High-Throughput Dissolution/Permeation Screening—A 96-Well Two-Compartment Microplate Approach


Abstract
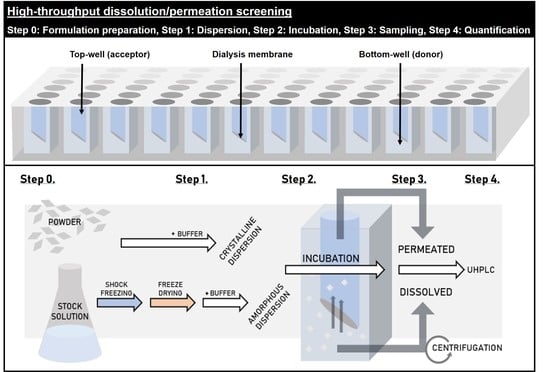
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Methods
2.2.1. Preparation of Dispersion Media and Acceptor Media
2.2.2. Solubilization of Tadalafil in Surfactant Solutions
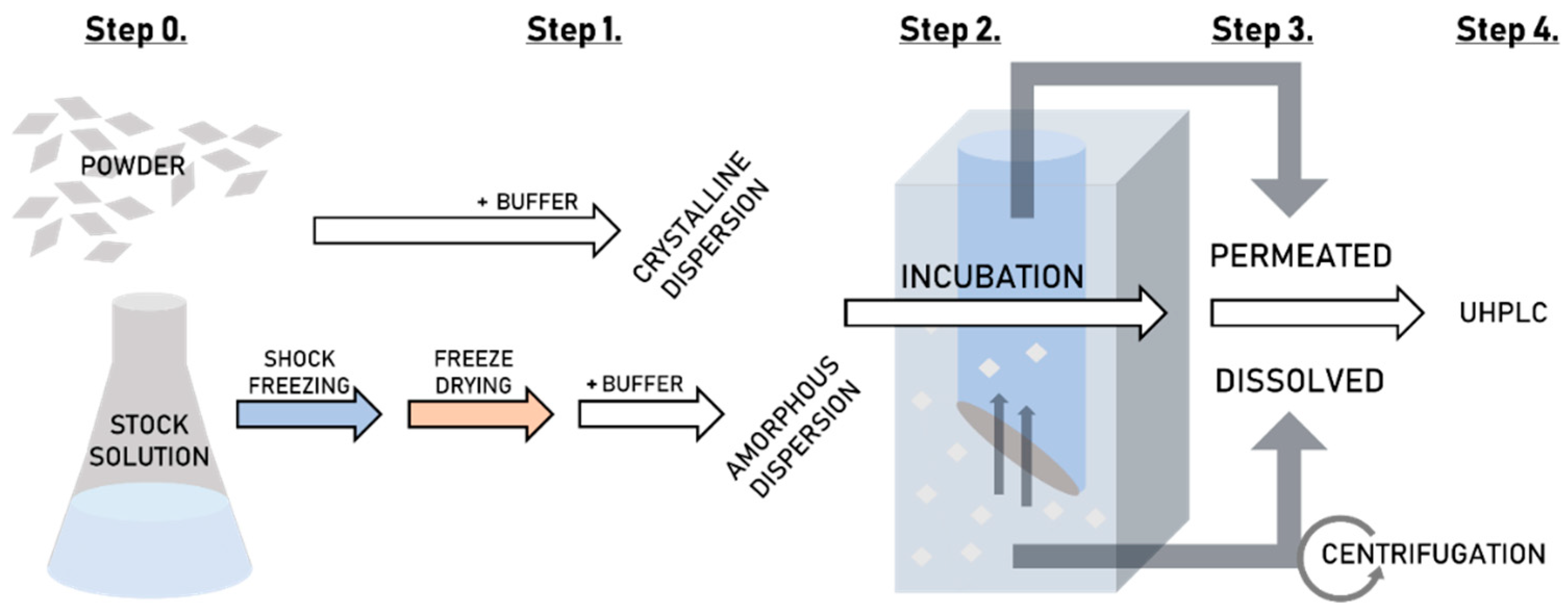
2.2.3. Preparation of Tadalafil Formulations and Their Aqueous Dispersions for High-Throughput Dissolution/Permeation Screening
2.2.4. X-Ray Powder Diffraction
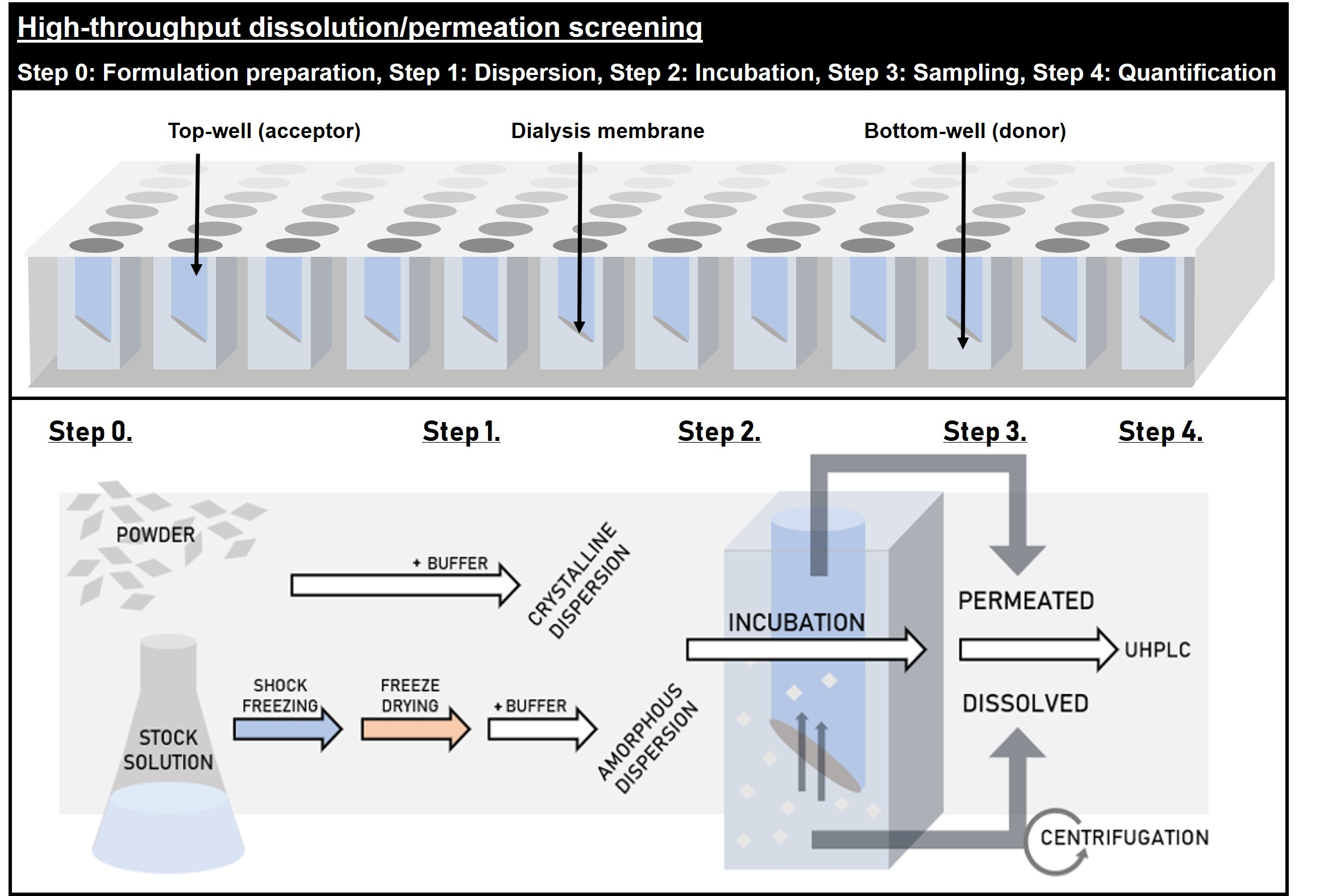
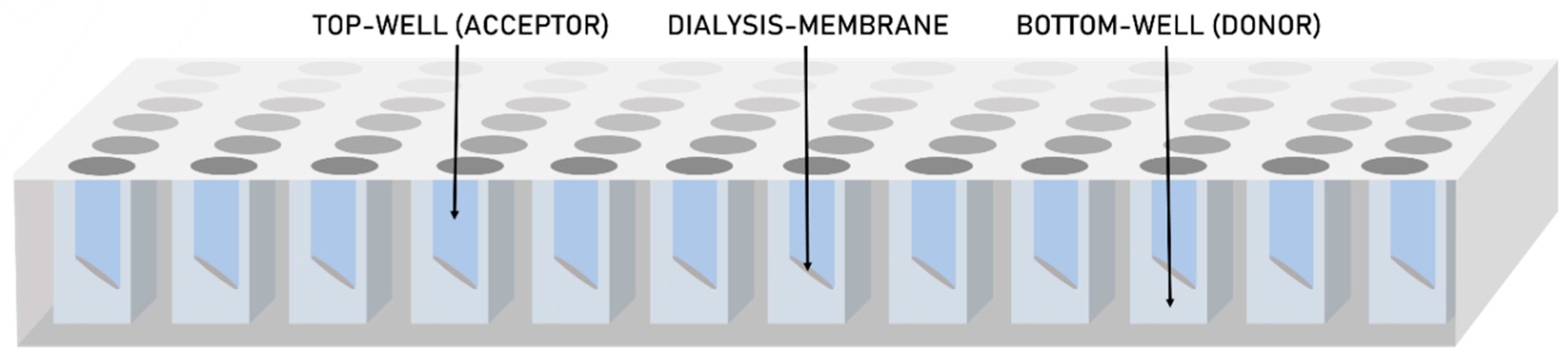
2.2.5. High-Throughput Dissolution/Permeation Screening
2.2.6. Non-Specific Adsorption of Tadalafil to Plastic Material
2.2.7. Quantification of Tadalafil by UHPLC-UV
2.2.8. Statistical Analysis
3. Results and Discussion
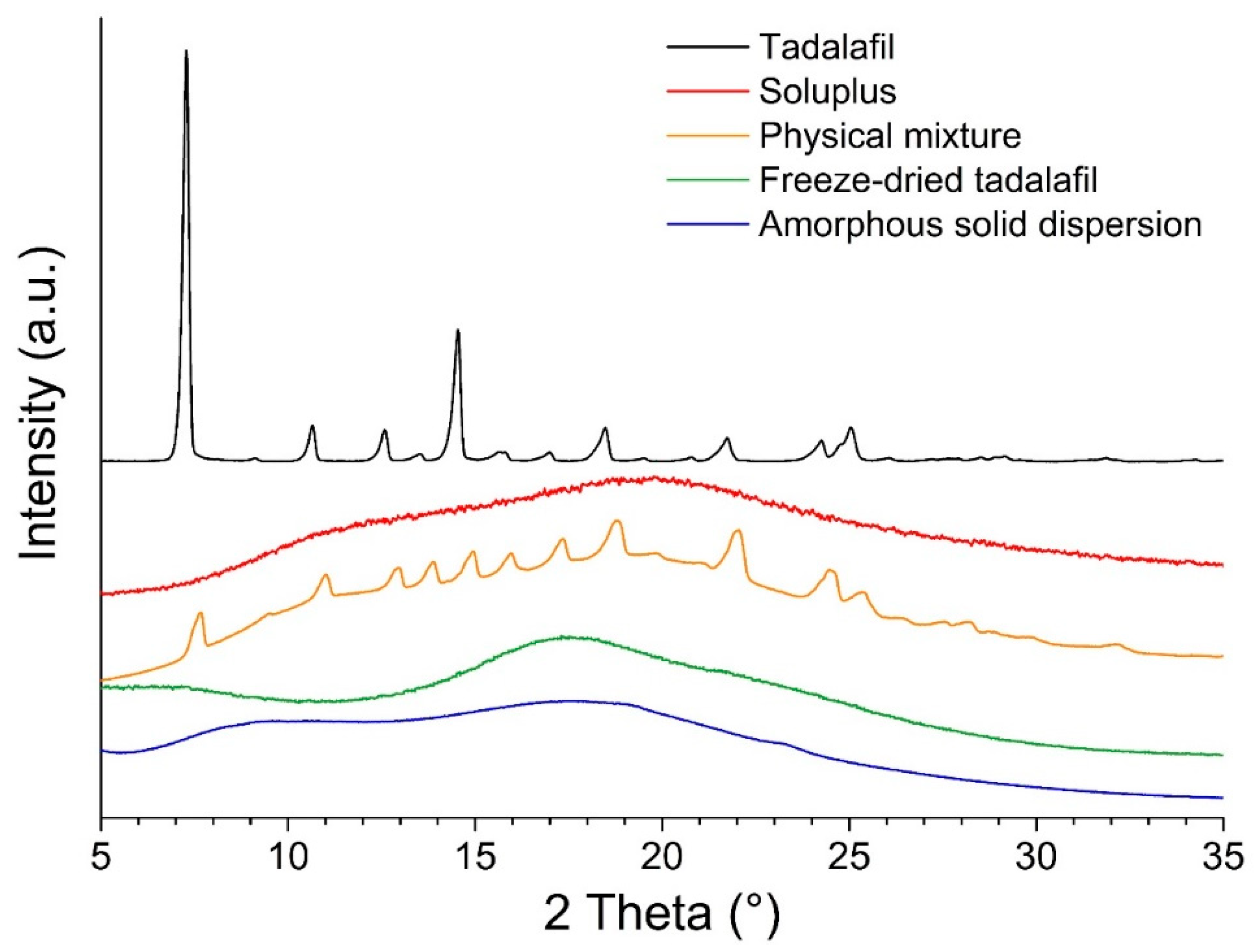
3.1. Sample Preparation and Solid-State Analysis by X-Ray Powder Diffraction
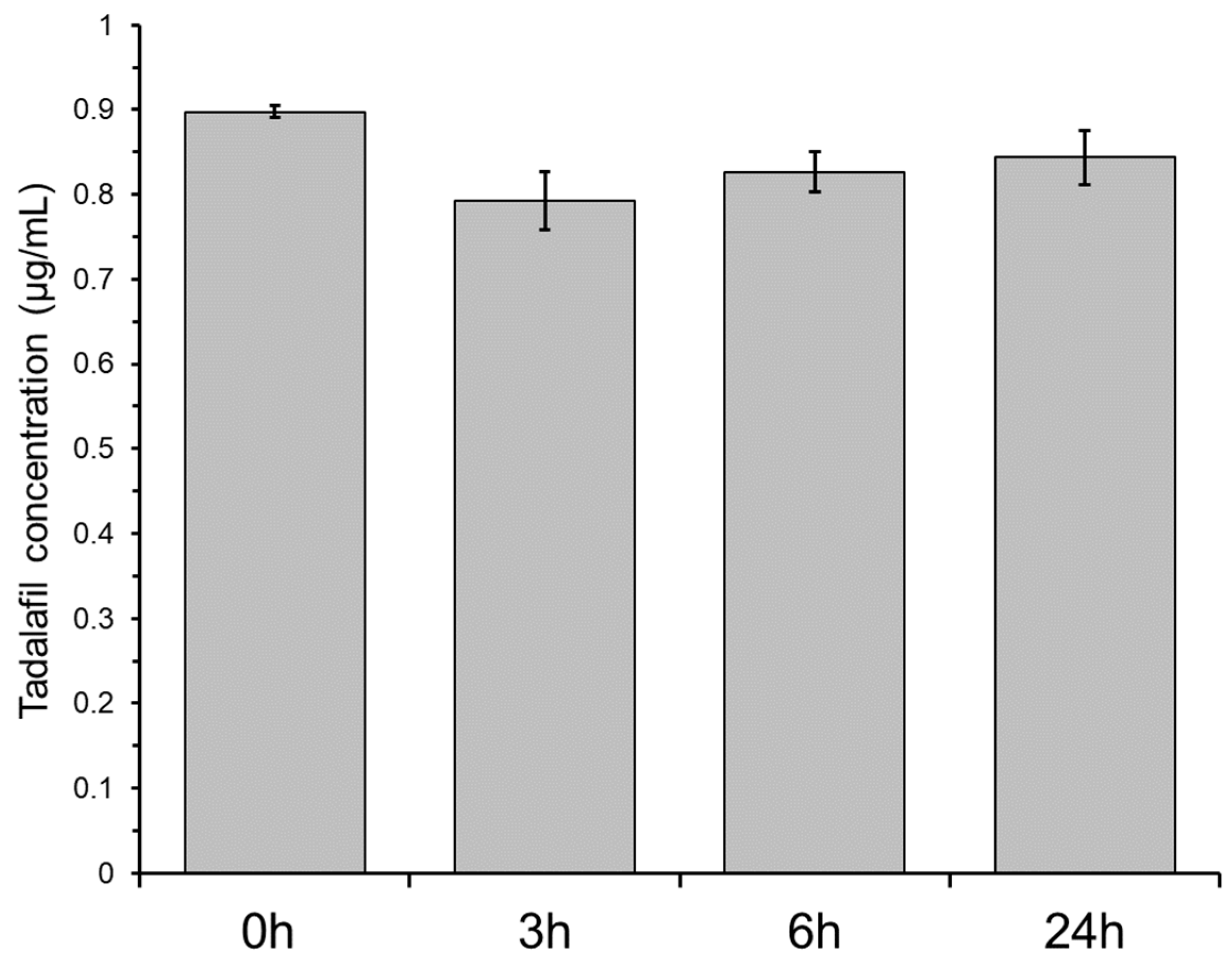
3.2. Non-Specific Adsorption of Tadalafil to Plastic Material
3.3. Preliminary Experiments—Incubation Time, Acceptor Media and Dispersion Media
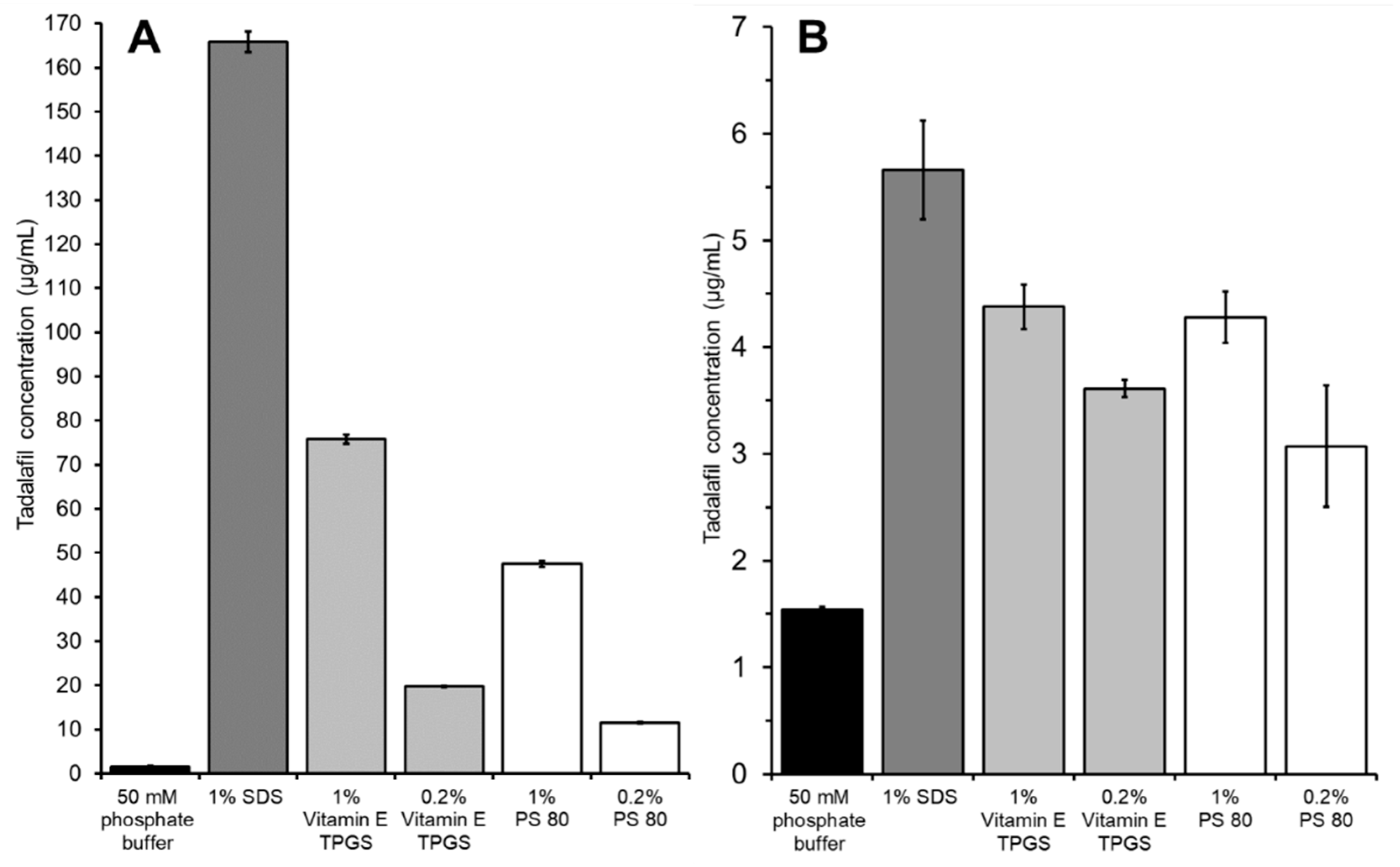
3.3.1. The Influence of Acceptor Media
3.3.2. The Influence of Incubation Time
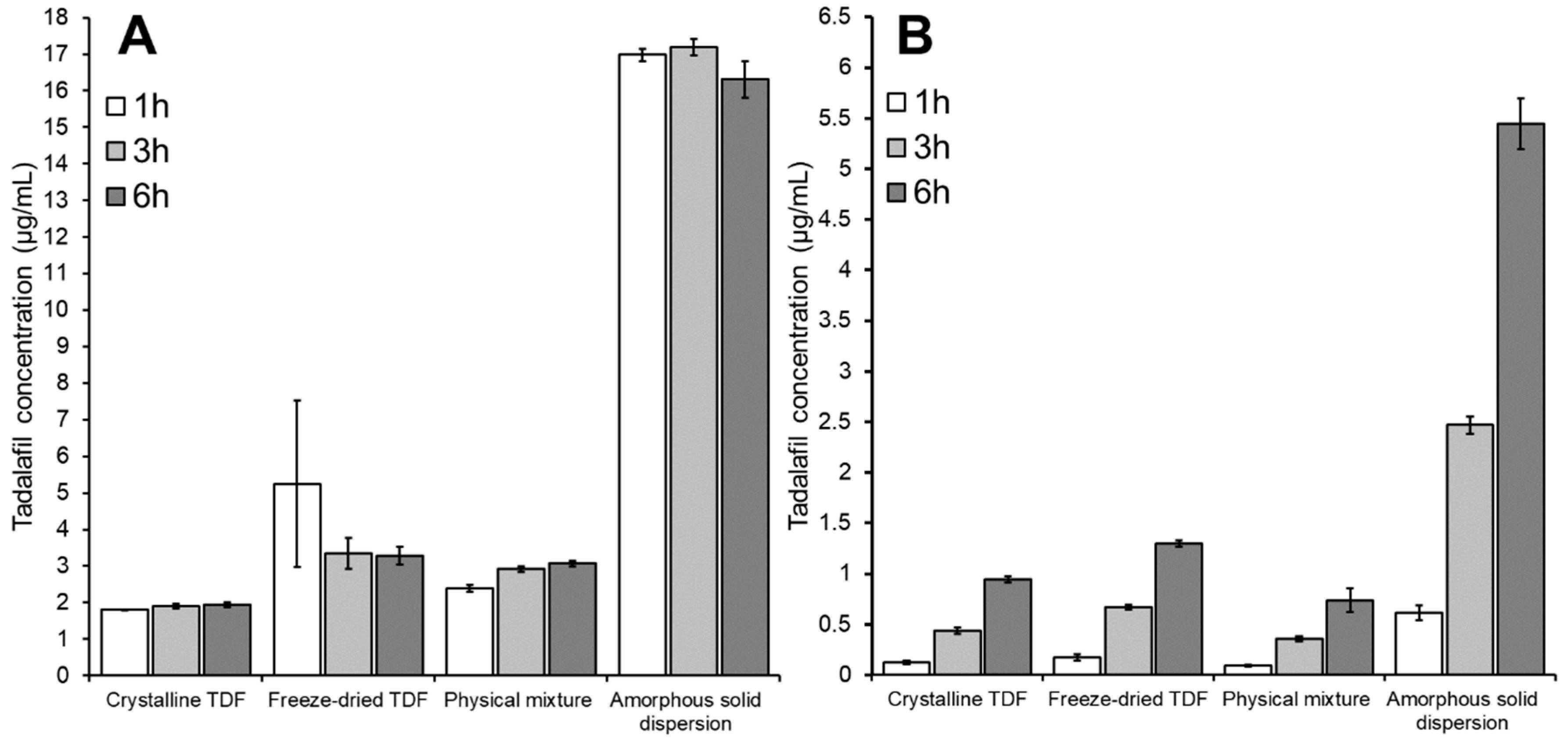
3.3.3. The influence of Dispersion Media
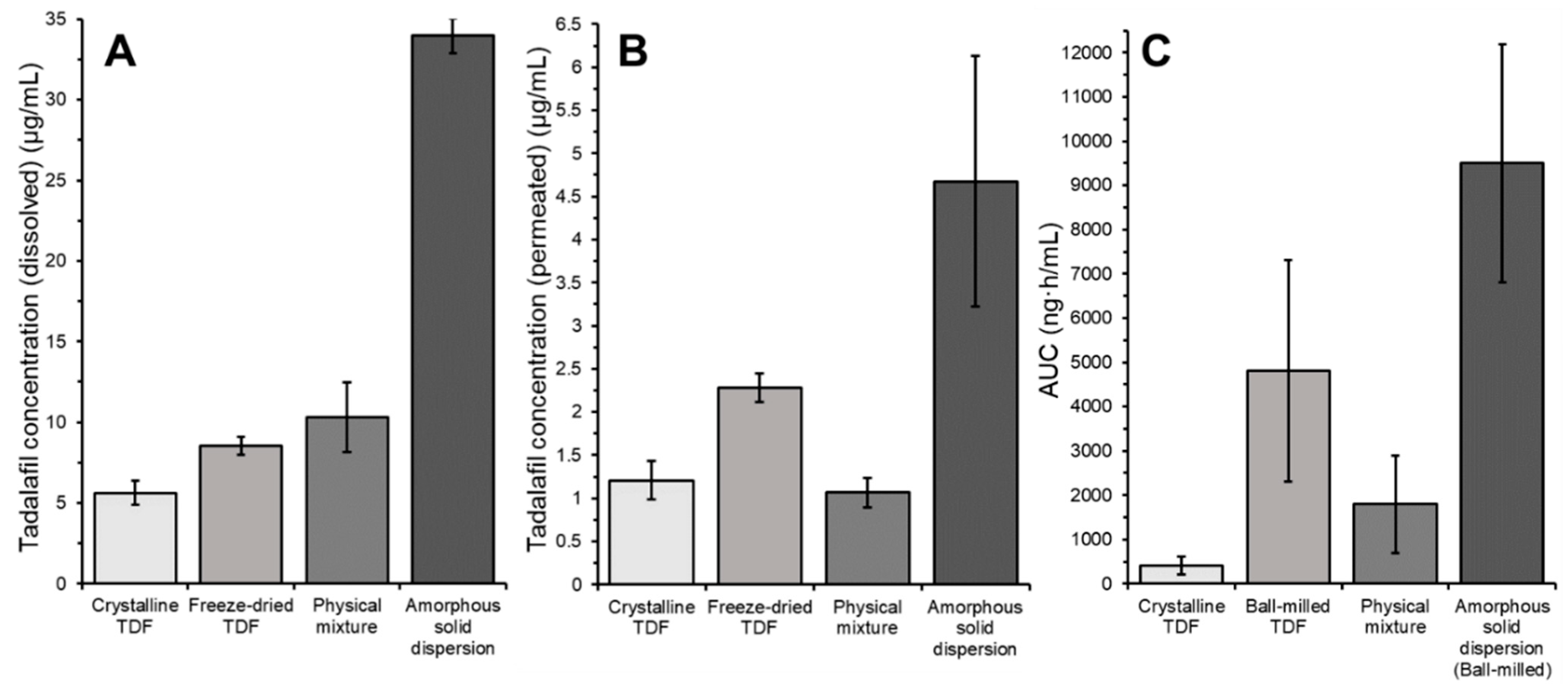
3.4. In Vitro In Vivo Correlation: Comparing the In Vitro High-Throughput Dissolution/Permeation Screening Results to In Vivo Oral Bioavailability Results
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Black, S.N.; Collier, E.A.; Davey, R.J.; Roberts, R.J. Structure, solubility, screening, and synthesis of molecular salts. J. Pharm. Sci. 2007, 96, 1053–1068. [Google Scholar] [CrossRef]
- Korn, C.; Balbach, S. Compound selection for development—Is salt formation the ultimate answer? Experiences with an extended concept of the “100 mg approach”. Eur. J. Pharm. Sci. 2014, 57, 257–263. [Google Scholar] [CrossRef]
- Wyttenbach, N.; Janas, C.; Siam, M.; Lauer, M.E.; Jacob, L.; Scheubel, E.; Page, S. Miniaturized screening of polymers for amorphous drug stabilization (SPADS): Rapid assessment of solid dispersion systems. Eur. J. Pharm. Biopharm. 2013, 84, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Kokubo, T.; Zhao, C.; Ohki, Y. Antiprecipitant Screening System for Basic Model Compounds using Bio-Relevant Media. JALA J. Assoc. Lab. Autom. 2010, 15, 306–312. [Google Scholar] [CrossRef]
- Yamashita, T.; Ozaki, S.; Kushida, I. Solvent shift method for anti-precipitant screening of poorly soluble drugs using biorelevant medium and dimethyl sulfoxide. Int. J. Pharm. 2011, 419, 170–174. [Google Scholar] [CrossRef]
- Warren, D.B.; Bergström, C.A.S.; Benameur, H.; Porter, C.J.H.; Pouton, C.W. Evaluation of the Structural Determinants of Polymeric Precipitation Inhibitors Using Solvent Shift Methods and Principle Component Analysis. Mol. Pharm. 2013, 10, 2823–2848. [Google Scholar] [CrossRef]
- Dai, W.-G.; Dong, L.C.; Li, S.; Pollock-Dove, C.; Chen, J.; Mansky, P.; Eichenbaum, G. Parallel screening approach to identify solubility-enhancing formulations for improved bioavailability of a poorly water-soluble compound using milligram quantities of material. Int. J. Pharm. 2007, 336, 1–11. [Google Scholar] [CrossRef]
- Shanbhag, A.; Rabel, S.; Nauka, E.; Casadevall, G.; Shivanand, P.; Eichenbaum, G.; Mansky, P. Method for screening of solid dispersion formulations of low-solubility compounds—Miniaturization and automation of solvent casting and dissolution testing. Int. J. Pharm. 2008, 351, 209–218. [Google Scholar] [CrossRef]
- Chiang, P.-C.; Ran, Y.; Chou, K.-J.; Cui, Y.; Sambrone, A.; Chan, C.; Hart, R. Evaluation of Drug Load and Polymer by Using a 96-Well Plate Vacuum Dry System for Amorphous Solid Dispersion Drug Delivery. AAPS PharmSciTech 2012, 13, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Banda, A.; Manchanda, A.; Zhang, W.; Alba, G.M.; Nagapudi, K. Comparative Assessment of Miniaturized Screening Approaches for Selection of Polymers for Amorphous Drug Stabilization. J. Pharm. Sci. 2018, 107, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Auch, C.; Harms, M.; Mäder, K. Melt-based screening method with improved predictability regarding polymer selection for amorphous solid dispersions. Eur. J. Pharm. Sci. 2018, 124, 339–348. [Google Scholar] [CrossRef]
- Van Eerdenbrugh, B.; Taylor, L.S. Small Scale Screening to Determine the Ability of Different Polymers to Inhibit Drug Crystallization upon Rapid Solvent Evaporation. Mol. Pharm. 2010, 7, 1328–1337. [Google Scholar] [CrossRef]
- Taresco, V.; Louzao, I.; Scurr, D.; Booth, J.; Treacher, K.; McCabe, J.; Turpin, E.; Laughton, C.A.; Alexander, C.; Burley, J.C.; et al. Rapid Nanogram Scale Screening Method of Microarrays to Evaluate Drug–Polymer Blends Using High-Throughput Printing Technology. Mol. Pharm. 2017, 14, 2079–2087. [Google Scholar] [CrossRef]
- Tho, I.; Liepold, B.; Rosenberg, J.; Maegerlein, M.; Brandl, M.; Fricker, G. Formation of nano/micro-dispersions with improved dissolution properties upon dispersion of ritonavir melt extrudate in aqueous media. Eur. J. Pharm. Sci. 2010, 40, 25–32. [Google Scholar] [CrossRef]
- Fischer, S.M.; Brandl, M.; Fricker, G. Effect of the non-ionic surfactant Poloxamer 188 on passive permeability of poorly soluble drugs across Caco-2 cell monolayers. Eur. J. Pharm. Biopharm. 2011, 79, 416–422. [Google Scholar] [CrossRef]
- Frank, K.J.; Rosenblatt, K.M.; Westedt, U.; Hölig, P.; Rosenberg, J.; Mägerlein, M.; Fricker, G.; Brandl, M. Amorphous solid dispersion enhances permeation of poorly soluble ABT-102: True supersaturation vs. apparent solubility enhancement. Int. J. Pharm. 2012, 437, 288–293. [Google Scholar] [CrossRef]
- Frank, K.J.; Westedt, U.; Rosenblatt, K.M.; Hölig, P.; Rosenberg, J.; Mägerlein, M.; Brandl, M.; Fricker, G. Impact of FaSSIF on the solubility and dissolution-/permeation rate of a poorly water-soluble compound. Eur. J. Pharm. Sci. 2012, 47, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.J.; Westedt, U.; Rosenblatt, K.M.; Hölig, P.; Rosenberg, J.; Mägerlein, M.; Fricker, G.; Brandl, M. What Is the Mechanism Behind Increased Permeation Rate of a Poorly Soluble Drug from Aqueous Dispersions of an Amorphous Solid Dispersion? J. Pharm. Sci. 2014, 103, 1779–1786. [Google Scholar] [CrossRef]
- Buckley, S.T.; Frank, K.J.; Fricker, G.; Brandl, M. Biopharmaceutical classification of poorly soluble drugs with respect to “enabling formulations”. Eur. J. Pharm. Sci. 2013, 50, 8–16. [Google Scholar] [CrossRef]
- Fong, S.Y.K.; Bauer-Brandl, A.; Brandl, M. Oral bioavailability enhancement through supersaturation: An update and meta-analysis. Expert Opin. Drug Deliv. 2017, 14, 403–426. [Google Scholar] [CrossRef]
- Bevernage, J.; Brouwers, J.; Annaert, P.; Augustijns, P. Drug precipitation–permeation interplay: Supersaturation in an absorptive environment. Eur. J. Pharm. Biopharm. 2012, 82, 424–428. [Google Scholar] [CrossRef]
- Hate, S.S.; Reutzel-Edens, S.M.; Taylor, L.S. Absorptive Dissolution Testing of Supersaturating Systems: Impact of Absorptive Sink Conditions on Solution Phase Behavior and Mass Transport. Mol. Pharm. 2017, 14, 4052–4063. [Google Scholar] [CrossRef]
- Sironi, D.; Christensen, M.; Rosenberg, J.; Bauer-Brandl, A.; Brandl, M. Evaluation of a dynamic dissolution/permeation model: Mutual influence of dissolution and barrier-flux under non-steady state conditions. Int. J. Pharm. 2017, 522, 50–57. [Google Scholar] [CrossRef]
- Tsume, Y.; Igawa, N.; Drelich, A.J.; Amidon, G.E.; Amidon, G.L. The Combination of GIS and Biphasic to Better Predict In Vivo Dissolution of BCS Class IIb Drugs, Ketoconazole and Raloxifene. J. Pharm. Sci. 2018, 107, 307–316. [Google Scholar] [CrossRef]
- Krupa, A.; Descamps, M.; Willart, J.-F.; Strach, B.; Wyska, E.; Jachowicz, R.; Danède, F. High-Energy Ball Milling as Green Process to Vitrify Tadalafil and Improve Bioavailability. Mol. Pharm. 2016, 13, 3891–3902. [Google Scholar] [CrossRef]
- Jacobsen, A.-C.; Elvang, P.A.; Bauer-Brandl, A.; Brandl, M. A dynamic in vitro permeation study on solid mono- and diacyl-phospholipid dispersions of celecoxib. Eur. J. Pharm. Sci. 2019, 127. [Google Scholar] [CrossRef]
- Bevernage, J.; Brouwers, J.; Clarysse, S.; Vertzoni, M.; Tack, J.; Annaert, P.; Augustijns, P. Drug supersaturation in simulated and human intestinal fluids representing different nutritional states. J. Pharm. Sci. 2010, 99, 4525–4534. [Google Scholar] [CrossRef]
- Bevernage, J.; Forier, T.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Excipient-Mediated Supersaturation Stabilization in Human Intestinal Fluids. Mol. Pharm. 2011, 8, 564–570. [Google Scholar] [CrossRef]
- Higashino, H.; Hasegawa, T.; Yamamoto, M.; Matsui, R.; Masaoka, Y.; Kataoka, M.; Sakuma, S.; Yamashita, S. In Vitro–in Vivo Correlation of the Effect of Supersaturation on the Intestinal Absorption of BCS Class 2 Drugs. Mol. Pharm. 2014, 11, 746–754. [Google Scholar] [CrossRef]
- Sironi, D.; Rosenberg, J.; Bauer-Brandl, A.; Brandl, M. Dynamic dissolution-/permeation-testing of nano- and microparticle formulations of fenofibrate. Eur. J. Pharm. Sci. 2017, 96, 20–27. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Tadalafil Concentration in Dispersion (mg/mL) | Soluplus® Concentration in Dispersion (mg/mL) |
|---|---|---|
| Crystalline TDF | 0.1 | |
| Freeze-dried TDF | 0.1 | |
| Physical mixture | 0.1 | 0.9 |
| Amorphous solid dispersion | 0.1 | 0.9 |
| Experimental Parameter to be Evaluated | Formulations | Dispersion Media | Acceptor Media | Incubation Time | Number of Experiments |
|---|---|---|---|---|---|
| Acceptor medium | Crystalline tadalafil (TDF) | 50 mM phosphate buffer | 50 mM phosphate buffer | 24 h | 6 |
| 1% sodium dodecyl sulfate (SDS) in phosphate buffer | |||||
| 0.2% Vitamin E TPGS in phosphate buffer | |||||
| 1% Vitamin E TPGS in phosphate buffer | |||||
| 0.2% PS 80 in phosphate buffer | |||||
| 1% PS 80 in phosphate buffer | |||||
| Incubation time | Crystalline TDF | 50 mM phosphate buffer | 1% Vitamin E TPGS in phosphate buffer | 1 h | 12 |
| Freeze-dried TDF | 3 h | ||||
| Physical mixture | 6 h | ||||
| Amorphous solid dispersion | |||||
| Dispersion medium | Crystalline TDF | 50 mM phosphate buffer | 1% Vitamin E TPGS in phosphate buffer | 6 h | 12 |
| Freeze-dried TDF | Simulated gastric fluid (SGF) | 1% Vitamin E TPGS in SGF | |||
| Physical mixture | FaSSIF | 1% Vitamin E TPGS in FaSSIF | |||
| Amorphous solid dispersion |
| Dispersion Medium | R2 (Dissolved Concentration vs. AUC) | R2 (Permeated Concentration vs. AUC) |
|---|---|---|
| 50 mM Phosphate buffer | 0.846 | 0.850 |
| SGF | 0.826 | 0.864 |
| FaSSIF | 0.823 | 0.940 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacobsen, A.-C.; Krupa, A.; Brandl, M.; Bauer-Brandl, A. High-Throughput Dissolution/Permeation Screening—A 96-Well Two-Compartment Microplate Approach. Pharmaceutics 2019, 11, 227. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11050227
Jacobsen A-C, Krupa A, Brandl M, Bauer-Brandl A. High-Throughput Dissolution/Permeation Screening—A 96-Well Two-Compartment Microplate Approach. Pharmaceutics. 2019; 11(5):227. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11050227
Chicago/Turabian StyleJacobsen, Ann-Christin, Anna Krupa, Martin Brandl, and Annette Bauer-Brandl. 2019. "High-Throughput Dissolution/Permeation Screening—A 96-Well Two-Compartment Microplate Approach" Pharmaceutics 11, no. 5: 227. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11050227