17-α Hydroxyprogesterone Nanoemulsifying Preconcentrate-Loaded Vaginal Tablet: A Novel Non-Invasive Approach for the Prevention of Preterm Birth


Abstract
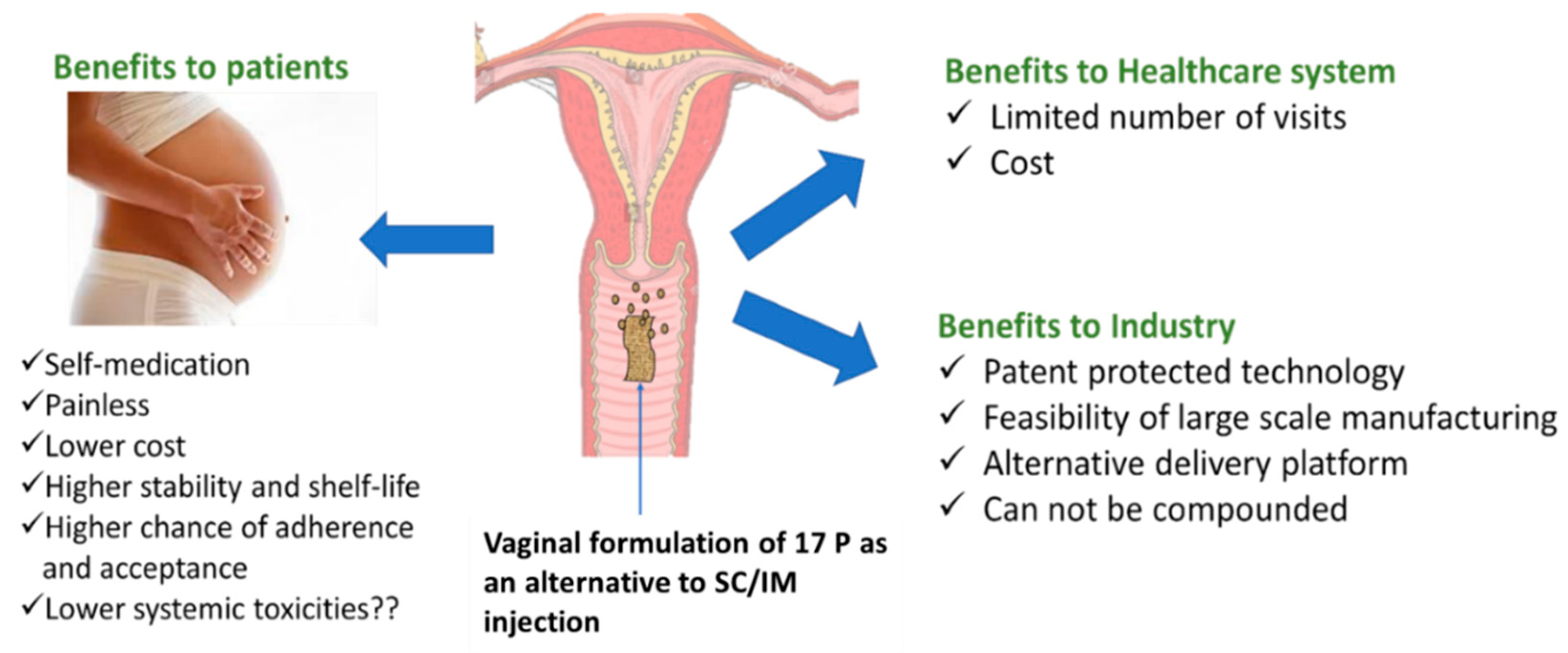
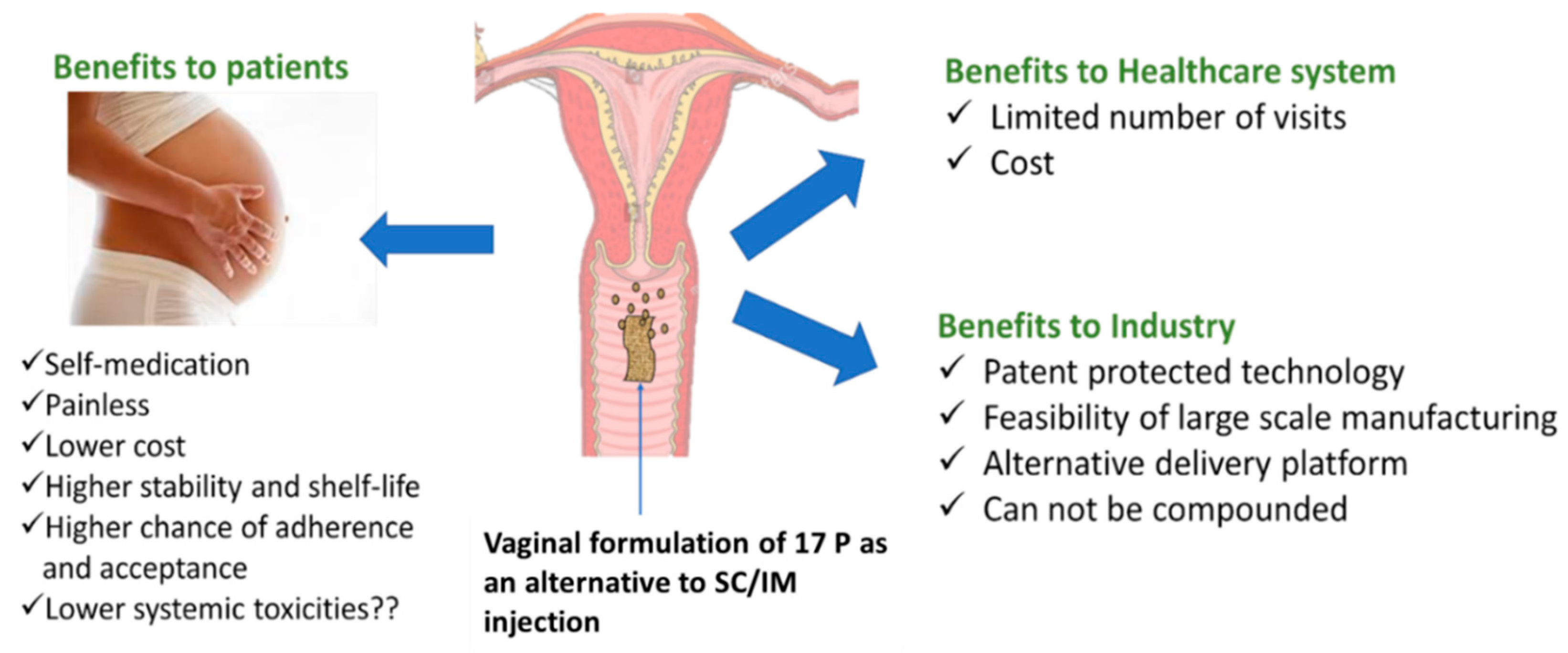
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Analytical Method
2.3. Preparation of Simulated Vaginal Fluid
2.4. Development of 17P-Loaded Self-Nanoemulsifying Preconcentrate (SNEDDS)
2.5. Preparation of Solid 17P-Loaded SNEDDS (S-SNEDDS)
2.6. Solid State Characterization of S-SNEDDS
2.7. Preparation and Characterization of a Vaginal Tablet
2.8. Characterization of Vaginal Tablet
2.9. In Vitro Drug Release
2.10. TNF-α Assay
2.11. Animals
2.12. In Vivo Efficacy Testing in a Preterm Birth Mouse Model
2.13. Statistics
3. Results
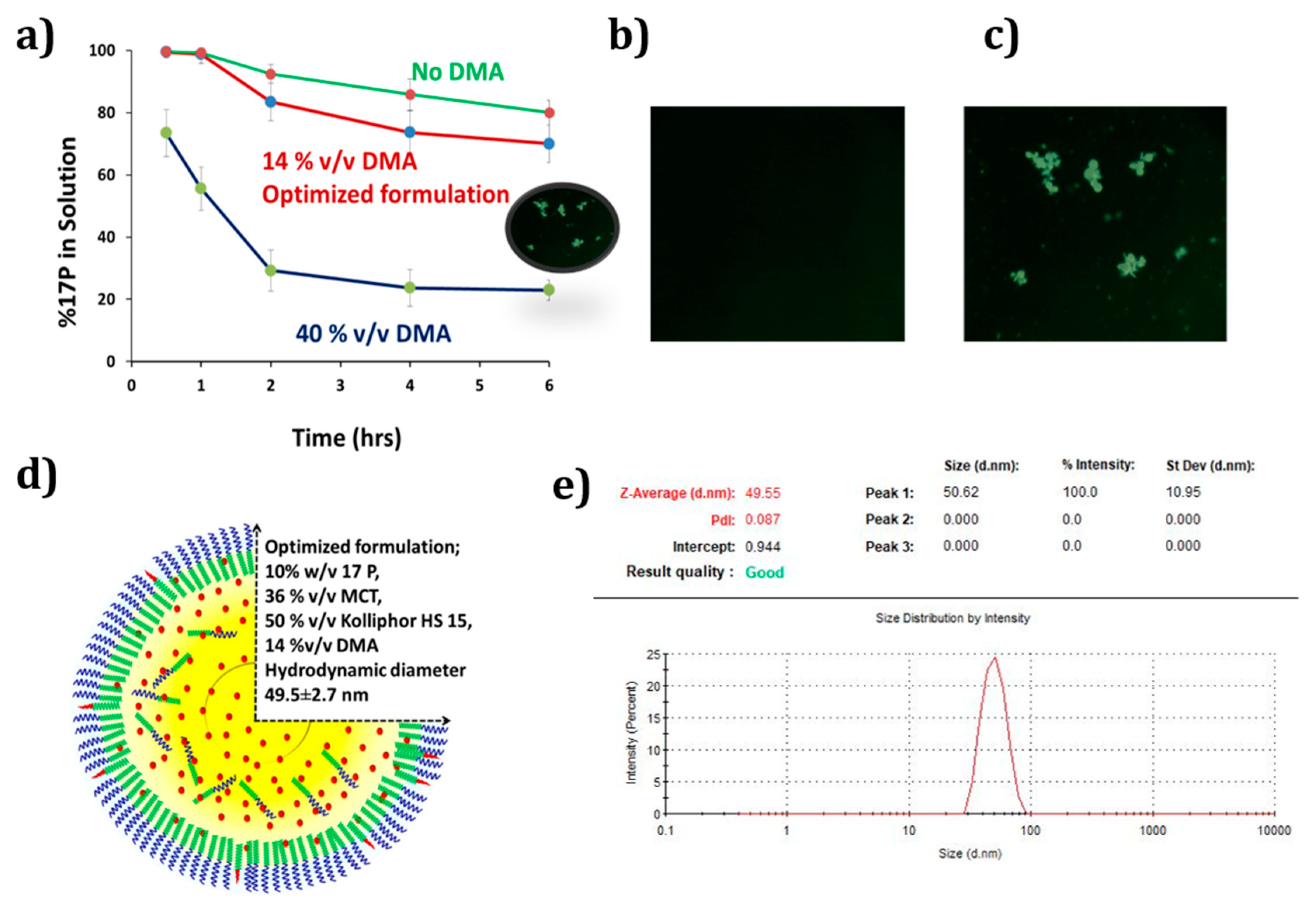
3.1. Development of Self-Nanoemulsifying Preconcentrate
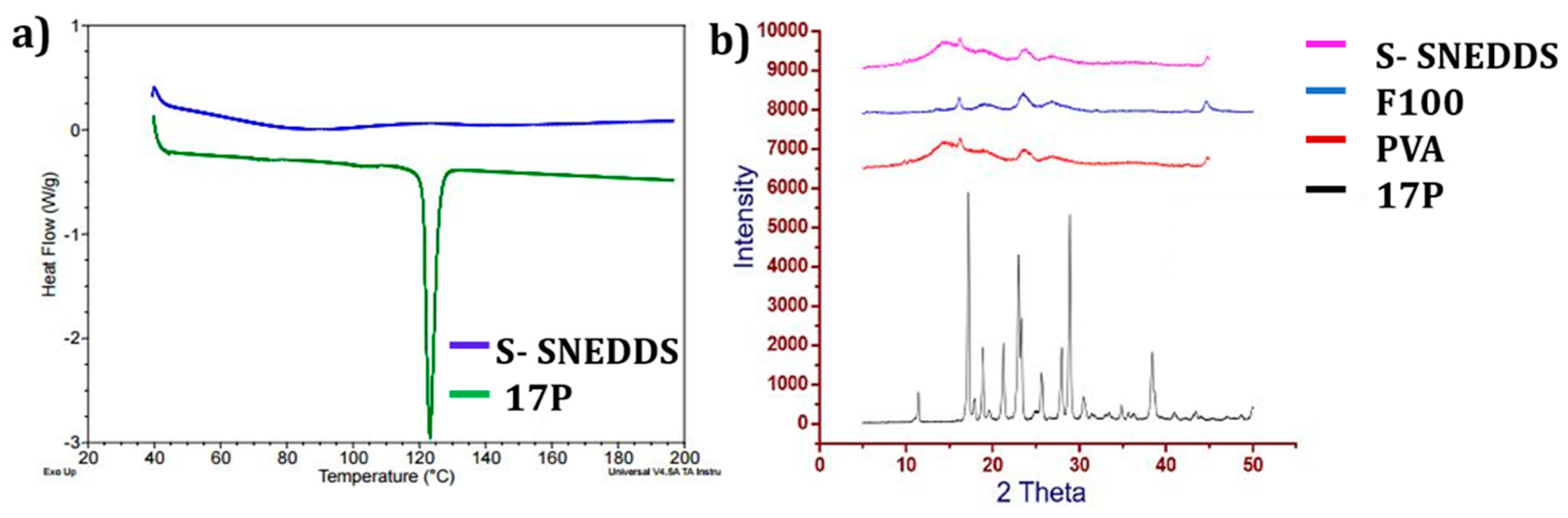
3.2. Solid State Characterization
3.3. Preparation and Characterization of Vaginal Tablet
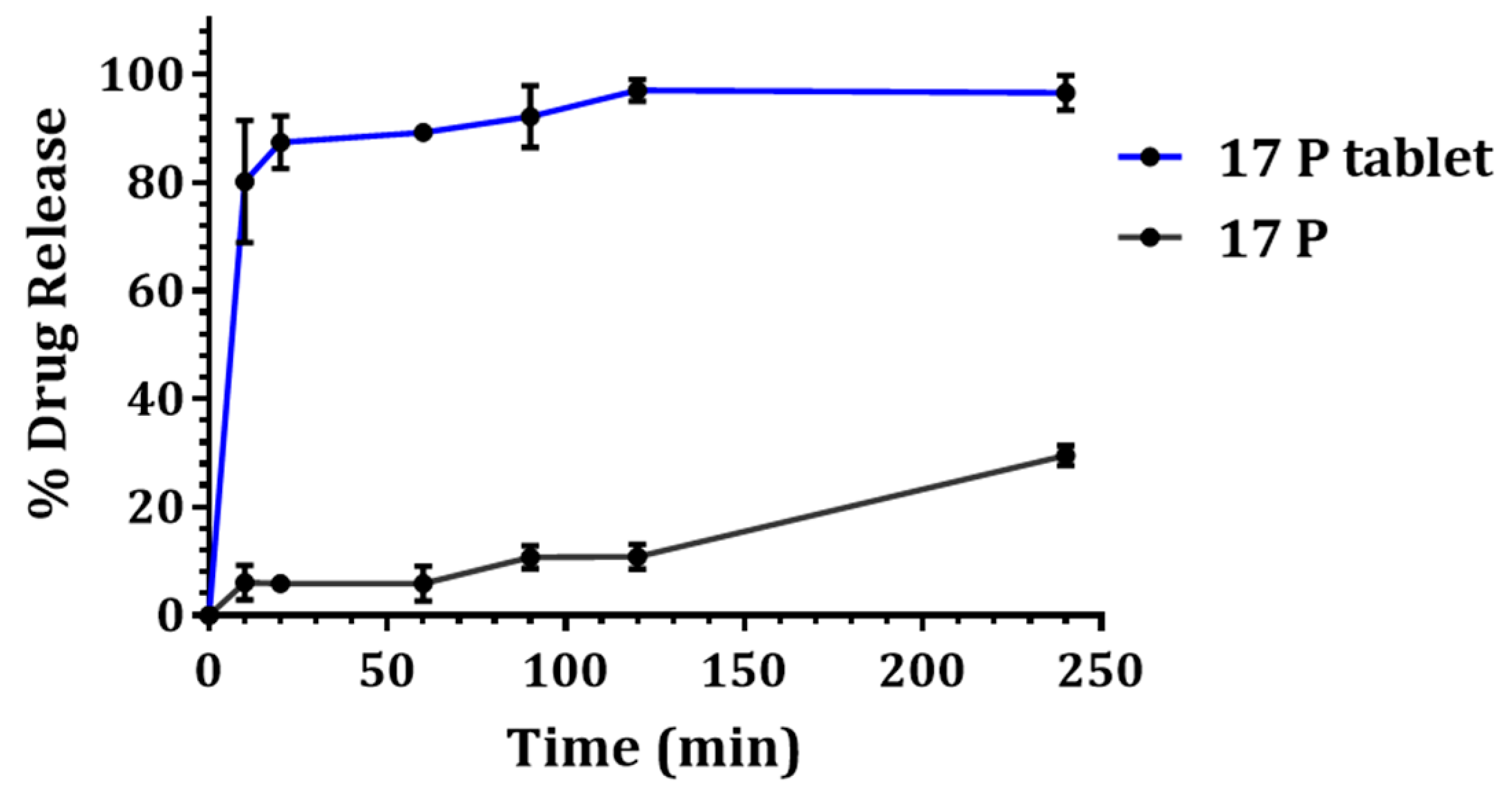
3.4. In Vitro Drug Release
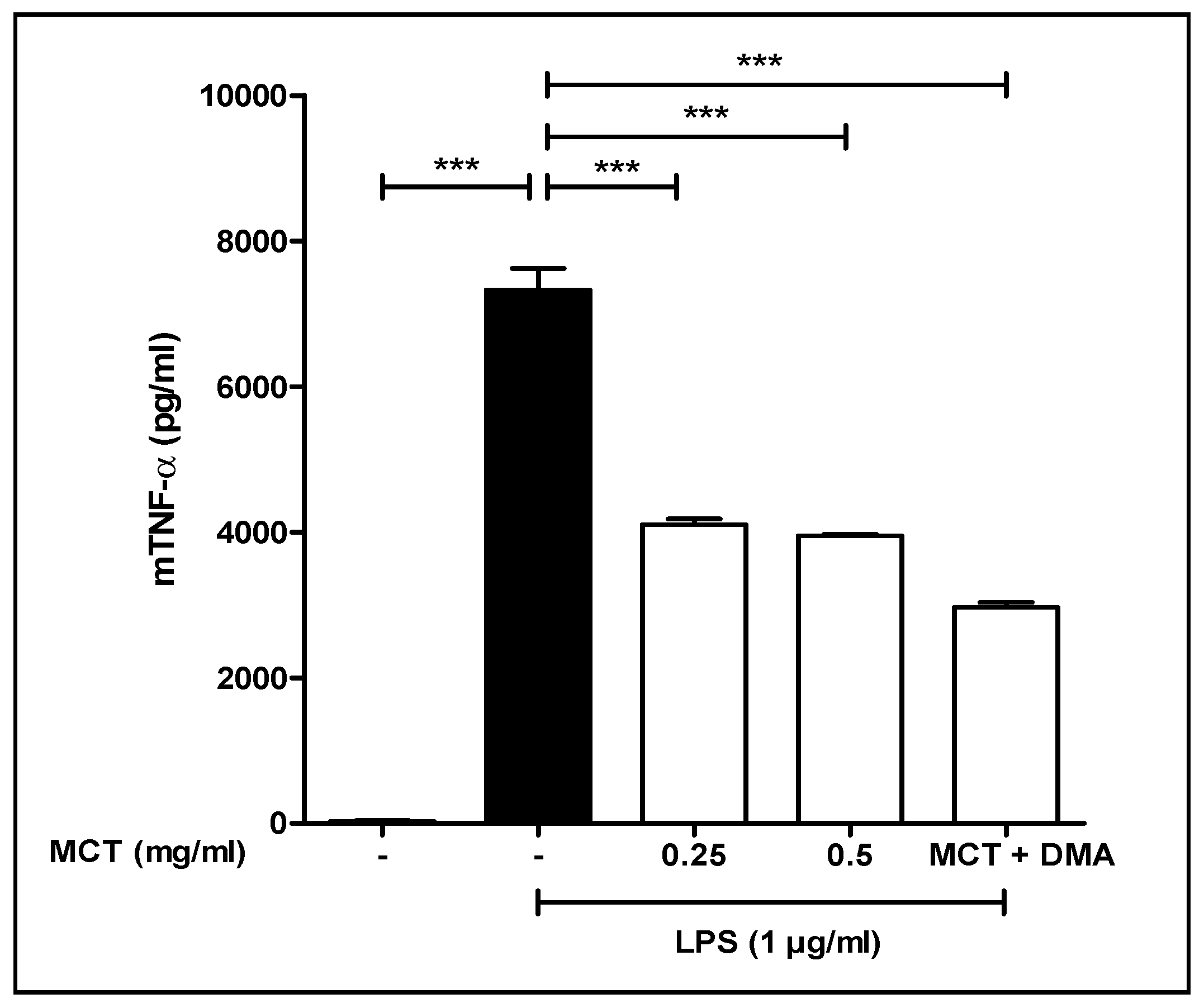
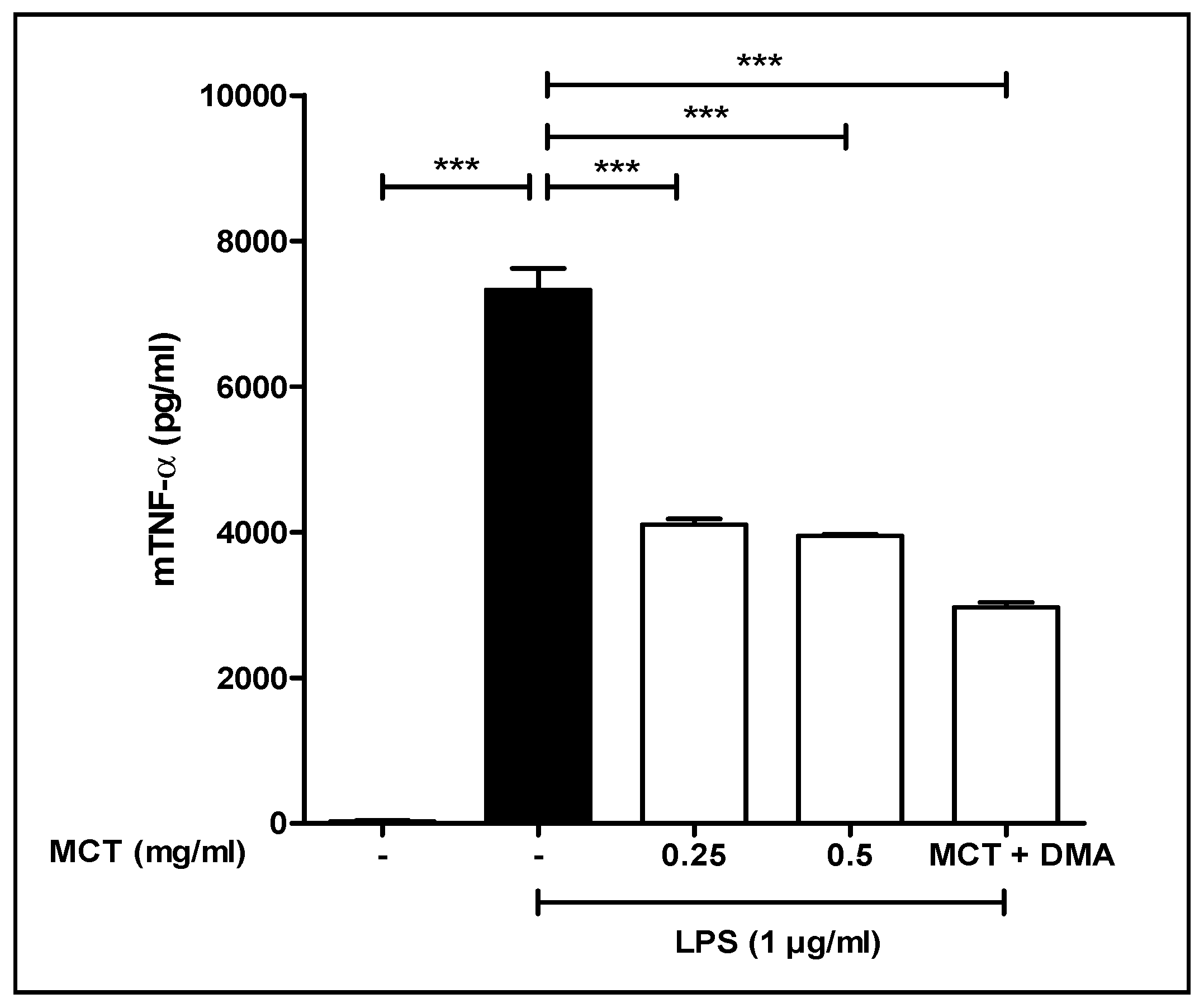
3.5. TNF-α Study
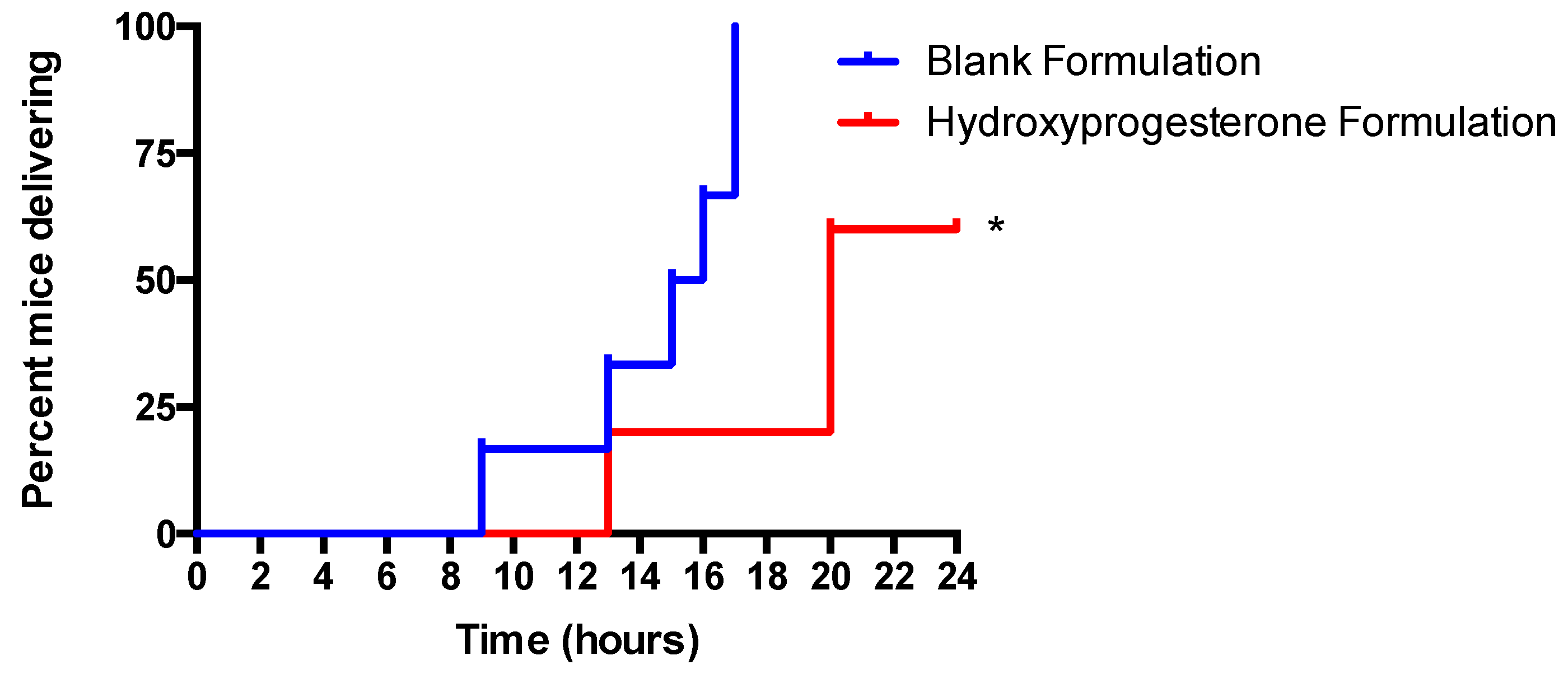
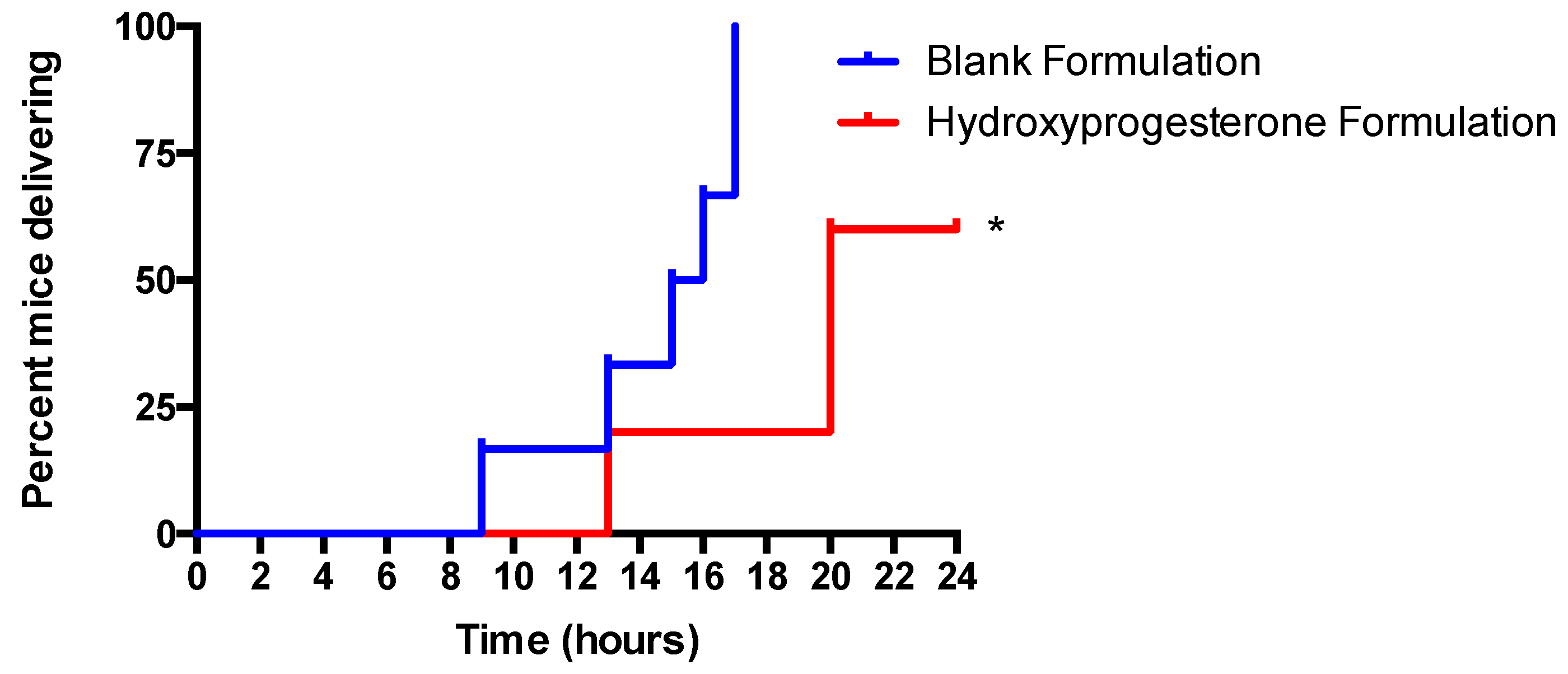
3.6. Vaginal Administration of a Hydroxyprogesterone Nanoformulation Delays the Onset of Preterm Labor (PTL) in LPS-Stimulated Pregnant Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beck, S.; Wojdyla, D.; Say, L.; Betran, A.P.; Merialdi, M.; Requejo, J.H.; Rubens, C.; Menon, R.; Van Look, P.F. The worldwide incidence of preterm birth: A systematic review of maternal mortality and morbidity. Bull. World Health Organ. 2010, 88, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Gotsch, F.; Gotsch, F.; Romero, R.; Erez, O.; Vaisbuch, E.; Kusanovic, J.P.; Mazaki-Tovi, S.; Kim, S.K.; Hassan, S.; Yeo, L. The preterm parturition syndrome and its implications for understanding the biology, risk assessment, diagnosis, treatment and prevention of preterm birth. J. Matern. Fetal Neonatal Med. 2009, 22 (Suppl. 2), 5–23. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.H.; Collins, J.W., Jr.; Wright, R.O. Racial/ethnic disparities in preterm birth: Clues from environmental exposures. Curr Opin Pediatr 2011, 23, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Dole, N.; Savitz, D.A.; Siega-Riz, A.M.; Hertz-Picciotto, I.; McMahon, M.J.; Buekens, P. Psychosocial factors and preterm birth among African American and White women in central North Carolina. Am. J. Public Health 2004, 94, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.A.; Thota, C.; Browne, P.C.; Diamond, M.P.; Al-Hendy, A. Why is Preterm Birth Stubbornly Higher in African-Americans? Obs. Gynecol. Int. J. 2014, 1. [Google Scholar] [CrossRef] [PubMed]
- Frey, H.A.; Klebanoff, M.A. The epidemiology, etiology, and costs of preterm birth. Semin. Fetal Neonatal Med. 2016, 21, 68–73. [Google Scholar] [CrossRef]
- Patel, Y.; Rumore, M.M. Hydroxyprogesterone caproate injection (makena) one year later: To compound or not to compound that is the question. P T 2012, 37, 405–411. [Google Scholar]
- Tavaniotou, A.; Smitz, J.; Bourgain, C.; Devroey, P. Comparison between different routes of progesterone administration as luteal phase support in infertility treatments. Hum. Reprod. Update 2000, 6, 139–148. [Google Scholar] [CrossRef]
- D’Arcangues, C. Once-a-month estrogen/progestogen injectables. Entre Nous Cph. Den. 1991, 15. [Google Scholar]
- Vidaeff, A.C.; Belfort, M.A. Critical appraisal of the efficacy, safety, and patient acceptability of hydroxyprogesterone caproate injection to reduce the risk of preterm birth. Patient Prefer. Adherence 2013, 7, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Meis, P.J.; Klebanoff, M.; Thom, E.; Dombrowski, M.P.; Sibai, B.; Moawad, A.H.; Spong, C.Y.; Hauth, J.C.; Miodovnik, M.; Varner, M.W. Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesterone caproate. N. Engl. J. Med. 2003, 348, 2379–2385. [Google Scholar] [CrossRef] [PubMed]
- Travanty, M.N.; Calawa, B.; Shalaby, W.S.; Jozwiakowski, M.J.; Haraldsen, K.B. Development and usability of a new subcutaneous auto-injector device to administer hydroxyprogesterone caproate to reduce the risk of recurrent preterm birth. Med. Devices (Auckl. Nz) 2018, 11, 241. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, H.; Cousens, S.; Chou, D.; Oestergaard, M.; Say, L.; Moller, A.B.; Kinney, M.; Lawn, J.; Born Too Soon Preterm Birth Action, G. Born too soon: The global epidemiology of 15 million preterm births. Reprod. Health 2013, 10 (Suppl. 1), S2. [Google Scholar] [CrossRef] [PubMed]
- Kollner, S.; Nardin, I.; Markt, R.; Griesser, J.; Prufert, F.; Bernkop-Schnurch, A. Self-emulsifying drug delivery systems: Design of a novel vaginal delivery system for curcumin. Eur. J. Pharm. Biopharm. 2017, 115, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, K.; Alamri, R.; Ahmad, A.; Raish, M.; Alanazi, F.K.; Hussain, M.D. Development of self-nanoemulsifying drug delivery systems for the enhancement of solubility and oral bioavailability of fenofibrate, a poorly water-soluble drug. Int. J. Nanomed. 2016, 11, 2829–2838. [Google Scholar] [CrossRef] [Green Version]
- Khandelwal, M. Vaginal progesterone in risk reduction of preterm birth in women with short cervix in the midtrimester of pregnancy. Int. J. Women’s Health 2012, 4, 481. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Conde-Agudelo, A.; El-Refaie, W.; Rode, L.; Brizot, M.L.; Cetingoz, E.; Serra, V.; Da Fonseca, E.; Abdelhafez, M.S.; Tabor, A.; et al. Vaginal progesterone decreases preterm birth and neonatal morbidity and mortality in women with a twin gestation and a short cervix: An updated meta-analysis of individual patient data. Ultrasound Obs. Gynecol. 2017, 49, 303–314. [Google Scholar] [CrossRef]
- Vanić, Ž.; Škalko-Basnet, N. Nanoformulations for Vaginal Therapy. In Nanotechnology Applied to Pharmaceutical Technology; Springer: Berlin, Germany, 2017; pp. 183–221. [Google Scholar]
- Katayama, M.; Nakane, R.; Matsuda, Y.; Kaneko, S.; Hara, I.; Sato, H. Determination of progesterone and 17-hydroxyprogesterone by high performance liquid chromatography after pre-column derivatization with 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionohydra zide. Analyst 1998, 123, 2339–2342. [Google Scholar] [CrossRef]
- Salminen, A.; Paananen, R.; Vuolteenaho, R.; Metsola, J.; Ojaniemi, M.; Autio-Harmainen, H.; Hallman, M. Maternal endotoxin-induced preterm birth in mice: Fetal responses in toll-like receptors, collectins, and cytokines. Pediatr. Res. 2008, 63, 280–286. [Google Scholar] [CrossRef]
- Rinaldi, S.F.; Catalano, R.D.; Wade, J.; Rossi, A.G.; Norman, J.E. Decidual neutrophil infiltration is not required for preterm birth in a mouse model of infection-induced preterm labor. J. Immunol. 2014, 192, 2315–2325. [Google Scholar] [CrossRef]
- Olgun, N.S.; Hanna, N.; Reznik, S.E. BQ-123 prevents LPS-induced preterm birth in mice via the induction of uterine and placental IL-10. Toxicol. Appl. Pharm. 2015, 282, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.; Ashby, C.R., Jr.; Olgun, N.S.; Sundaram, S.; Salami, O.; Munnangi, S.; Pekson, R.; Mahajan, P.; Reznik, S.E. Inhibition of sphingosine kinase prevents lipopolysaccharide-induced preterm birth and suppresses proinflammatory responses in a murine model. Am. J. Pathol. 2015, 185, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Pekson, R.; Poltoratsky, V.; Gorasiya, S.; Sundaram, S.; Ashby, C.R., Jr.; Vancurova, I.; Reznik, S.E. N, N-Dimethylacetamide Significantly Attenuates LPS-and TNFα-Induced Proinflammatory Responses Via Inhibition of the Nuclear Factor Kappa B Pathway. Mol. Med. 2016, 22, 747. [Google Scholar] [CrossRef]
- Sundaram, S.; Ashby, C.R.; Pekson, R.; Sampat, V.; Sitapara, R.; Mantell, L.; Chen, C.-H.; Yen, H.; Abhichandani, K.; Munnangi, S. N, N-dimethylacetamide regulates the proinflammatory response associated with endotoxin and prevents preterm birth. Am. J. Pathol. 2013, 183, 422–430. [Google Scholar] [CrossRef]
- Cicinelli, E.; de Ziegler, D. Transvaginal progesterone: Evidence for a new functional ’portal system’ flowing from the vagina to the uterus. Hum. Reprod. Update 1999, 5, 365–372. [Google Scholar] [CrossRef] [PubMed]
- De Ziegler, D.; Bulletti, C.; De Monstier, B.; Jaaskelainen, A.S. The first uterine pass effect. Ann. N. Y. Acad. Sci. 1997, 828, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Miles, R.A.; Paulson, R.J.; Lobo, R.A.; Press, M.F.; Dahmoush, L.; Sauer, M.V. Pharmacokinetics and endometrial tissue levels of progesterone after administration by intramuscular and vaginal routes: A comparative study. Fertil. Steril. 1994, 62, 485–490. [Google Scholar] [CrossRef]
- Paulson, R.J.; Collins, M.G.; Yankov, V.I. Progesterone pharmacokinetics and pharmacodynamics with 3 dosages and 2 regimens of an effervescent micronized progesterone vaginal insert. J. Clin. Endocrinol. Metab. 2014, 99, 4241–4249. [Google Scholar] [CrossRef]
- Ensign, L.M.; Tang, B.C.; Wang, Y.Y.; Tse, T.A.; Hoen, T.; Cone, R.; Hanes, J. Mucus-penetrating nanoparticles for vaginal drug delivery protect against herpes simplex virus. Sci. Transl. Med. 2012, 4, 138ra179. [Google Scholar] [CrossRef]
- Cicinelli, E.; Schonauer, L.M.; Galantino, P.; Matteo, M.G.; Cassetta, R.; Pinto, V. Mechanisms of uterine specificity of vaginal progesterone. Hum. Reprod. 2000, 15 (Suppl. 1), 159–165. [Google Scholar] [CrossRef]
- Bulletti, C.; de Ziegler, D.; Flamigni, C.; Giacomucci, E.; Polli, V.; Bolelli, G.; Franceschetti, F. Targeted drug delivery in gynaecology: The first uterine pass effect. Hum. Reprod. 1997, 12, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Cicinelli, E.; de Ziegler, D.; Bulletti, C.; Matteo, M.G.; Schonauer, L.M.; Galantino, P. Direct transport of progesterone from vagina to uterus. Obstet. Gynecol. 2000, 95, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Chollet, J.L.; Jozwiakowski, M.J. Quality investigation of hydroxyprogesterone caproate active pharmaceutical ingredient and injection. Drug Dev. Ind. Pharm. 2012, 38, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Caritis, S.N.; Feghali, M.N.; Grobman, W.A.; Rouse, D.J.; Eunice Kennedy Shriver National Institute of Child Health; Human Development Maternal–Fetal Medicine Units Network. What we have learned about the role of 17-alpha-hydroxyprogesterone caproate in the prevention of preterm birth. Semin. Perinatol. 2016, 40, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Gudeman, J.; Jozwiakowski, M.; Chollet, J.; Randell, M. Potential risks of pharmacy compounding. Drugs R D 2013, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Beltsos, A.N.; Sanchez, M.D.; Doody, K.J.; Bush, M.R.; Domar, A.D.; Collins, M.G. Patients’ administration preferences: Progesterone vaginal insert (Endometrin(R)) compared to intramuscular progesterone for Luteal phase support. Reprod. Health 2014, 11, 78. [Google Scholar] [CrossRef]
- Check, J.H. Luteal Phase Support in assisted reproductive technology treatment: Focus on Endometrin(R) (progesterone) vaginal insert. Ther. Clin. Risk Manag. 2009, 5, 403–407. [Google Scholar] [CrossRef]
- Heine, P.; Sellar, L.; Whitten, S.; Bajaj, P. A questionnaire-based audit to assess overall experience and convenience among patients using vaginal progesterone tablets (Lutigest((R))) for luteal phase support during IVF treatment. Patient Relat. Outcome Meas. 2017, 8, 169–179. [Google Scholar] [CrossRef]
- Kim, R.M.; Jang, D.J.; Kim, Y.C.; Yoon, J.H.; Min, K.A.; Maeng, H.J.; Cho, K.H. Flurbiprofen-Loaded Solid SNEDDS Preconcentrate for the Enhanced Solubility, In-Vitro Dissolution and Bioavailability in Rats. Pharmaceutics 2018, 10, 247. [Google Scholar] [CrossRef]
- Nasr, A.; Gardouh, A.; Ghorab, M. Novel Solid Self-Nanoemulsifying Drug Delivery System (S-SNEDDS) for Oral Delivery of Olmesartan Medoxomil: Design, Formulation, Pharmacokinetic and Bioavailability Evaluation. Pharmaceutics 2016, 8, 20. [Google Scholar] [CrossRef]
- Nikolakakis, I.; Partheniadis, I. Self-Emulsifying Granules and Pellets: Composition and Formation Mechanisms for Instant or Controlled Release. Pharmaceutics 2017, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Doddapaneni, R.; Patki, M.; Sekar, V.; Bagde, A.; Singh, M. Erlotinib-Valproic Acid Liquisolid Formulation: Evaluating Oral Bioavailability and Cytotoxicity in Erlotinib-Resistant Non-small Cell Lung Cancer Cells. AAPS PharmSciTech 2019, 20, 135. [Google Scholar] [CrossRef] [PubMed]
- Gumaste, S.G.; Dalrymple, D.M.; Serajuddin, A.T. Development of Solid SEDDS, V: Compaction and Drug Release Properties of Tablets Prepared by Adsorbing Lipid-Based Formulations onto Neusilin(R) US2. Pharm. Res. 2013, 30, 3186–3199. [Google Scholar] [CrossRef] [PubMed]
- Beringhs, A.O.; Minatovicz, B.C.; Zhang, G.G.Z.; Chaudhuri, B.; Lu, X. Impact of Porous Excipients on the Manufacturability and Product Performance of Solid Self-Emulsifying Drug Delivery Systems. AAPS PharmSciTech 2018, 19, 3298–3310. [Google Scholar] [CrossRef] [PubMed]
- Patki, M.; Patel, K. Development of a solid supersaturated self-nanoemulsifying preconcentrate (S-superSNEP) of fenofibrate using dimethylacetamide and a novel co-processed excipient. Drug Dev. Ind. Pharm. 2019, 45, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Fitaihi, R.A.; Aleanizy, F.S.; Elsamaligy, S.; Mahmoud, H.A.; Bayomi, M.A. Role of chitosan on controlling the characteristics and antifungal activity of bioadhesive fluconazole vaginal tablets. Saudi Pharm. J. 2018, 26, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Cevher, E.; Acma, A.; Sinani, G.; Aksu, B.; Zloh, M.; Mulazimoglu, L. Bioadhesive tablets containing cyclodextrin complex of itraconazole for the treatment of vaginal candidiasis. Int. J. Biol. Macromol. 2014, 69, 124–136. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batch | Pressure (lbs) | S-SNEDDS (mg) | KG 1000 (mg) | Hardness (kg) |
|---|---|---|---|---|
| F1 | 2500 | 150 | 170 | - |
| F2 | 6000 | 150 | 170 | 1.6 |
| F3 | 7000 | 150 | 170 | 3.1 |
| F4 | 2500 | 150 | 200 | - |
| F5 | 6000 | 150 | 200 | 4.7 |
| F6 | 7000 | 150 | 200 | 8.1 |
| Composition | Weight/Tablet |
|---|---|
| S-SNEDDS | 150 mg |
| Microcrystalline cellulose (KG-1000) | 200 mg |
| Kollidon CL | 18.5 mg |
| Magnesium stearate | 1.5 mg |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patki, M.; Giusto, K.; Gorasiya, S.; Reznik, S.E.; Patel, K. 17-α Hydroxyprogesterone Nanoemulsifying Preconcentrate-Loaded Vaginal Tablet: A Novel Non-Invasive Approach for the Prevention of Preterm Birth. Pharmaceutics 2019, 11, 335. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11070335
Patki M, Giusto K, Gorasiya S, Reznik SE, Patel K. 17-α Hydroxyprogesterone Nanoemulsifying Preconcentrate-Loaded Vaginal Tablet: A Novel Non-Invasive Approach for the Prevention of Preterm Birth. Pharmaceutics. 2019; 11(7):335. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11070335
Chicago/Turabian StylePatki, Manali, Kiersten Giusto, Samir Gorasiya, Sandra E. Reznik, and Ketan Patel. 2019. "17-α Hydroxyprogesterone Nanoemulsifying Preconcentrate-Loaded Vaginal Tablet: A Novel Non-Invasive Approach for the Prevention of Preterm Birth" Pharmaceutics 11, no. 7: 335. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11070335