Development of Remdesivir as a Dry Powder for Inhalation by Thin Film Freezing

 , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Dry Powder for Inhalation Using Thin Film Freezing
2.3. Drug Quantification (HPLC)
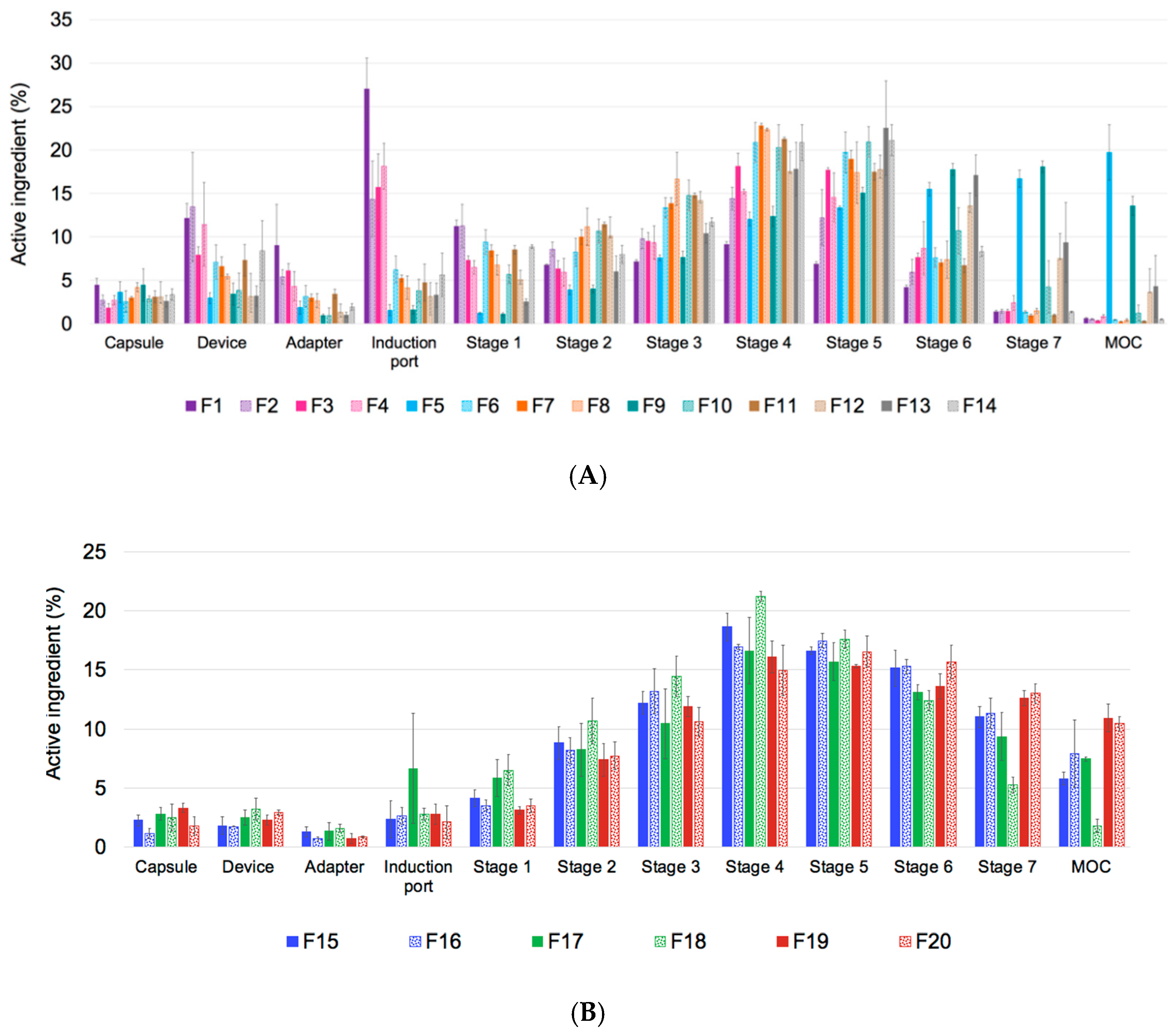
2.4. In Vitro Aerosol Performance
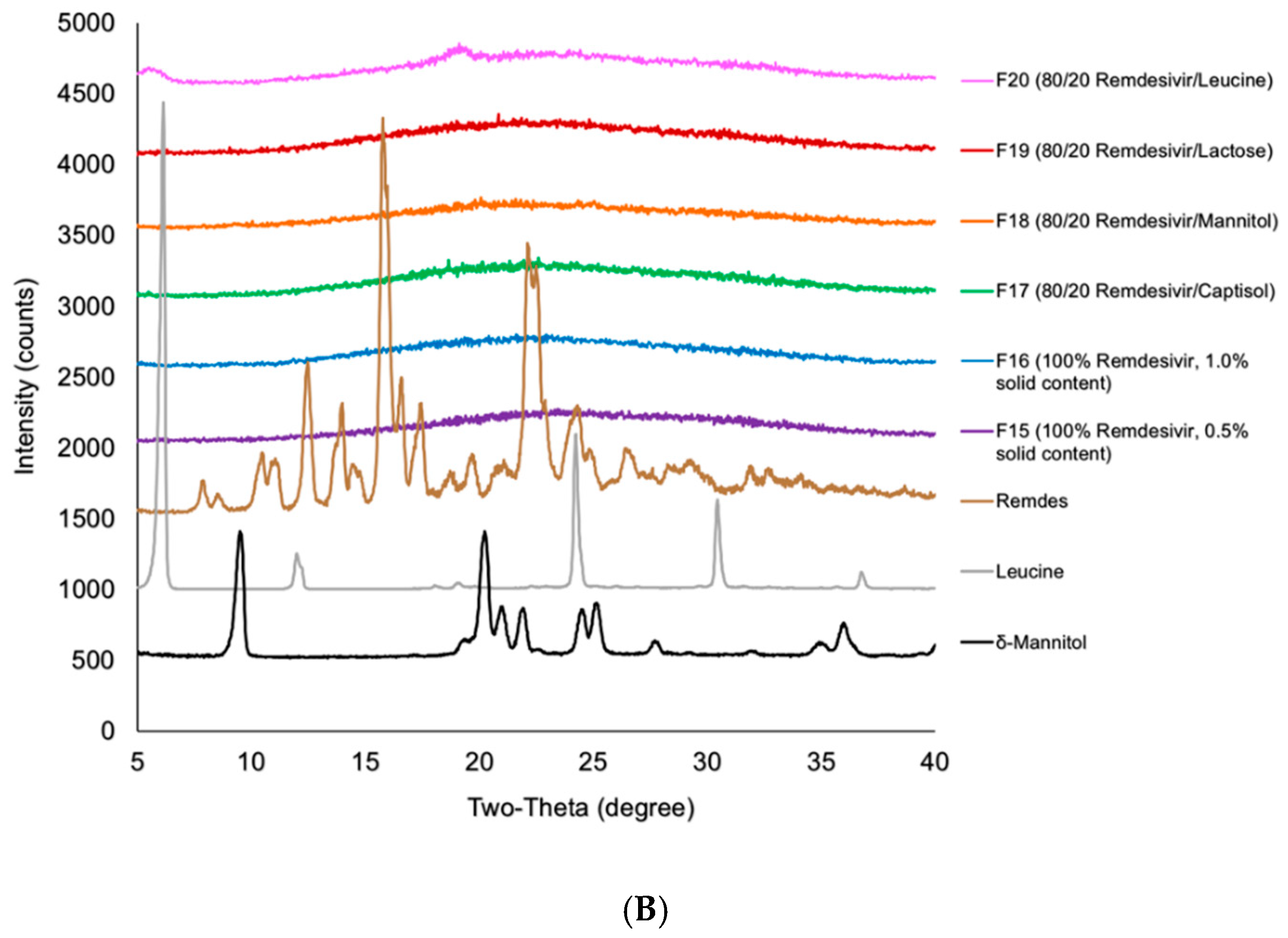
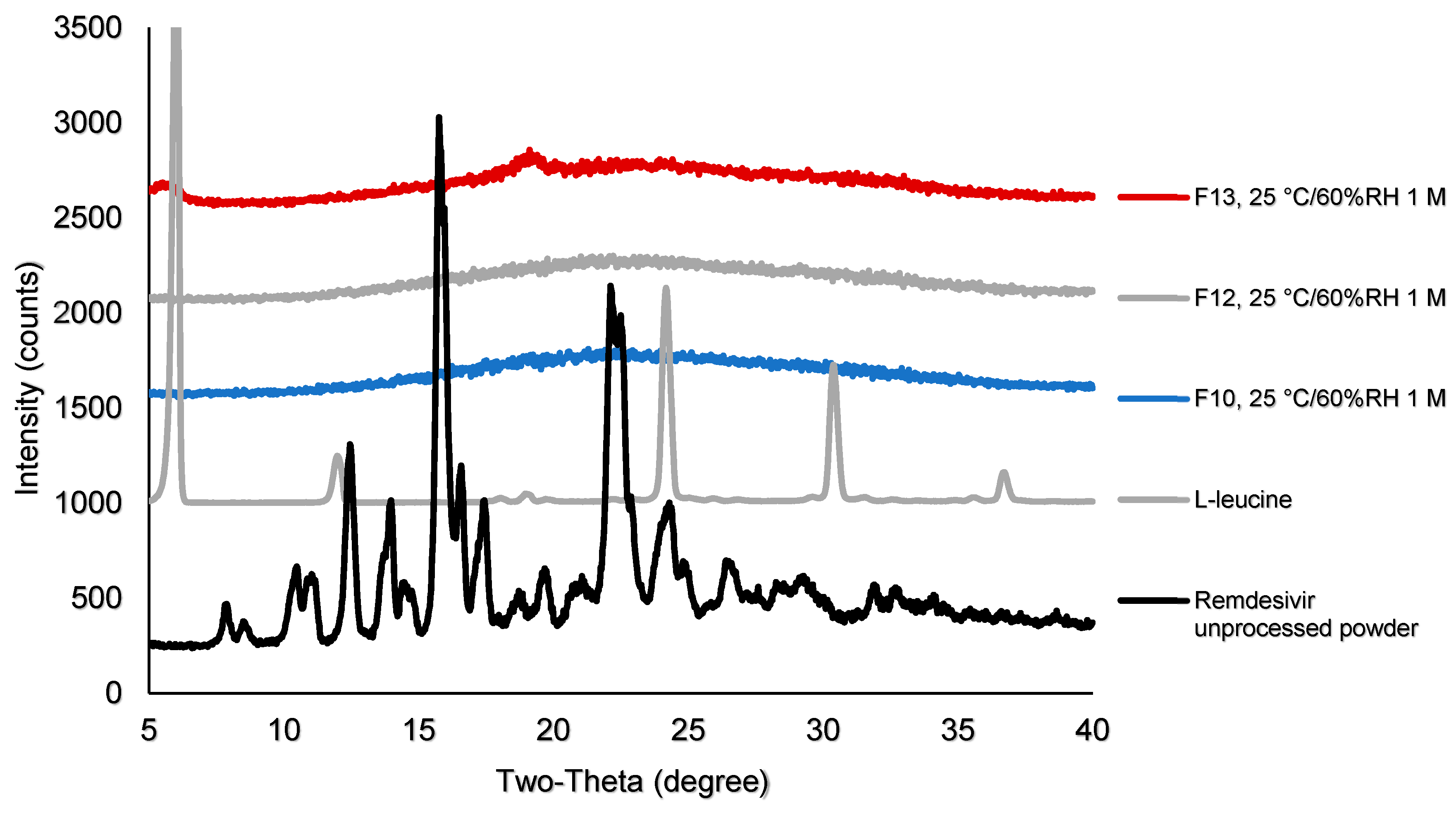
2.5. X-ray Powder Diffraction (XRPD)
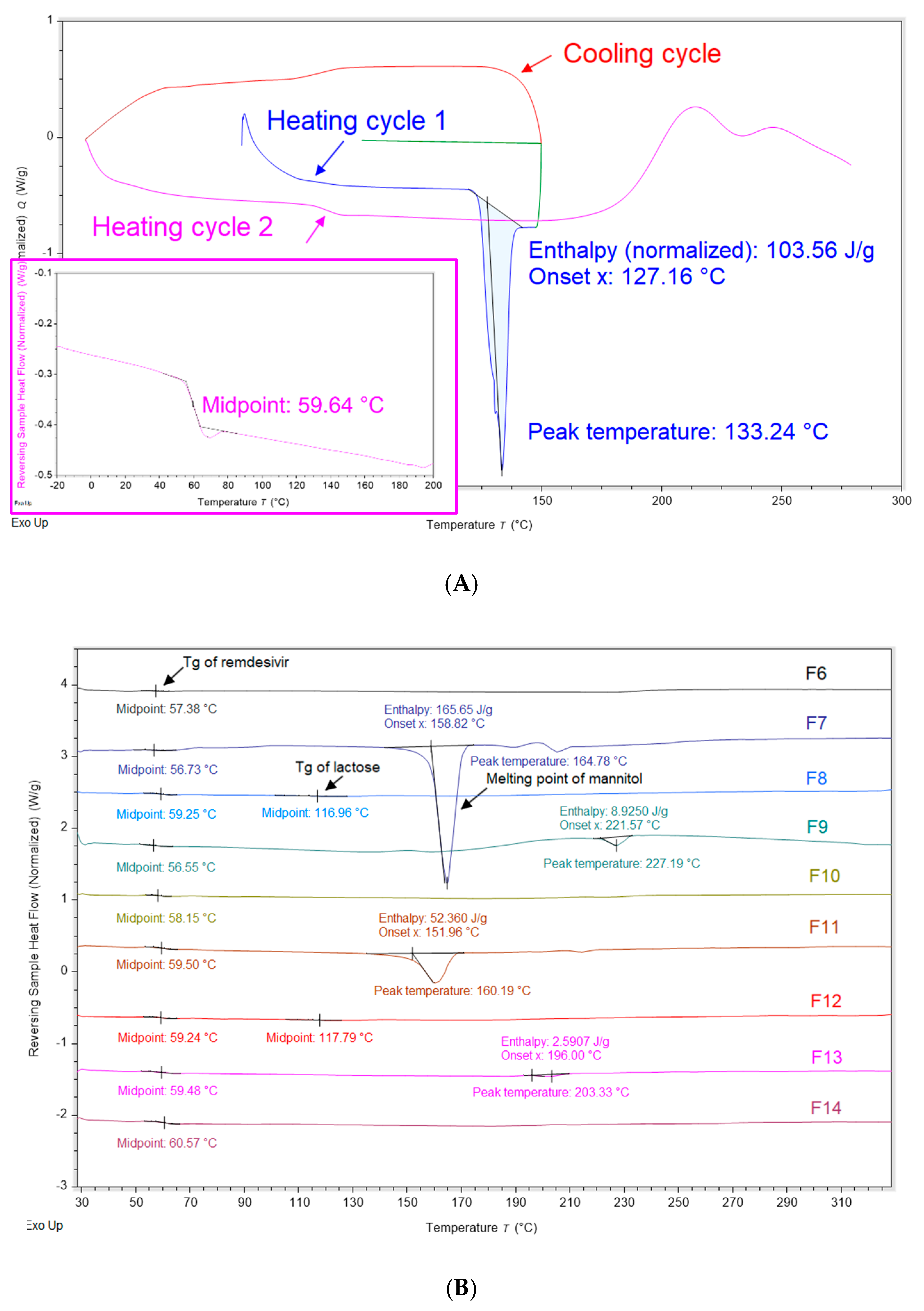
2.6. Modulated Differential Scanning Calorimetry (mDSC)
2.7. Scanning Electron Microscopy (SEM)
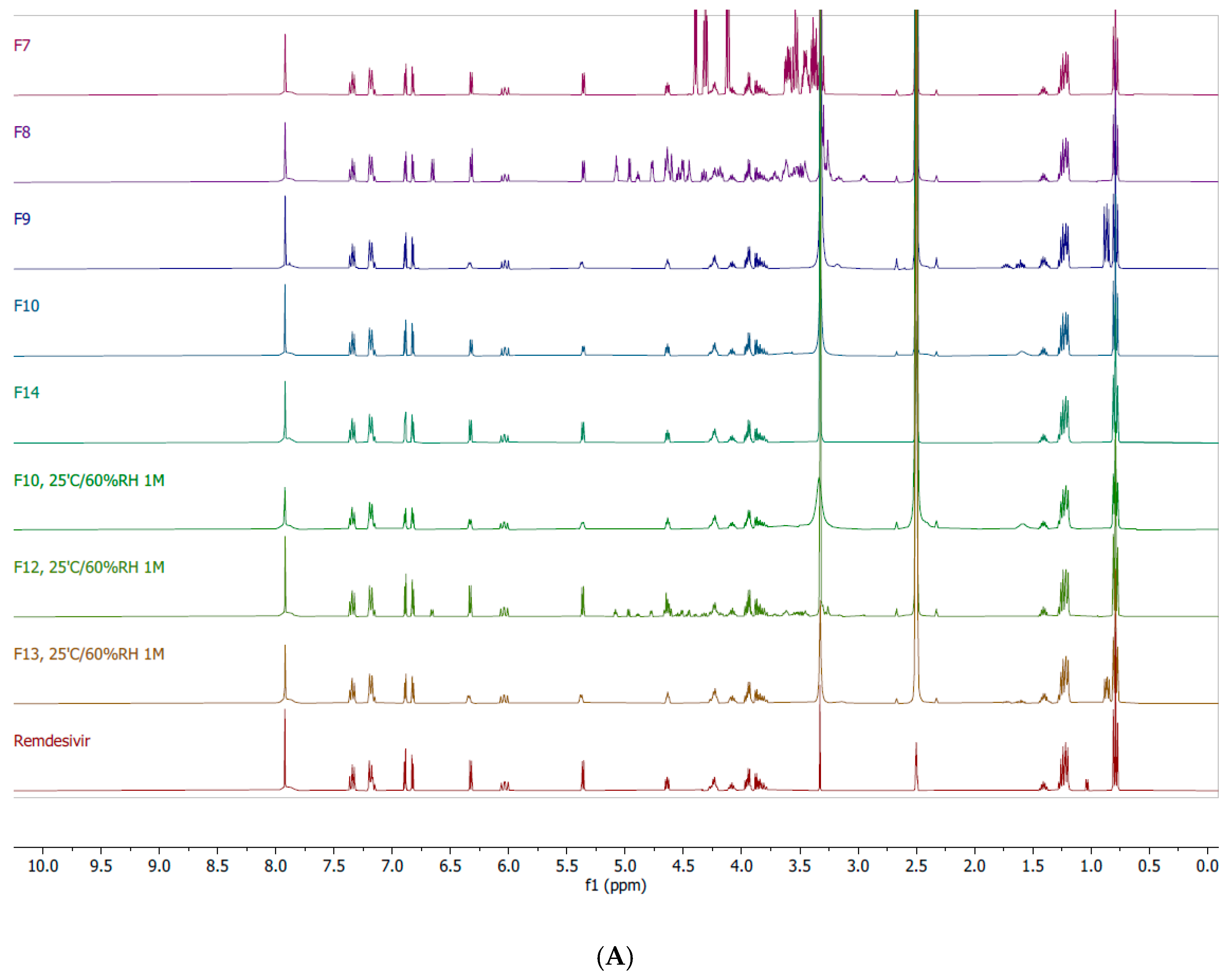
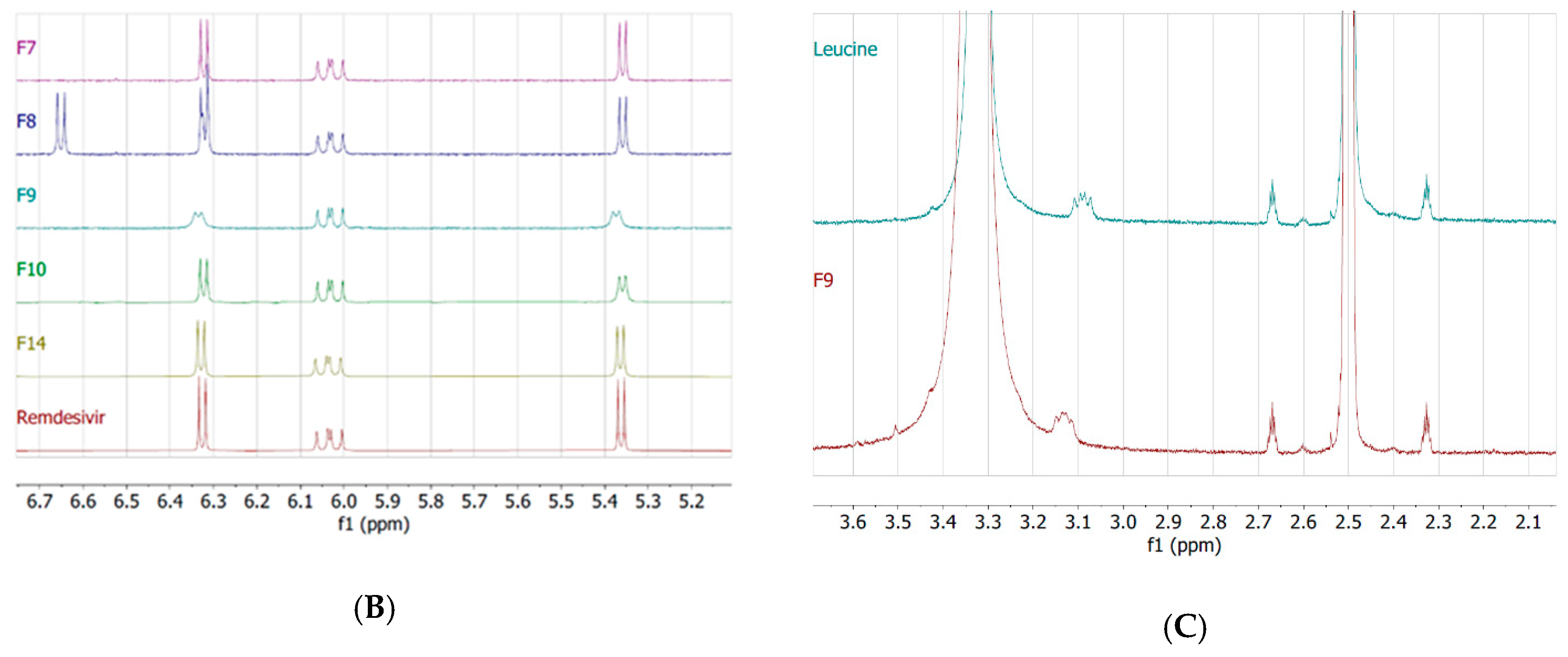
2.8. Nuclear Magnetic Resonance (NMR)
2.9. Preparation of Simulated Lung Fluid (SLF)
2.10. Solubility Testing
2.11. Dissolution Testing
2.12. Stability Study
2.13. In Vivo Pharmacokinetic Study
2.13.1. Single-Dose Dry Powder Insufflation
2.13.2. Analysis of Remdesivir and Its Metabolites in Plasma and Lung Tissue
2.13.3. Pharmacokinetic Analysis
2.14. Statistical Analysis
3. Results
3.1. Physical Properties of TFF Remdesivir
3.2. Aerodynamic Properties of TFF Remdesivir
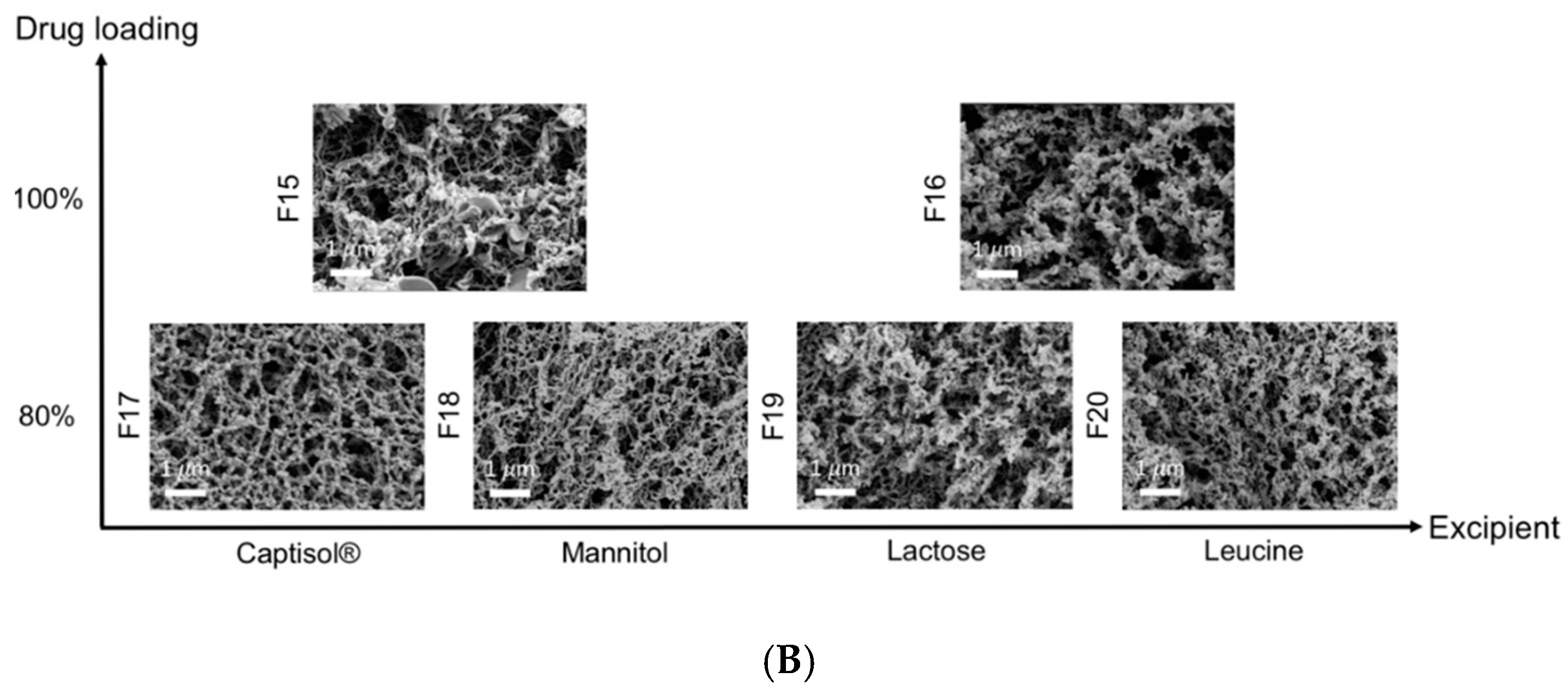
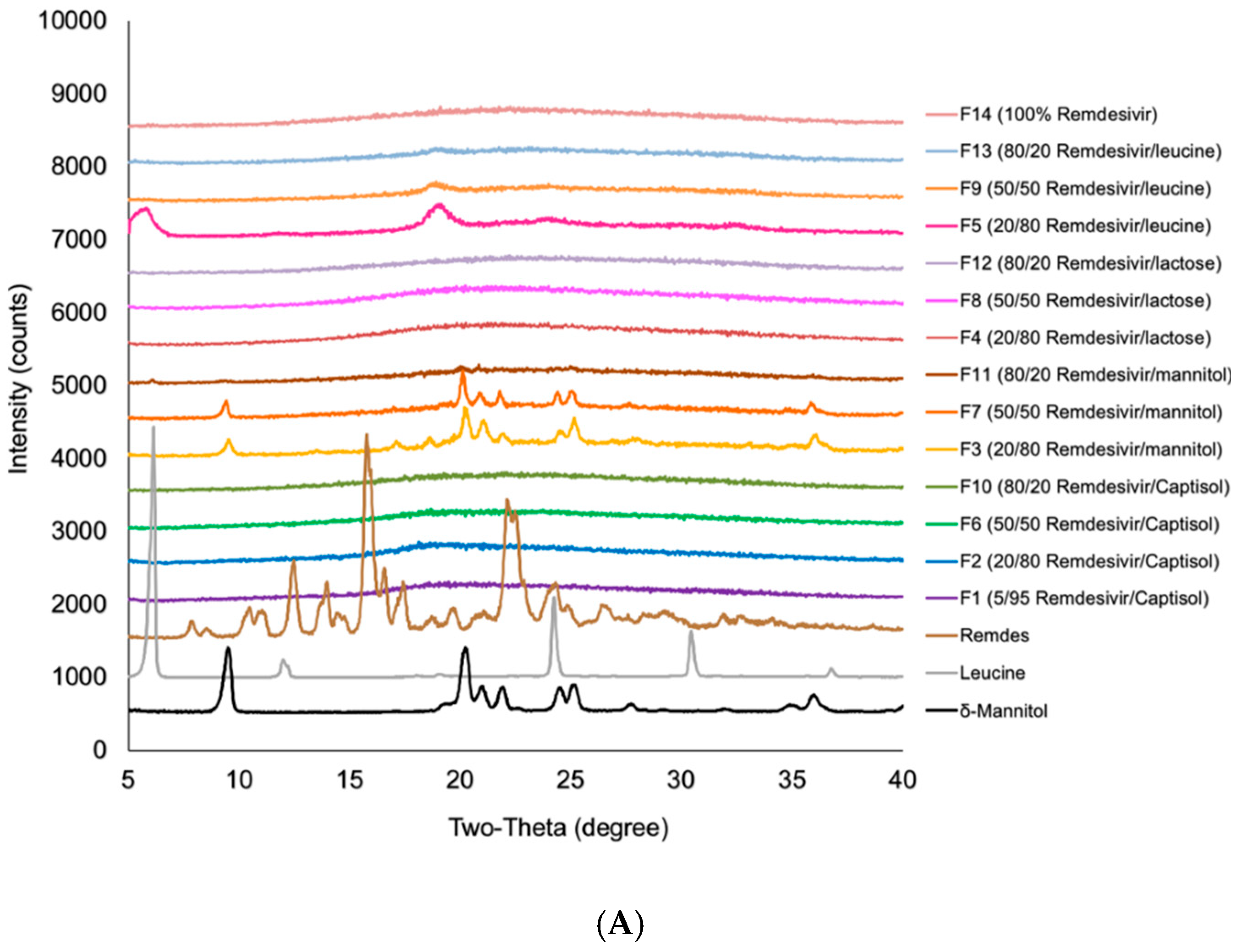
3.3. Interactions Between TFF Remdesivir and Excipients
3.4. Stability Study of TFF Remdesivir
3.4.1. Chemical Stability of TFF Remdesivir Compositions
3.4.2. Physical Stability of TFF Remdesivir Compositions
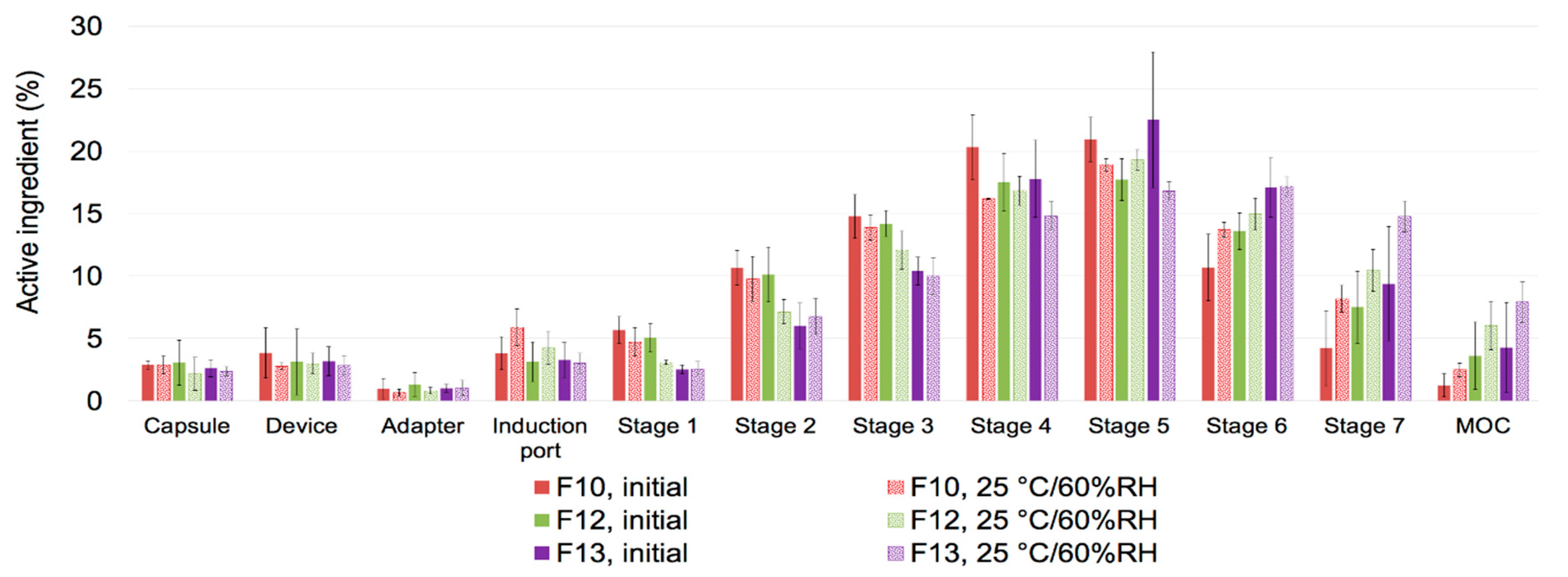
3.4.3. Aerosol Performance after Storage
3.5. Solubility in Simulated Lung Fluid
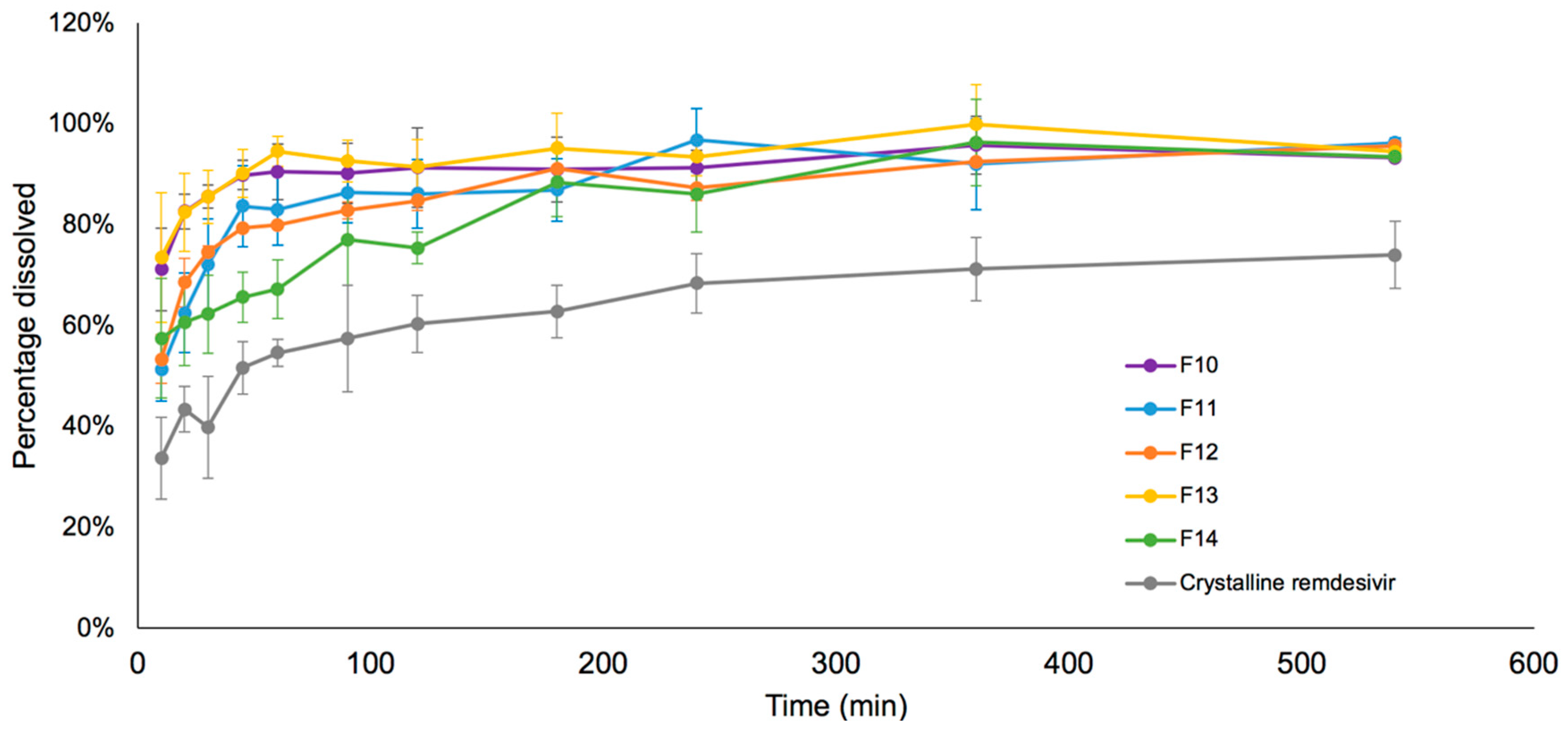
3.6. Drug Release in Simulated Lung Fluid
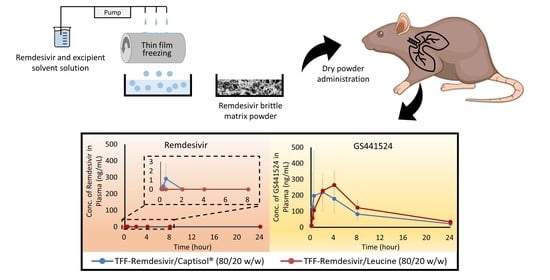
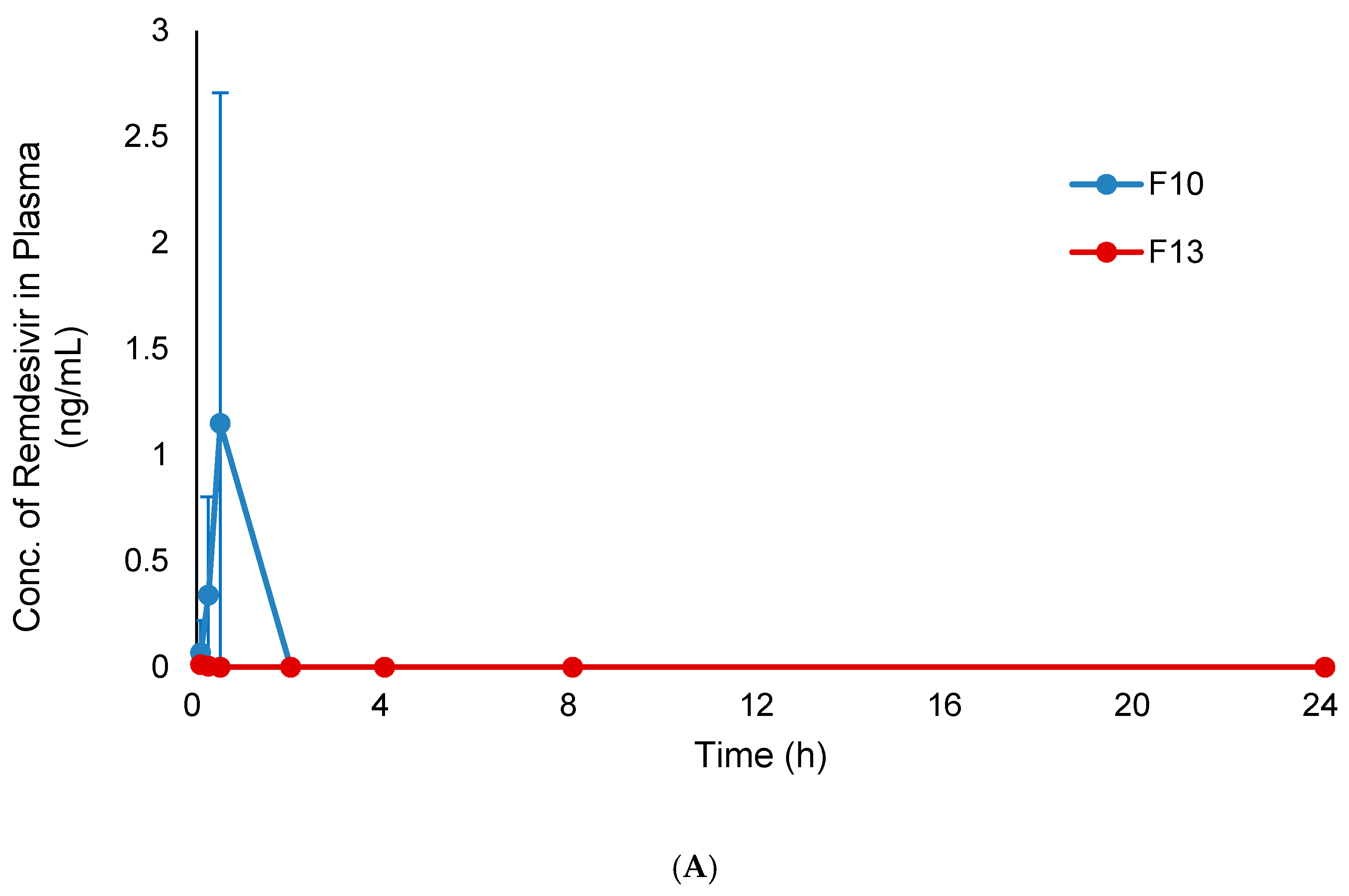
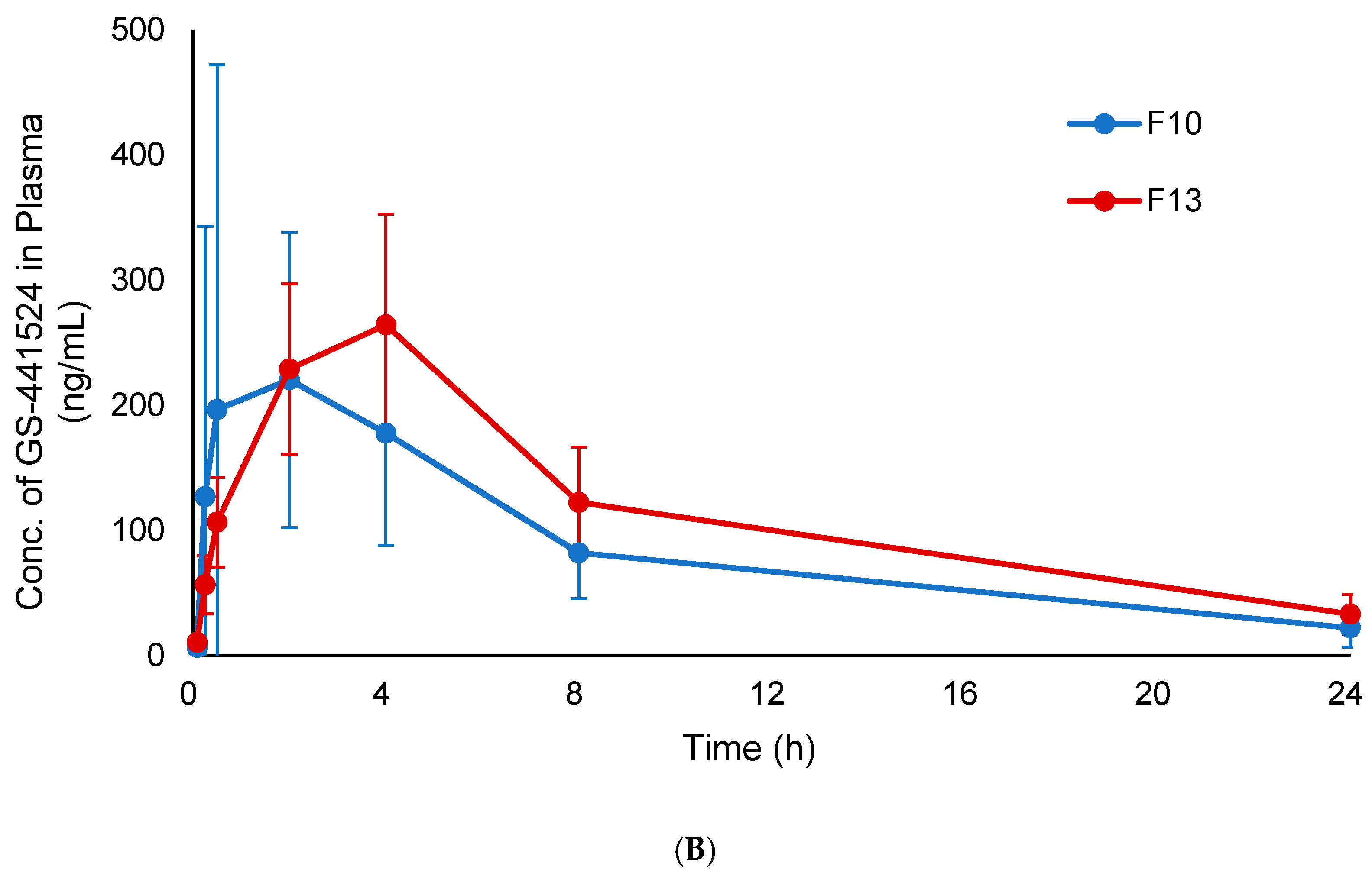
3.7. In Vivo Pharmacokinetic Study
4. Discussion
4.1. Thin Film Freezing Produces High Potency Remdesivir Dry Powders for Inhalation with High Aerosol Performance
4.2. Physical and Chemical Stability of Remdesivir Dry Powder Produced Using TFF
4.3. Remdesivir in TFF Powder Compositions Can Be Dissolved, Absorbed, and Metabolized to GS-441524 in the Lungs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dong, E.; Du, H.; Gardner, L. COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU); Johns Hopkins University Press: Baltimore, MD, USA, 2020. [Google Scholar]
- Singhal, T. A Review of Coronavirus Disease-2019 (COVID-19). Indian J. Pediatr. 2020, 87, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Summary on Compassionate Use: Remdesivir Gilead; European Medicines Agency: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Ko, W.C.; Rolain, J.M.; Lee, N.Y.; Chen, P.L.; Huang, C.T.; Lee, P.I.; Hsueh, P.R. Arguments in favour of remdesivir for treating SARS-CoV-2 infections. Int. J. Antimicrob. Agents 2020, 55, 105933. [Google Scholar] [CrossRef] [PubMed]
- Amirian, S.E.; Levy, J.K. Current knowledge about the antivirals remdesivir (GS-5734) and GS-441524 as therapeutic options for coronaviruses. One Health 2020, 9, 100128. [Google Scholar] [CrossRef] [PubMed]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Gotte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, B.N.; Feldmann, F.; Schwarz, B.; Meade-White, K.; Porter, D.; Schulz, J.; van Doremalen, N.; Leighton, I.; Yinda, C.K.; Perez-Perez, L. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 2020, 585, 273–276. [Google Scholar] [CrossRef]
- Lo, M.K.; Jordan, R.; Arvey, A.; Sudhamsu, J.; Shrivastava-Ranjan, P.; Hotard, A.L.; Flint, M.; McMullan, L.K.; Siegel, D.; Clarke, M.O.; et al. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. Sci. Rep. 2017, 7, 43395. [Google Scholar] [CrossRef]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef]
- Ferner, R.E.; Aronson, J.K. Remdesivir in covid-19. BMJ 2020, 369, m1610. [Google Scholar] [CrossRef] [Green Version]
- National Library of Medicine (US). A Trial of Remdesivir in Adults with Mild and Moderate COVID-19. Identifier NCT04252664. February 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04252664 (accessed on 19 October 2020).
- National Library of Medicine (US). A Trial of Remdesivir in Adults with Severe COVID-19. Identifier NCT04257656. February 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04257656 (accessed on 19 October 2020).
- Stella, V.J.; Rajewski, R.A. Sulfobutylether-beta-cyclodextrin. Int. J. Pharm. 2020, 583, 119396. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Roy, S.; Kumar, A.; Mahmood, S.; Khodapanah, N.; Thomas, S.; Agatemor, C.; Ghosal, K. physicochemical characterization, molecular docking, and in vitro dissolution of glimepiride-captisol inclusion complexes. ACS Omega 2020, 5, 19968–19977. [Google Scholar] [CrossRef] [PubMed]
- Beig, A.; Agbaria, R.; Dahan, A. The use of captisol (SBE7-beta-CD) in oral solubility-enabling formulations: Comparison to HPbetaCD and the solubility-permeability interplay. Eur. J. Pharm. Sci. 2015, 77, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Sun, D. Remdesivir for treatment of COVID-19: Combination of pulmonary and IV administration May Offer Aditional Benefit. AAPS J. 2020, 22, 77. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.B.; Hansen, P.R.; Taboureau, O.; Thomsen, R.; Jürgens, G. Pulmonary administration of remdesivir in the treatment of COVID-19. AAPS J. 2020, 22, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Labiris, N.R.; Dolovich, M.B. Pulmonary drug delivery. Part I: Physiological factors affecting therapeutic effectiveness of aerosolized medications. Br. J. Clin. Pharmacol. 2003, 56, 588–599. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Inactive Ingredient Search for Approved Drug Products; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2020.
- Tolman, J.A.; Nelson, N.A.; Son, Y.J.; Bosselmann, S.; Wiederhold, N.P.; Peters, J.I.; McConville, J.T.; Williams, R.O., 3rd. Characterization and pharmacokinetic analysis of aerosolized aqueous voriconazole solution. Eur. J. Pharm. Biopharm. 2009, 72, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Tolman, J.A.; Nelson, N.A.; Bosselmann, S.; Peters, J.I.; Coalson, J.J.; Wiederhold, N.P.; Williams, R.O., 3rd. Dose tolerability of chronically inhaled voriconazole solution in rodents. Int. J. Pharm. 2009, 379, 25–31. [Google Scholar] [CrossRef]
- Beinborn, N.A.; Du, J.; Wiederhold, N.P.; Smyth, H.D.; Williams, R.O., 3rd. Dry powder insufflation of crystalline and amorphous voriconazole formulations produced by thin film freezing to mice. Eur. J. Pharm. Biopharm. 2012, 81, 600–608. [Google Scholar] [CrossRef]
- Gardenhire, D.S.; Burnett, D.; Strickland, S.; Myers, T.R. A Guide to Aerosol Delivery Devices for Respiratory Therapists, 4th ed.; American Association for Respiratory Care: Irving, TX, USA, 2017. [Google Scholar]
- Overhoff, K.A.; Johnston, K.P.; Tam, J.; Engstrom, J.; Williams, R.O. Use of thin film freezing to enable drug delivery: A review. J. Drug Deliv. Sci. Technol. 2009, 19, 89–98. [Google Scholar] [CrossRef]
- Wang, Y.B.; Watts, A.B.; Williams, R.O. Effect of processing parameters on the physicochemical and aerodynamic properties of respirable brittle matrix powders. J. Drug Deliv. Sci. Technol. 2014, 24, 390–396. [Google Scholar] [CrossRef]
- Watts, A.B.; Wang, Y.B.; Johnston, K.P.; Williams, R.O., 3rd. Respirable low-density microparticles formed in situ from aerosolized brittle matrices. Pharm. Res. 2013, 30, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.B.; Watts, A.B.; Peters, J.I.; Liu, S.; Batra, A.; Williams, R.O., 3rd. In vitro and in vivo performance of dry powder inhalation formulations: Comparison of particles prepared by thin film freezing and micronization. AAPS PharmSciTech 2014, 15, 981–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longest, P.W.; Hindle, M. Small airway absorption and microdosimetry of inhaled corticosteroid particles after deposition. Pharm. Res. 2017, 34, 2049–2065. [Google Scholar] [CrossRef]
- Overhoff, K.A.; Engstrom, J.D.; Chen, B.; Scherzer, B.D.; Milner, T.E.; Johnston, K.P.; Williams, R.O., 3rd. Novel ultra-rapid freezing particle engineering process for enhancement of dissolution rates of poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2007, 65, 57–67. [Google Scholar] [CrossRef]
- Hassoun, M.; Royall, P.G.; Parry, M.; Harvey, R.D.; Forbes, B. Design and development of a biorelevant simulated human lung fluid. J. Drug Deliv. Sci. Technol. 2018, 47, 485–491. [Google Scholar] [CrossRef]
- Arora, D.; Shah, K.A.; Halquist, M.S.; Sakagami, M. In vitro aqueous fluid-capacity-limited dissolution testing of respirable aerosol drug particles generated from inhaler products. Pharm. Res. 2010, 27, 786–795. [Google Scholar] [CrossRef]
- Brunaugh, A.D.; Seo, H.; Warnken, Z.; Ding, L.; Seo, S.H.; Smyth, H.D.C. Broad-Spectrum, Patient-Adaptable Inhaled Niclosamide-Lysozyme Particles are Efficacious Against Coronaviruses in Lethal Murine Infection Models. bioRxiv 2020, 310490. [Google Scholar] [CrossRef]
- Rohrschneider, M.; Bhagwat, S.; Krampe, R.; Michler, V.; Breitkreutz, J.; Hochhaus, G. Evaluation of the transwell system for characterization of dissolution behavior of inhalation drugs: Effects of membrane and surfactant. Mol. Pharm. 2015, 12, 2618–2624. [Google Scholar] [CrossRef]
- Cares-Pacheco, M.G.; Vaca-Medina, G.; Calvet, R.; Espitalier, F.; Letourneau, J.J.; Rouilly, A.; Rodier, E. Physicochemical characterization of D-mannitol polymorphs: The challenging surface energy determination by inverse gas chromatography in the infinite dilution region. Int. J. Pharm. 2014, 475, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Molina, C.; Kaialy, W.; Chen, Q.; Commandeur, D.; Nokhodchi, A. Agglomerated novel spray-dried lactose-leucine tailored as a carrier to enhance the aerosolization performance of salbutamol sulfate from DPI formulations. Drug Deliv. Transl. Res. 2018, 8, 1769–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilcer, G.; Amighi, K. Formulation strategy and use of excipients in pulmonary drug delivery. Int. J. Pharm. 2010, 392, 1–19. [Google Scholar] [CrossRef] [PubMed]
- United States Securities and Exchange Commission. Annual Report Pursuant to Section 13 Or 15(D) OF THE Securities Exchange Act of 1934; For the fiscal year ended 31 December 2019; Form 10-K Savara Inc.; United States Securities and Exchange Commission: Washington, DC, USA, 2020.
- National Library of Medicine (US). A Study of AeroVanc for the Treatment of MRSA Infection in CF Patients. Identifier NCT03181932. June 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03181932 (accessed on 19 October 2020).
- Sahakijpijarn, S.; Moon, C.; Ma, X.; Su, Y.; Koleng, J.J.; Dolocan, A.; Williams, R.O. Using thin film freezing to minimize excipients in inhalable tacrolimus dry powder formulations. Int. J. Pharm. 2020, 586, 119490. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.; Sahakijpijarn, S.; Koleng, J.J.; Williams, R.O. Processing design space is critical for voriconazole nanoaggregates for dry powder inhalation produced by thin film freezing. J. Drug Deliv. Sci. Technol. 2019, 54, 1–11. [Google Scholar] [CrossRef]
- Paajanen, M.; Katainen, J.; Raula, J.; Kauppinen, E.I.; Lahtinen, J. Direct evidence on reduced adhesion of Salbutamol sulphate particles due to L-leucine coating. Powder Technol. 2009, 192, 6–11. [Google Scholar] [CrossRef]
- Mangal, S.; Park, H.; Nour, R.; Shetty, N.; Cavallaro, A.; Zemlyanov, D.; Thalberg, K.; Puri, V.; Nicholas, M.; Narang, A.S.; et al. Correlations between surface composition and aerosolization of jet-milled dry powder inhaler formulations with pharmaceutical lubricants. Int. J. Pharm. 2019, 568, 118504. [Google Scholar] [CrossRef]
- Thompson, J.W.; Kaiser, T.J.; Jorgenson, J.W. Viscosity measurements of methanol-water and acetonitrile-water mixtures at pressures up to 3500 bar using a novel capillary time-of-flight viscometer. J. Chromatogr. A 2006, 1134, 201–209. [Google Scholar] [CrossRef]
- Besbes, R.; Ouerfelli, N.; Latrous, H. Density, dynamic viscosity, and derived properties of binary mixtures of 1,4 dioxane with water at T=298.15 K. J. Mol. Liq. 2009, 145, 1–4. [Google Scholar] [CrossRef]
- Beinborn, N.A.; Lirola, H.L.; Williams, R.O., 3rd. Effect of process variables on morphology and aerodynamic properties of voriconazole formulations produced by thin film freezing. Int. J. Pharm. 2012, 429, 46–57. [Google Scholar] [CrossRef]
- Wyttenbach, N.; Kuentz, M. Glass-forming ability of compounds in marketed amorphous drug products. Eur. J. Pharm. Biopharm. 2017, 112, 204–208. [Google Scholar] [CrossRef]
- Blaabjerg, L.I.; Bulduk, B.; Lindenberg, E.; Lobmann, K.; Rades, T.; Grohganz, H. Influence of glass forming ability on the physical stability of supersaturated amorphous solid dispersions. J. Pharm. Sci. 2019, 108, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Panini, P.; Rampazzo, M.; Singh, A.; Vanhoutte, F.; van den Mooter, G. Myth or Truth: The Glass Forming Ability Class III Drugs Will Always Form Single-Phase Homogenous Amorphous Solid Dispersion Formulations. Pharmaceutics 2019, 11, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.M.; Neef, N. Interpretation and prediction of inhaled drug particle accumulation in the lung and its associated toxicity. Xenobiotica 2011, 42, 86–93. [Google Scholar] [CrossRef]
- Sinswat, P.; Overhoff, K.A.; McConville, J.T.; Johnston, K.P.; Williams, R.O., 3rd. Nebulization of nanoparticulate amorphous or crystalline tacrolimus--single-dose pharmacokinetics study in mice. Eur. J. Pharm. Biopharm. 2008, 69, 1057–1066. [Google Scholar] [CrossRef]
- Jiang, N.; Wang, S.; Cheng, Z.; Liu, W. In vitro and in vivo evaluation of porous lactose/mannitol carriers for solubility enhancement of poorly water-soluble drugs. Dry. Technol. 2020, 38, 889–902. [Google Scholar] [CrossRef]
- Van der Merwe, J.; Steenekamp, J.; Steyn, D.; Hamman, J. The role of functional excipients in solid oral dosage forms to overcome poor drug dissolution and bioavailability. Pharmaceutics 2020, 12, 393. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Gundampati, R.; Kumar, T. NMR methods to characterize protein-ligand interactions. Nat. Prod. Commun. 2019, 14, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hibbert, F.; Emsley, J. Hydrogen bonding and chemical reactivity. Adv. Phys. Org. Chem. 1990, 26, 255–379. [Google Scholar]
- Konrat, R.; Tollinger, M.; Kontaxis, G.; Krautler, B. NMR techniques to study hydrogen bonding in aqueous solution. Mon. Fur. Chem. 1999, 130, 961–982. [Google Scholar]
- Mangal, S.; Nie, H.; Xu, R.; Guo, R.; Cavallaro, A.; Zemlyanov, D.; Zhou, Q.T. Physico-chemical properties, aerosolization and dissolution of co-spray dried azithromycin particles with l-leucine for inhalation. Pharm. Res. 2018, 35, 28. [Google Scholar] [CrossRef] [Green Version]
- Löbmann, K.; Grohganz, H.; Laitinen, R.; Strachan, C.; Rades, T. Amino acids as co-amorphous stabilizers for poorly water soluble drugs—Part 1: Preparation, stability and dissolution enhancement. Eur. J. Pharm. Biopharm. 2013, 85, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, S.F.; O’Malley, S.; Mosher, G.L. Improved aqueous solubility of crystalline astaxanthin (3,3’-dihydroxy-beta, beta-carotene-4,4’-dione) by Captisol (sulfobutyl ether beta-cyclodextrin). J. Pharm. Sci. 2003, 92, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Yang, K. What do we know about remdesivir drug interactions? Dlin. Transl. Sci. 2020, 13, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.; Thorn, H.; Sjogren, E.; Holmsten, L.; Rubin, K.; Lennernas, H. Pulmonary Dissolution of Poorly Soluble Compounds Studied in an ex Vivo Rat Lung Model. Mol. Pharm. 2019, 16, 3053–3064. [Google Scholar] [CrossRef] [PubMed]
- Pruijssers, A.J.; George, A.S.; Schafer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H., 3rd; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir Inhibits SARS-CoV-2 in Human Lung Cells and Chimeric SARS-CoV Expressing the SARS-CoV-2 RNA Polymerase in Mice. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef] [PubMed]
- Yan, V.C.; Muller, F.L. Advantages of the parent nucleoside GS-441524 over remdesivir for Covid-19 treatment. ACS Med. Chem. Lett. 2020, 11, 1361–1366. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Drug Loading (% w/w) | Excipient | Solid Content (% w/v) | Solvent (v/v) | Processing Temperature (°C) |
|---|---|---|---|---|---|
| F1 | 5 | Captisol® | 1.00 | Acetonitrile/water (50/50) | −100 |
| F2 | 20 | Captisol® | 1.00 | Acetonitrile/water (50/50) | −100 |
| F3 | 20 | Mannitol | 1.00 | Acetonitrile/water (50/50) | −100 |
| F4 | 20 | Lactose | 1.00 | Acetonitrile/water (50/50) | −100 |
| F5 | 20 | Leucine | 0.75 | Acetonitrile/water (50/50) | −100 |
| F6 | 50 | Captisol® | 0.30 | Acetonitrile/water (50/50) | −100 |
| F7 | 50 | Mannitol | 0.30 | Acetonitrile/water (50/50) | −100 |
| F8 | 50 | Lactose | 0.30 | Acetonitrile/water (50/50) | −100 |
| F9 | 50 | Leucine | 0.30 | Acetonitrile/water (50/50) | −100 |
| F10 | 80 | Captisol® | 0.30 | Acetonitrile/water (50/50) | −100 |
| F11 | 80 | Mannitol | 0.30 | Acetonitrile/water (50/50) | −100 |
| F12 | 80 | Lactose | 0.30 | Acetonitrile/water (50/50) | −100 |
| F13 | 80 | Leucine | 0.30 | Acetonitrile/water (50/50) | −100 |
| F14 | 100 | - | 0.25 | Acetonitrile/water (50/50) | −100 |
| F15 | 100 | - | 0.50 | 1,4-dioxane/water (50/50) | −100 |
| F16 | 100 | - | 1.00 | 1,4-dioxane/water (50/50) | −100 |
| F17 | 80 | Captisol® | 1.00 | 1,4-dioxane/water (50/50) | −100 |
| F18 | 80 | Mannitol | 1.00 | 1,4-dioxane/water (50/50) | −100 |
| F19 | 80 | Lactose | 1.00 | 1,4-dioxane/water (50/50) | −100 |
| F20 | 80 | Leucine | 1.00 | 1,4-dioxane/water (50/50) | −100 |
| Formulations | MMAD (μm) | GSD | FPF<5μm (%, Recovered Dose) | FPF<3μm (%, Recovered Dose) | FPF<5μm (%, Delivered Dose) | FPF<3μm (%, Delivered Dose) | EF (%, Recovered Dose) |
|---|---|---|---|---|---|---|---|
| F1 | 3.10 ± 0.04 | 2.88 ± 0.03 | 30.73 ± 1.11 | 22.16 ± 1.01 | 36.87 ± 0.78 | 27.78 ± 0.84 | 83.35 ± 1.83 |
| F2 | 2.59 ± 0.15 | 2.56 ± 0.28 | 45.84 ± 6.03 | 35.65 ± 5.97 | 55.00 ± 5.44 | 42.68 ± 4.88 | 83.32 ± 6.70 |
| F3 | 2.03 ± 0.10 | 2.76 ± 0.07 | 56.21 ± 2.16 | 46.69 ± 1.11 | 62.27 ± 2.72 | 51.72 ± 1.52 | 90.28 ± 0.67 |
| F4 | 2.02 ± 0.37 | 2.69 ± 0.10 | 52.38 ± 4.65 | 43.09 ± 6.64 | 60.98 ± 3.16 | 50.08 ± 5.50 | 85.86 ± 5.40 |
| F5 | 0.74 ± 0.06 | 3.50 ± 0.13 | 86.03 ± 2.82 | 78.64 ± 2.80 | 92.10 ± 1.76 | 84.19 ± 2.00 | 93.40 ± 1.73 |
| F6 | 2.22 ± 0.14 | 2.73 ± 0.22 | 65.19 ± 1.18 | 51.98 ± 2.16 | 72.17 ± 2.04 | 57.56 ± 3.11 | 90.37 ± 2.49 |
| F7 | 2.32 ± 0.07 | 2.57 ± 0.03 | 65.90 ± 0.91 | 51.65 ± 1.27 | 73.08 ± 0.22 | 57.28 ± 0.79 | 90.17 ± 0.97 |
| F8 | 2.40 ± 0.39 | 2.28 ± 0.11 | 68.43 ± 4.33 | 51.56 ± 7.23 | 75.74 ± 4.60 | 57.08 ± 7.92 | 90.34 ± 0.82 |
| F9 | 0.82 ± 0.07 | 3.19 ± 0.10 | 85.83 ± 3.96 | 78.32 ± 4.16 | 92.99 ± 1.11 | 84.84 ± 1.73 | 92.28 ± 3.23 |
| F10 | 2.16 ± 0.21 | 2.42 ± 0.06 | 71.48 ± 5.52 | 57.01 ± 6.25 | 78.08 ± 5.51 | 62.25 ± 5.65 | 91.48 ± 2.09 |
| F11 | 2.44 ± 0.06 | 2.53 ± 0.06 | 64.21 ± 3.53 | 48.90 ± 3.06 | 71.62 ± 3.10 | 54.54 ± 2.68 | 89.61 ± 1.30 |
| F12 | 2.03 ± 0.11 | 2.51 ± 0.12 | 70.32 ± 3.39 | 55.54 ± 2.70 | 77.24 ± 2.94 | 61.02 ± 2.85 | 91.02 ± 1.55 |
| F13 | 1.45 ± 0.07 | 2.17 ± 0.01 | 82.71 ± 2.54 | 73.34 ± 3.17 | 89.68 ± 0.91 | 79.51 ± 1.85 | 92.21 ± 1.94 |
| F14 | 2.09 ± 0.07 | 2.79 ± 0.13 | 65.80 ± 2.68 | 53.95 ± 1.26 | 74.44 ± 1.29 | 61.05 ± 0.43 | 88.37 ± 2.16 |
| F15 | 1.53 ± 0.16 | 2.70 ± 0.10 | 81.66 ± 0.80 | 69.08 ± 1.63 | 85.11 ± 1.20 | 72.00 ± 2.11 | 95.95 ± 0.56 |
| F16 | 1.42 ± 0.20 | 2.77 ± 0.18 | 84.25 ± 1.87 | 71.00 ± 3.91 | 86.77 ± 1.67 | 73.11 ± 3.86 | 97.10 ± 0.40 |
| F17 | 1.55 ± 0.16 | 2.99 ± 0.43 | 74.59 ± 7.03 | 63.74 ± 3.76 | 78.76 ± 6.75 | 67.30 ± 3.37 | 94.67 ± 0.97 |
| F18 | 2.03 ± 0.20 | 2.48 ± 0.07 | 75.34 ± 1.06 | 60.44 ± 3.13 | 79.94 ± 2.72 | 64.15 ± 4.60 | 94.30 ± 1.94 |
| F19 | 1.28 ± 0.10 | 2.92 ± 0.17 | 82.40 ± 1.18 | 70.49 ± 1.14 | 87.28 ± 0.57 | 74.55 ± 0.56 | 94.41 ± 0.83 |
| F20 | 1.29 ± 0.11 | 3.15 ± 0.18 | 82.26 ± 3.86 | 70.68 ± 4.39 | 86.09 ± 3.51 | 73.97 ± 4.15 | 95.53 ± 0.60 |
| Formulation | Condition | MMAD (μm) | GSD | FPF<5μm (%, Recovered Dose) | FPF<3μm (%, Recovered Dose) | FPF<5μm (%, Delivered Dose) | FPF<3μm (%, Delivered Dose) | EF (%, Recovered Dose) |
|---|---|---|---|---|---|---|---|---|
| F10 | Initial | 1.99 ± 0.19 | 2.49 ± 0.13 | 74.84 ± 4.70 | 59.64 ± 4.37 | 80.22 ± 3.08 | 63.93 ± 2.55 | 93.22 ± 2.71 |
| 25 °C/60% RH, 1 M | 1.70 ± 0.16 | 2.67 ± 0.13 | 75.85 ± 1.45 | 61.61 ± 2.89 | 80.40 ± 1.79 | 65.31 ± 3.26 | 94.35 ± 0.54 | |
| F12 | Initial | 1.78 ± 0.23 | 2.72 ± 0.30 | 76.88 ± 6.37 | 62.30 ± 6.15 | 81.79 ± 3.70 | 66.25 ± 4.14 | 93.88 ± 3.61 |
| 25 °C/60% RH, 1 M | 1.41 ± 0.20 | 2.65 ± 0.16 | 81.46 ± 2.32 | 69.45 ± 2.83 | 85.89 ± 1.16 | 73.21 ± 2.03 | 94.84 ± 1.52 | |
| F13 | Initial | 1.33 ± 0.10 | 2.54 ± 0.42 | 83.07 ± 1.35 | 72.76 ± 2.81 | 88.19 ± 2.43 | 77.26 ± 3.87 | 94.22 ± 1.09 |
| 25 °C/60% RH, 1 M | 1.19 ± 0.22 | 2.96 ± 0.16 | 81.23 ± 4.16 | 72.46 ± 3.07 | 87.60 ± 1.82 | 76.54 ± 3.74 | 94.69 ± 0.61 |
| Formulations | Tmax (h) | Cmax (ng/mL) | AUC0–24 (ng·h/mL) | AUCinf (ng·h/mL) |
|---|---|---|---|---|
| F10 | 2 | 220.4 ± 118.0 | 2115.3 ± 979.6 | 2397.8 ± 1178.3 |
| F13 | 4 | 264.3 ± 88.5 | 2788.5 ± 857.1 | 3204.9 ± 967.6 |
| Formulation | Rat | Wet Lung (mg) | Remdesivir | GS-441524 | ||
|---|---|---|---|---|---|---|
| Total Weight (μg) | In wet Lung (ng/mg) | Total Weight (μg) | In Wet Lung (ng/mg) | |||
| F10 | A | 1164.5 | 16.5 | 14.2 | 6.2 | 5.3 |
| B | 1494.8 | 11.3 | 7.5 | 6.4 | 4.3 | |
| C | 1255.2 | 13.6 | 10.9 | 10.8 | 8.6 | |
| D | 1194.0 | 0.1 | 0.1 | 0.2 | 0.2 | |
| E | 1176.3 | 16.2 | 13.8 | 12.2 | 10.4 | |
| Average ± SD | - | 11.6 ± 6.7 | 9.3 ± 5.8 | 7.2 ± 4.7 | 5.8 ± 4.0 | |
| F13 | F | 1025.1 | 12.4 | 11.2 | 12.5 | 11.3 |
| G | 1126.8 | 7.7 | 7.6 | 15.3 | 14.9 | |
| H | 1254.5 | 4.0 | 3.5 | 18.7 | 16.6 | |
| I | 1263.1 | 3.4 | 2.7 | 18.7 | 14.9 | |
| J | 1164.5 | 0.6 | 0.5 | 1.2 | 1.0 | |
| Average ± SD | - | 5.6 ± 4.5 | 5.1 ± 4.3 | 13.4 ± 7.3 | 11.7 ± 6.3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahakijpijarn, S.; Moon, C.; Koleng, J.J.; Christensen, D.J.; Williams, R.O., III. Development of Remdesivir as a Dry Powder for Inhalation by Thin Film Freezing. Pharmaceutics 2020, 12, 1002. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12111002
Sahakijpijarn S, Moon C, Koleng JJ, Christensen DJ, Williams RO III. Development of Remdesivir as a Dry Powder for Inhalation by Thin Film Freezing. Pharmaceutics. 2020; 12(11):1002. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12111002
Chicago/Turabian StyleSahakijpijarn, Sawittree, Chaeho Moon, John J. Koleng, Dale J. Christensen, and Robert O. Williams, III. 2020. "Development of Remdesivir as a Dry Powder for Inhalation by Thin Film Freezing" Pharmaceutics 12, no. 11: 1002. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12111002