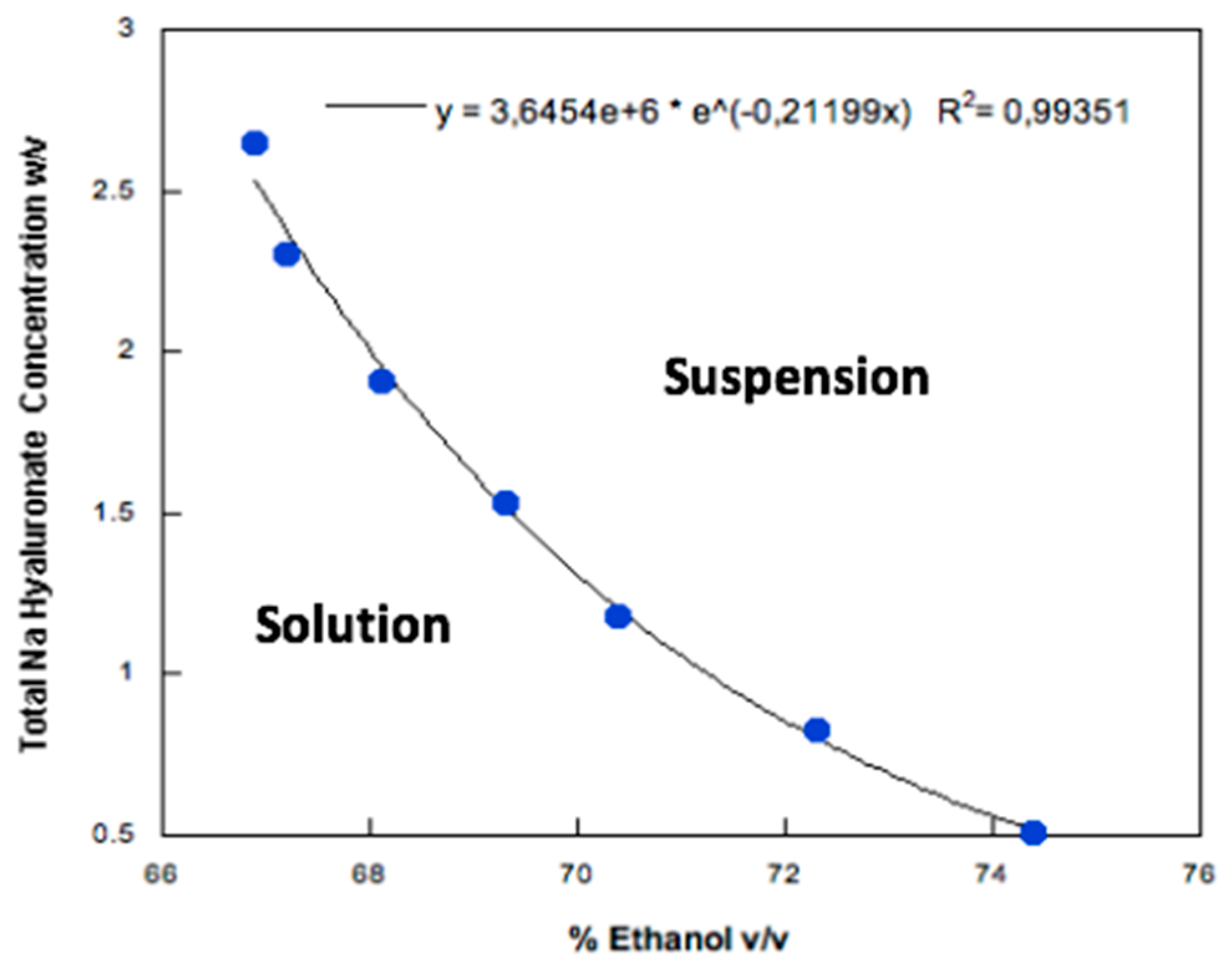
3.1. Particle Production
3.2. Powder Production, Thermal and Solid-State Analysis
3.3. Encapsulation Efficiency
3.4. Aerodynamic Performance
3.5. Preliminary Stability Assessment
3.6. De-aggregation Kinetics
3.7. Morphology Characterization
3.8. Dissolution
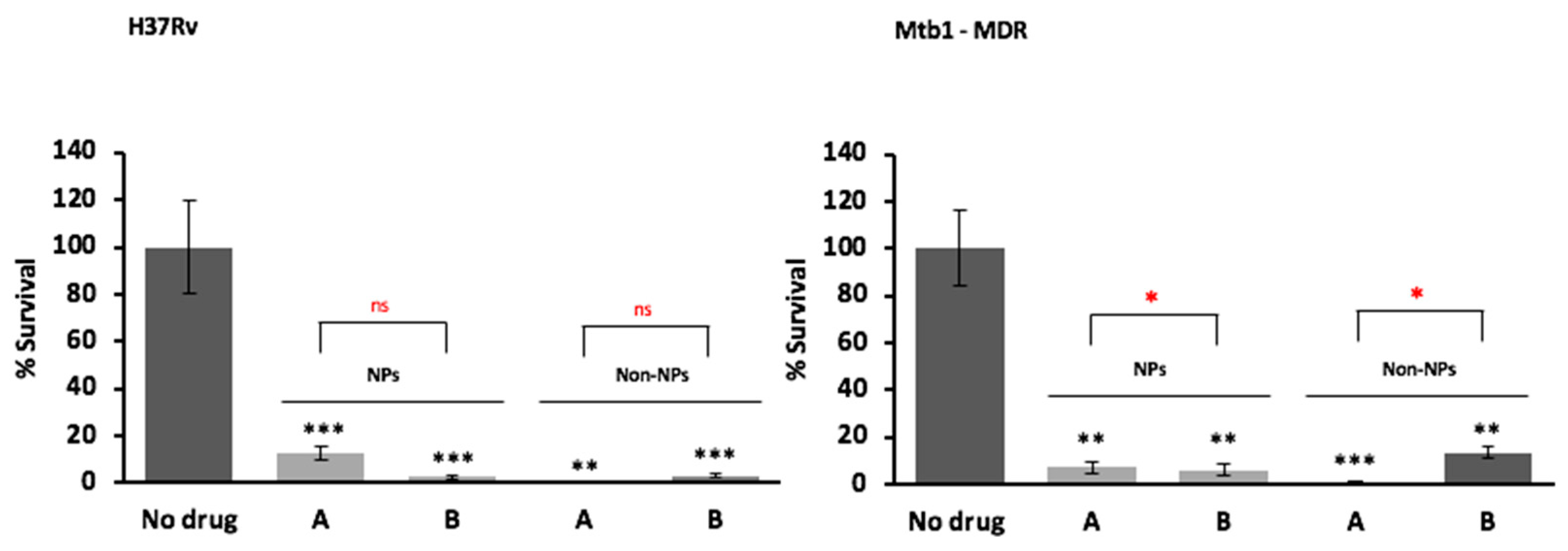
3.9. In Vitro Antimicrobial Activity against M. tuberculosis
3.10. Cytotoxicity Evaluation
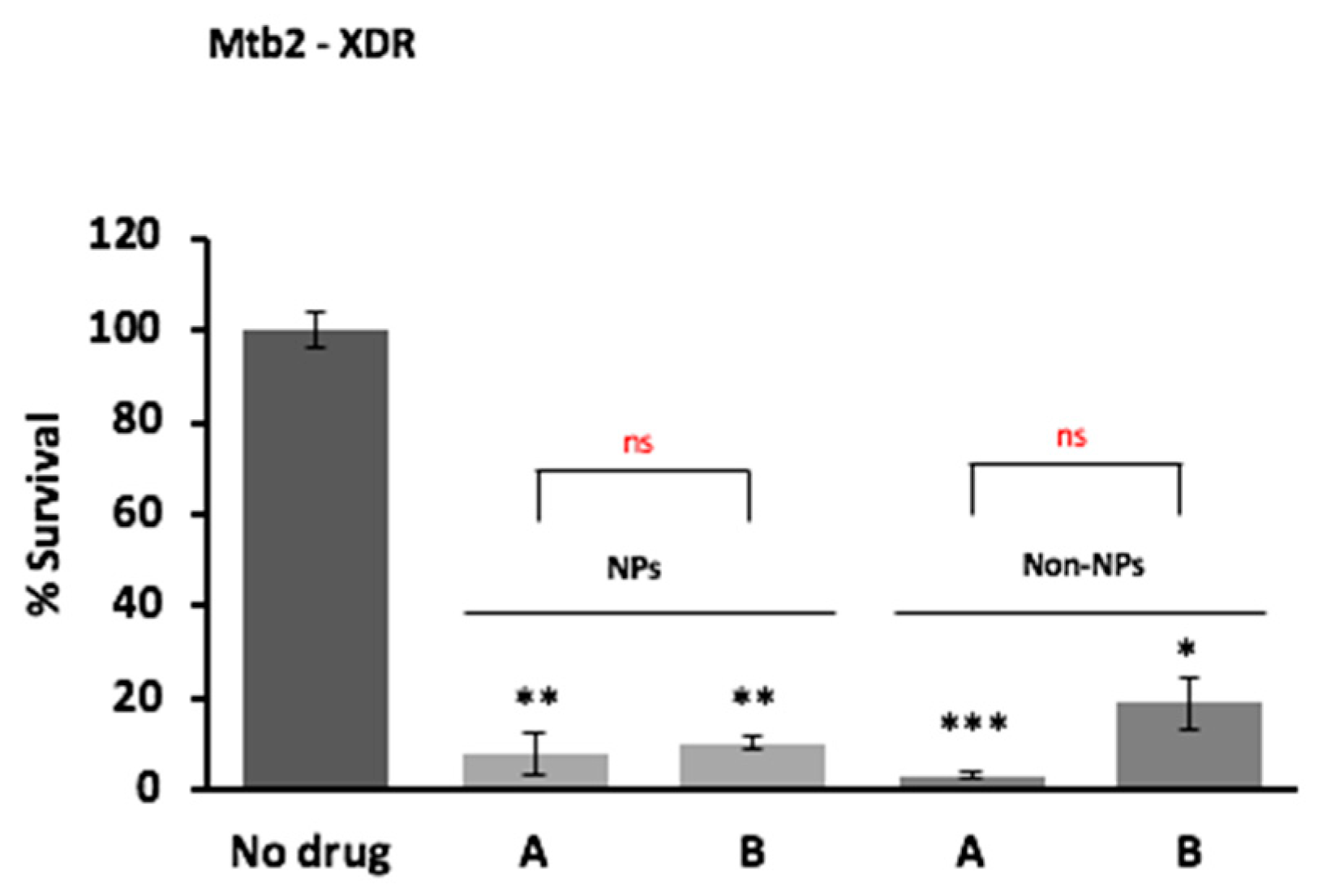
3.11. Ex Vivo Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2018; World Health Organization: Geneva, Switzerland, 2018; pp. 1–277. [Google Scholar]
- Johnson, M.M.; Odell, J.A. Nontuberculous mycobacterial pulmonary infections. J. Thorac. Dis. 2014, 6, 210–220. [Google Scholar] [PubMed]
- Chan, E.D.; Bai, X.; Kartalija, M.; Orme, I.M.; Ordway, D.J. Host immune response to rapidly growing mycobacteria, an emerging cause of chronic lung disease. Am. J. Respir. Cell. Mol. Biol. 2010, 43, 387–393. [Google Scholar] [CrossRef]
- Griffith, D.E.; Aksamit, T.R. Understanding nontuberculous mycobacterial lung disease: It’s been a long time coming. F1000Research 2016, 5, 2797–2798. [Google Scholar] [CrossRef]
- Helguera-Repetto, A.C.; Chacon-Salinas, R.; Cerna-Cortes, J.F.; Rivera-Gutierrez, S.; Ortiz-Navarrete, V.; Estrada-Garcia, I.; Gonzalez-y-Merchand, J.A. Differential Macrophage Response to Slow-and Fast-Growing Pathogenic Mycobacteria. BioMed Res. Int. 2014. [Google Scholar] [CrossRef]
- Stewart, G.R.; Robertson, B.D.; Young, D.B. Tuberculosis: A problem with persistence. Nat. Rev. Microbiol. 2003, 1, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Lezcano, O.M.; González-Cortés, C.; Reyes-Ruvalcaba, D.; Diez-Tascón, C. CCL20 is overexpressed in Mycobacterium tuberculosis-infected monocytes and inhibits the production of reactive oxygen species (ROS). Clin. Exp. Immunol. 2010, 162, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Crossman, D.K.; Mai, D.; Guidry, L.; Voskuil, M.I.; Renfrow, M.B.; Steyn, A.J.C. Mycobacterium tuberculosis WhiB3 Maintains Redox Homeostasis by Regulating Virulence Lipid Anabolism to Modulate Macrophage Response. PLoS Pathog. 2009, 5, e1000545. [Google Scholar] [CrossRef] [PubMed]
- Tufariello, J.M.; Chan, J.; Flynn, J.L. Latent tuberculosis: Mechanisms of host and bacillus that contribute to persistent infection. Lancet Infect. Dis. 2003, 3, 578–590. [Google Scholar] [CrossRef]
- Muttil, P.; Wang, C.; Hickey, A.J. Inhaled drug delivery for tuberculosis therapy. Pharm. Res. 2009, 26, 2401–2416. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.; Viveiros, M. Why thioridazine in combination with antibiotics cures extensively drug-resistant Mycobacterium tuberculosis infections. Int. J. Antimicrob. Agents 2012, 39, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Louw, G.E.; Warren, R.M.; Gey van Pittius, N.C.; McEvoy, C.R.E.; Van Helden, P.D.; Victor, T.C. A balancing act: Efflux/influx in mycobacterial drug resistance. Antimicrob. Agents Chemother. 2009, 53, 3181–3189. [Google Scholar] [CrossRef]
- Adams, K.N.; Szumowski, J.D.; Ramakrishnan, L. Verapamil, and its metabolite norverapamil, inhibit macrophage-induced, bacterial efflux pump-mediated tolerance to multiple anti-tubercular drugs. J. Infect. Dis. 2014, 210, 456–466. [Google Scholar] [CrossRef]
- Machado, D.; Perdigão, J.; Portugal, I.; Pieroni, M.; Silva, P.A.; Couto, I.; Viveiros, M. Efflux Activity Differentially Modulates the Levels of Isoniazid and Rifampicin Resistance among Multidrug Resistant and Monoresistant Mycobacterium tuberculosis Strains. Antibiotics 2018, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.S.; Lopes, E.; Azzali, E.; Machado, D.; Coelho, T.; da Silva, P.E.A.; Viveiros, M.; Pieroni, M.; Couto, I. An Experimental Model for the Rapid Screening of Compounds with Potential Use Against Mycobacteria. Assay Drug Dev. Technol. 2016, 14, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Parumasivam, T.; Chan, J.G.Y.; Pang, A.; Quan, D.H.; Triccas, J.A.; Britton, W.J.; Chan, H.K. In vitro evaluation of novel inhalable dry powders consisting of thioridazine and rifapentine for rapid tuberculosis treatment. Eur. J. Pharm. Biopharm. 2016, 107, 205–214. [Google Scholar] [CrossRef]
- Machado, D.; Couto, I.; Perdigão, J.; Rodrigues, L.; Portugal, I.; Baptista, P.; Veigas, B.; Amaral, L.; Viveiros, M. Contribution of efflux to the emergence of isoniazid and multidrug resistance in Mycobacterium tuberculosis. PLoS ONE 2012, 7, e34538. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gardete, S.; Jansen, R.S.; Shetty, A.; Dick, T.; Rhee, K.Y.; Dartois, V. Verapamil Targets Membrane Energetics in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62, 495. [Google Scholar] [CrossRef]
- Rinkenberger, R.L.; Prystowsky, E.N.; Heger, J.J.; Troup, P.J.; Jackman, W.M.; Zipes, D.P. Effects of intravenous and chronic oral verapamil administration in patients with supraventricular tachyarrhythmias. Circulation 1980, 62, 996–1010. [Google Scholar] [CrossRef]
- Boshoff, H.I.M.; Barry, C.E. Tuberculosis-metabolism and respiration in the absence of growth. Nat. Rev. Microbiol. 2005, 3, 70–80. [Google Scholar] [CrossRef]
- Hickey, A.J.; Misra, A.; Fourie, P.B. Dry powder antibiotic aerosol product development: Inhaled therapy for tuberculosis. J. Pharm. Sci. 2013, 102, 3900–3907. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y.; Biggs, D.L.; Manning, M.C.; Randolph, T.W.; Christians, U.; Hybertson, B.M.; Ng, K.-Y. Microparticle-based lung delivery of INH decreases INH metabolism and targets alveolar macrophages. J. Control. Release 2005, 107, 288–299. [Google Scholar]
- El-Sherbiny, I.M.; El-Baz, N.M.; Yacoub, M.H. Inhaled nano-and microparticles for drug delivery. Glob. Cardiol. Sci. Pract. 2015, 2, 1–14. [Google Scholar] [CrossRef]
- Maretti, E.; Costantino, L.; Rustichelli, C.; Leo, E.; Croce, M.A.; Buttini, F.; Truzzi, E.; Iannuccelli, V. Surface engineering of Solid Lipid Nanoparticle assemblies by methyl α-d-mannopyranoside for the active targeting to macrophages in anti-tuberculosis inhalation therapy. Int. J. Pharm. 2017, 528, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Geiser, M. Update on macrophage clearance of inhaled micro- and nanoparticles. J. Aerosol. Med. Pulm. Drug Deliv. 2010, 23, 207–217. [Google Scholar] [CrossRef]
- Todoroff, J.; Vanbever, R. Fate of nanomedicines in the lungs. Curr. Opin. Colloid Interface Sci. 2011, 16, 246–254. [Google Scholar] [CrossRef]
- Vyas, S.; Quraishi, S.; Gupta, S.; Jaganathan, K. Aerosolized liposome-based delivery of amphotericin B to alveolar macrophages. Int. J. Pharm. 2005, 296, 12–25. [Google Scholar] [CrossRef]
- Mortensen, N.P.; Durham, P.; Hickey, A.J. The role of particle physico-chemical properties in pulmonary drug delivery for tuberculosis therapy. J. Microencapsul. 2014, 31, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Hoppentocht, M.; Hagedoorn, P.; Frijlink, H.W.; de Boer, A.H. Developments and strategies for inhaled antibiotic drugs in tuberculosis therapy: A critical evaluation. Eur. J. Pharm. Biopharm. 2014, 86, 23–30. [Google Scholar]
- Ellis, T.; Chiappi, M.; García-Trenco, A.; Al-Ejji, M.; Sarkar, S.; Georgiou, T.K.; Shaffer, M.S.P.; Tetley, T.D.; Schwander, S.; Ryan, M.P.; et al. Multimetallic Microparticles Increase the Potency of Rifampicin against Intracellular Mycobacterium tuberculosis. ACS Nano 2018, 12, 5228–5240. [Google Scholar] [CrossRef]
- Cunha, L.; Rodrigues, S.; Rosa da Costa, A.; Faleiro, M.; Buttini, F.; Grenha, A. Inhalable Fucoidan Microparticles Combining Two Antitubercular Drugs with Potential Application in Pulmonary Tuberculosis Therapy. Polymers 2018, 10, 636. [Google Scholar] [CrossRef]
- Lesley, J.; Hascall, V.C.; Tammi, M.; Hyman, R. Hyaluronan binding by cell surface CD44. J. Biol. Chem. 2000, 275, 26967–26975. [Google Scholar] [CrossRef]
- Ghosh, S.; Hoselton, S.A.; Dorsam, G.P.; Schuh, J.M. Hyaluronan fragments as mediators of inflammation in allergic pulmonary disease. Immunobiology 2015, 220, 575–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Sarfraz, M.K.; Clas, S.-D.; Roa, W.; Löbenberg, R. Hyaluronic Acid-Tocopherol Succinate-Based Self-Assembling Micelles for Targeted Delivery of Rifampicin to Alveolar Macrophages. J. Biomed. Nanotechnol. 2015, 11, 1312–1329. [Google Scholar] [CrossRef] [PubMed]
- Rayahin, J.E.; Buhrman, J.S.; Zhang, Y.; Koh, T.J.; Gemeinhart, R.A. High and Low Molecular Weight Hyaluronic Acid Differentially Influence Macrophage Activation. ACS Biomat. Sci. Eng. 2015, 1, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, F.; Balducci, A.G.; Kumar, A.; Sonvico, F.; Forbes, B.; Bettini, R.; Buttini, F. Engineered sodium hyaluronate respirable dry powders for pulmonary drug delivery. Int. J. Pharm. 2017, 517, 286–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.G.Y.; Chan, H.-K.; Prestidge, C.A.; Denman, J.A.; Young, P.M.; Traini, D. A novel dry powder inhalable formulation incorporating three first-line anti-tubercular antibiotics. Eur. J. Pharm. Biopharm. 2013, 83, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Karimi, K.; Katona, G.; Csóka, I.; Ambrus, R. Physicochemical stability and aerosolization performance of dry powder inhalation system containing ciprofloxacin hydrochloride. J. Pharm. Biomed. Anal. 2018, 148, 73–79. [Google Scholar] [CrossRef]
- Caviedes, L.; Delgado, J.; Gilman, R.H. Tetrazolium Microplate Assay as a Rapid and Inexpensive Colorimetric Method for Determination of Antibiotic Susceptibility of Mycobacterium tuberculosis. J. Clin. Microbiol. 2002, 40, 1873–1874. [Google Scholar] [CrossRef] [Green Version]
- Coelho, T.; Machado, D.; Couto, I.; Maschmann, R.; Ramos, D.; von Groll, A.; Rossetti, M.L.; Silva, P.A.; Viveiros, M. Enhancement of antibiotic activity by efflux inhibitors against multidrug resistant Mycobacterium tuberculosis clinical isolates from Brazil. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Vestal, A.L. Procedure for Isolation and Identification of Mycobacteria. U.S. Department of Health, Education, and Welfare Publication No. (CDC) 77-8230; Centers for Disease Control and Prevention: Atlanta, GA, USA, 1977.
- Machado, D.; Pires, D.; Perdigão, J.; Couto, I.; Portugal, I.; Martins, M.; Amaral, L.; Anes, E.; Viveiros, M. Ion Channel Blockers as Antimicrobial Agents, Efflux Inhibitors, and Enhancers of Macrophage Killing Activity against Drug Resistant Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0149326. [Google Scholar] [CrossRef]
- Machado, D.; Fernandes, L.; Costa, S.S.; Cannalire, R.; Manfroni, G.; Tabarrini, O.; Couto, I.; Sabatini, S.; Viveiros, M. Mode of action of the 2-phenylquinoline efflux inhibitor PQQ4R against Escherichia coli. PeerJ 2017, 5, e3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Necas, J.; Bartosikova, L.; Brauner, P.; Kolar, J. Hyaluronic acid (hyaluronan): A review. Veterinární Med. 2008, 53, 397–411. [Google Scholar] [CrossRef]
- Kluin, O.S.; Busscher, H.J.; Neut, D.; van der Mei, H.C. Poly (trimethylene carbonate) as a carrier for rifampicin and vancomycin to target therapy-recalcitrant staphylococcal biofilms. J. Orthop. Res. 2016, 34, 1828–1837. [Google Scholar] [CrossRef]
- Yoshida, M.I.; Gomes, E.C.L.; Soares, C.D.V.; Cunha, A.F.; Oliveira, M.A. Thermal Analysis Applied to Verapamil Hydrochloride Characterization in Pharmaceutical Formulations. Molecules 2010, 15, 2439–2452. [Google Scholar] [CrossRef] [Green Version]
- Schütz, E.; Ha, H.R.; Bühler, F.R.; Follath, F. Serum concentration and antihypertensive effect of slow-release verapamil. J. Cardiovasc. Pharmacol. 1982, 4 (Suppl. 3), S346–S349. [Google Scholar]
- Böttger, E.C. Drug Resistance in Mycobacterium Tuberculosis: Molecular Mechanisms and Laboratory Susceptibility Testing, Antituberculosis Chemotherapy; Karger: Basel, Switzerland, 2011; Volume 40, pp. 128–144. [Google Scholar]
- Balducci, A.G.; Bettini, R.; Colombo, P.; Buttini, F. Drug Delivery Strategies for Pulmonary Administration of Antibiotics. In Pulmonary Drug Delivery: Advances and Challenges; John Wiley and Sons, Ltd.: Chichester, UK, 2015; Volume 6, pp. 241–262. [Google Scholar]
- Eedara, B.B.; Rangnekar, B.; Sinha, S.; Doyle, C.; Cavallaro, A.; Das, S.C. Development and characterization of high payload combination dry powders of anti-tubercular drugs for treating pulmonary tuberculosis. Eur. J. Pharm. Sci. 2018, 118, 216–226. [Google Scholar] [CrossRef]
- Chan, J.G.Y.; Tyne, A.S.; Pang, A.; Chan, H.-K.; Young, P.M.; Britton, W.J.; Duke, C.C.; Traini, D. A Rifapentine-Containing Inhaled Triple Antibiotic Formulation for Rapid Treatment of Tubercular Infection. Pharm. Res. 2013, 31, 1239–1253. [Google Scholar] [CrossRef]
- Gupta, S.; Tyagi, S.; Almeida, D.V.; Maiga, M.C.; Ammerman, N.C.; Bishai, W.R. Acceleration of Tuberculosis Treatment by Adjunctive Therapy with Verapamil as an Efflux Inhibitor. Am. J. Respir. Crit. Care Med. 2013, 188, 600–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popa, V.T.; Somani, P.; Simon, V. The Effect of Inhaled Verapamil on Resting Bronchial Tone and Airway Contractions Induced by Histamine and Acetylcholine in Normal and Asthmatic Subjects. Am. J. Respir. Crit. Care Med. 1984, 130, 1006–1013. [Google Scholar]
- Pennock, G.D.; Dalton, W.S.; Roeske, W.R.; Appleton, C.P.; Mosley, K.; Plezia, P.; Miller, T.P.; Salmon, S.E. Systemic Toxic Effects Associated with High-Dose Verapamil Infusion and Chemotherapy Administration. JNCI J. Natl. Cancer Inst. 1991, 83, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Harman, E.; Hill, M.; Pieper, J.A.; Hendeles, L. Inhaled Verapamil-induced Bronchoconstriction in Mild Asthma. Chest 1991, 100, 17–22. [Google Scholar] [CrossRef]
- MCintyre, E.; Fitzgibbon, B.; Otto, H.; Minson, R.; Alpers, J.; Ruffin, R. Inhaled verapamil in histamine-induced bronchoconstriction. J. Allergy Clin. Immunol. Pract 1983, 71, 375–381. [Google Scholar] [CrossRef]
- Bhutani, H.; Singh, S.; Vir, S.; Bhutani, K.K.; Kumar, R.; Chakraborti, A.K.; Jindal, K.C. LC and LC-MS study of stress decomposition behaviour of isoniazid and establishment of validated stability-indicating assay method. J. Pharm. Biomed. Anal. 2007, 43, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Florey, K.; Brewer, G.A. Analytical profiles of drug substances: Isoniazid. Acad. Pharm. Sci. 2006, 88, 112–116. [Google Scholar]
- Pereira, G.G.; Detoni, C.B.; Balducci, A.G.; Rondelli, V.; Colombo, P.; Guterres, S.S.; Sonvico, F. Hyaluronate nanoparticles included in polymer films for the prolonged release of vitamin E for the management of skin wounds. Eur. J. Pharm. Sci. 2016, 83, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.-H.; Dong, X.-M.; Liu, C.-G. In Vitro Investigation of Self-Assembled Nanoparticles Based on Hyaluronic Acid-Deoxycholic Acid Conjugates for Controlled Release Doxorubicin: Effect of Degree of Substitution of Deoxycholic Acid. Int. J. Mol. Sci. 2015, 16, 7195–7209. [Google Scholar] [CrossRef] [Green Version]
- Rawal, T.; Parmar, R.; Tyagi, R.K.; Butani, S. Rifampicin loaded chitosan nanoparticle dry powder presents an improved therapeutic approach for alveolar tuberculosis. Colloids Surf. B 2017, 154, 321–330. [Google Scholar] [CrossRef]
- Ge, Z.; Ma, R.; Xu, G.; Chen, Z.; Zhang, D.; Wang, Q.; Hei, L.; Ma, W. Development and In Vitro Release of Isoniazid and Rifampicin-Loaded Bovine Serum Albumin Nanoparticles. Med. Sci. Monit. 2018, 24, 473–478. [Google Scholar] [CrossRef]
- Qin, H.; Wang, C.M.; Dong, Q.Q.; Zhang, L.; Zhang, X.; Ma, Z.Y.; Han, Q.R. Preparation and characterization of magnetic Fe3O4–chitosan nanoparticles loaded with isoniazid. J. Magn. Magn. Mater. 2015, 381, 120–126. [Google Scholar] [CrossRef]
- Nagheh, Z.; Irani, S.; Mirfakhraie, R.; Dinarvand, R. SN38-PEG-PLGA-verapamil nanoparticles inhibit proliferation and downregulate drug transporter ABCG2 gene expression in colorectal cancer cells. Progr. Biomater. 2017, 6, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, F.; Balducci, A.G.; Rossi, A.; Sonvico, F.; Colombo, P.; Buttini, F. “Pierce and inhale” design in capsule based dry powder inhalers: Effect of capsule piercing and motion on aerodynamic performance of drugs. Int. J. Pharm. 2015, 487, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Oh, N. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomed. 2014, 9, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, P.; Bettini, R.; Santi, P.; De Ascentiis, A.; Peppas, N.A. Analysis of the swelling and release mechanisms from drug delivery systems with emphasis on drug solubility and water transport. J. Control. Release 1996, 39, 231–237. [Google Scholar] [CrossRef]
- Kundawala, A.J.; Patel, V.A.; Patel, H.V.; Choudhary, D. Isoniazid loaded chitosan microspheres for pulmonary delivery: Preparation and characterization. Der Pharm. Sin. 2011, 2, 88–97. [Google Scholar]
- Wang, W.; Zhou, Q.T.; Sun, S.-P.; Denman, J.A.; Gengenbach, T.R.; Barraud, N.; Rice, S.A.; Li, J.; Yang, M.; Chan, H.-K. Effects of Surface Composition on the Aerosolisation and Dissolution of Inhaled Antibiotic Combination Powders Consisting of Colistin and Rifampicin. AAPS J. 2015, 18, 372–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parumasivam, T.; Chan, J.G.Y.; Pang, A.; Quan, D.H.; Triccas, J.A.; Britton, W.J.; Chan, H.K. In Vitro Evaluation of Inhalable Verapamil-Rifapentine Particles for Tuberculosis Therapy. Mol. Pharm. 2016, 13, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Floroiu, A.; Klein, M.; Krämer, J.; Lehr, C.-M. Towards Standardized Dissolution Techniques for In Vitro Performance Testing of Dry Powder Inhalers. Dissolut. Technol. 2018, 25, 6–18. [Google Scholar] [CrossRef]
- Makino, K.; Nakajima, T.; Shikamura, M.; Ito, F.; Ando, S.; Kochi, C.; Inagawa, H.; Soma, G.-I.; Terada, H. Efficient intracellular delivery of rifampicin to alveolar macrophages using rifampicin-loaded PLGA microspheres: Effects of molecular weight and composition of PLGA on release of rifampicin. Colloids Surf. B 2004, 36, 35–42. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Nominal (% w/w) | Assay (% of the Nominal Value) | ||
|---|---|---|---|---|
| Time zero | Month 1/25 °C | 24 h/50 °C | ||
| Isoniazid | 18 | 98.4 ± 0.03 | 90.4 ± 0.02 | 81.3 ± 0.02 |
| Rifampicin | 35 | 100 ± 0.03 | 100 ± 0.01 | 96.9 ± 0.03 |
| Verapamil | 12 | 100 ± 0.02 | 100 ± 0.01 | 99.2 ± 0.04 |
| Drug | RS01® | Turbospin® | ||||||
|---|---|---|---|---|---|---|---|---|
| ED (mg) | EF (%) | FPD (mg) | FPF (%) | ED (mg) | EF (%) | FPD (mg) | FPF (%) | |
| Isoniazid | 0.44 ± 0.14 | 49.35 ± 15.32 | 0.40 ± 0.16 | 90.62 ± 8.15 | 0.63 ± 0.17 | 88.72 ± 3.35 | 0.47 ± 0.10 | 76.88 ± 7.48 |
| Rifampicin | 0.87 ± 0.26 | 47.86 ± 14.19 | 0.83 ± 0.29 | 93.55 ± 5.69 | 1.23 ± 0.20 | 74.75 ± 6.23 | 0.96 ± 0.15 | 77.68 ± 4.87 |
| Verapamil | 0.31 ± 0.10 | 50.89 ± 16.16 | 0.30 ± 0.11 | 93.89 ± 5.72 | 0.37 ± 0.04 | 68.43 ± 3.14 | 0.30 ± 0.05 | 80.54 ± 5.01 |
| Drug | RS01® | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 Month/25 °C | 24 h/50 °C | |||||||
| ED (mg) | EF (%) | FPD (mg) | FPF (%) | ED (mg) | EF (%) | FPD (mg) | FPF (%) | |
| Isoniazid | 0.50 ± 0.05 | 61.99 ± 5.92 | 0.46 ± 0.04 | 92.34 ± 1.28 | 0.42 ± 0.01 | 58.75 ± 1.96 | 0.37 ± 0.01 | 87.95 ± 0.57 |
| Rifampicin | 1.03 ± 0.11 | 58.56 ± 6.07 | 0.93 ± 0.08 | 90.81 ± 1.21 | 0.95 ± 0.06 | 55.81 ± 3.11 | 0.80 ± 0.05 | 83.62 ± 0.83 |
| Verapamil | 0.37 ± 0.03 | 62.83 ± 5.35 | 0.34 ± 0.03 | 92.26 ± 0.85 | 0.35 ± 0.02 | 59.54 ± 4.24 | 0.31 ± 0.03 | 88.81 ± 1.33 |
| Specimen | MIC (µg/mL) | ||
|---|---|---|---|
| H37Rv INHS/RIFS | Mtb1-MDR | Mtb2-XDR | |
| SD Powder A * | 0.25 | 32 | 32 |
| SD Powder B ** | 0.25 | 32 | 32 |
| RIF | 0.5 | 512 | 512 |
| INH | 0.1 | 12.8 | 12.8 |
| VER | 512 | 512 | 512 |
| HA | >256 | >256 | >256 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, I.; Buttini, F.; Sonvico, F.; Affaticati, F.; Martinelli, F.; Annunziato, G.; Machado, D.; Viveiros, M.; Pieroni, M.; Bettini, R. Sodium Hyaluronate Nanocomposite Respirable Microparticles to Tackle Antibiotic Resistance with Potential Application in Treatment of Mycobacterial Pulmonary Infections. Pharmaceutics 2019, 11, 203. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11050203
Rossi I, Buttini F, Sonvico F, Affaticati F, Martinelli F, Annunziato G, Machado D, Viveiros M, Pieroni M, Bettini R. Sodium Hyaluronate Nanocomposite Respirable Microparticles to Tackle Antibiotic Resistance with Potential Application in Treatment of Mycobacterial Pulmonary Infections. Pharmaceutics. 2019; 11(5):203. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11050203
Chicago/Turabian StyleRossi, Irene, Francesca Buttini, Fabio Sonvico, Filippo Affaticati, Francesco Martinelli, Giannamaria Annunziato, Diana Machado, Miguel Viveiros, Marco Pieroni, and Ruggero Bettini. 2019. "Sodium Hyaluronate Nanocomposite Respirable Microparticles to Tackle Antibiotic Resistance with Potential Application in Treatment of Mycobacterial Pulmonary Infections" Pharmaceutics 11, no. 5: 203. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11050203