Large-Scale Production of Lentiviral Vectors: Current Perspectives and Challenges

 and
and 
Abstract
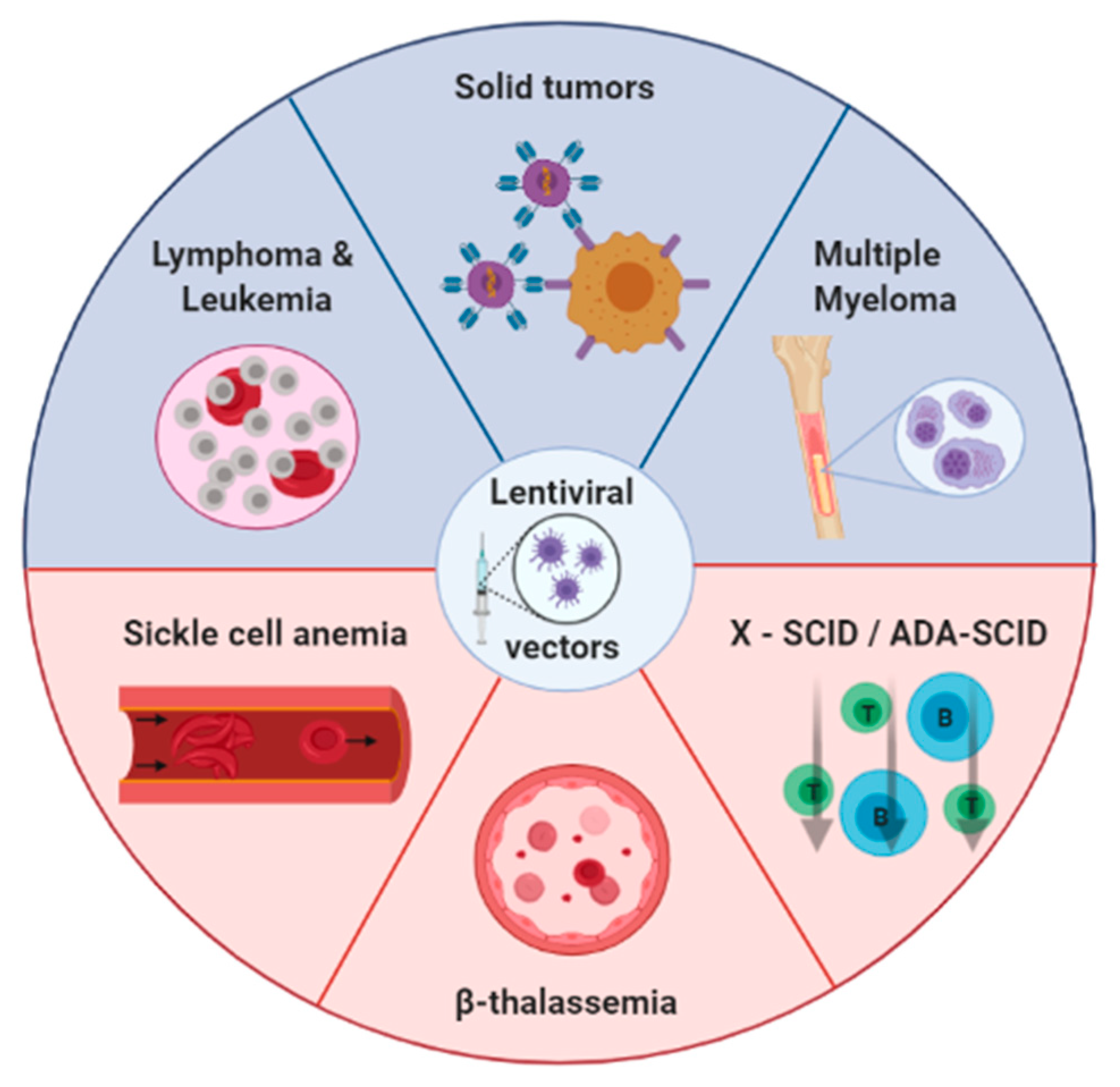
:1. Introduction
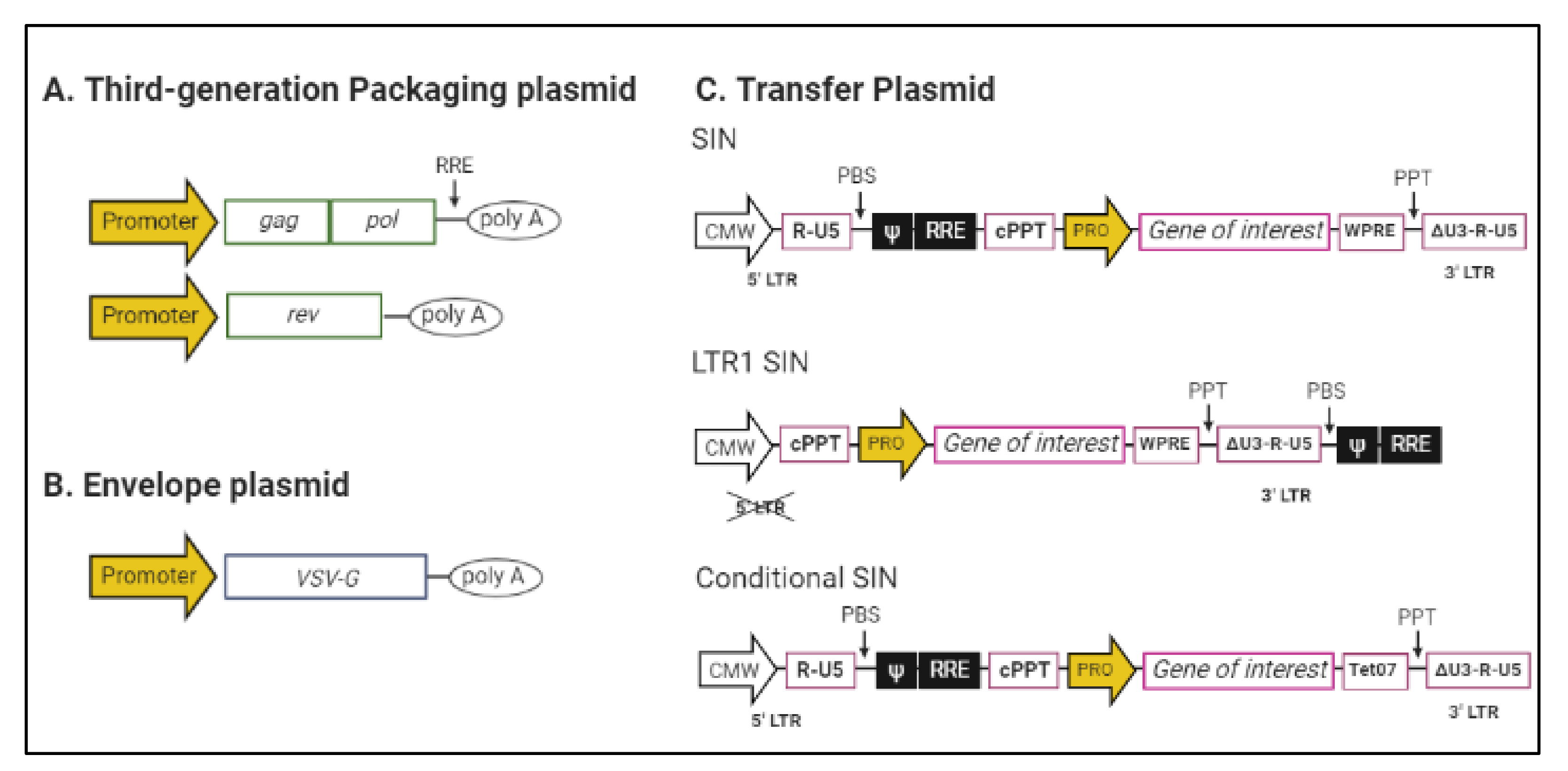
2. Lentiviral Vectors
3. Lentiviral Vectors Production
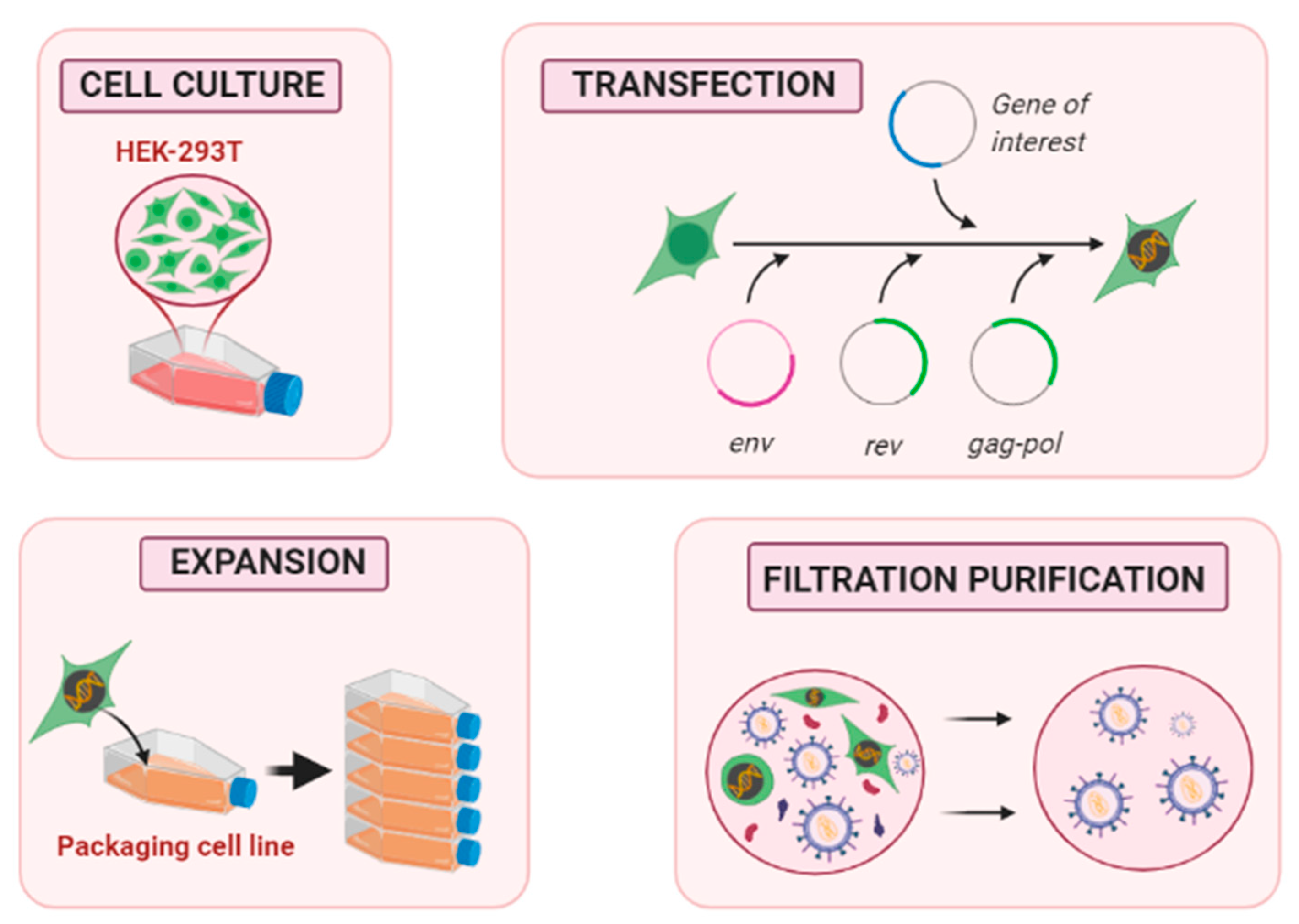
3.1. Upstream Process
3.1.1. Packaging Cell Line Culture
Constitutive PCL
Inducible PCL
3.1.2. Methods for Gene Transfection
3.1.3. How to Improve the Lentiviral Vectors Production
Adherent Cells Culture
Suspension Cells Culture
3.2. Downstream Processing of LV
3.2.1. Clarification
3.2.2. Concentration and Purification
Tangential Flow Filtration
Chromatography
3.2.3. Polishing and Storage
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LV | Lentiviral vector |
| RV | Retroviral vector |
| FDA | Food and Drug Administration |
| EMA | European Medicine Agency |
| GMP | Good manufacturing practices |
| PCL | Packaging cell line |
| HIV | Human immunodeficiency virus |
| HEK | Human embryonic kidney |
| VSV-G | Vesicular stomatitis virus envelope glycoprotein |
| PEI | Polyethyleneimine |
| CF | Cell factory |
References
- Anguela, X.M.; High, K.A. Entering the Modern Era of Gene Therapy. Annu. Rev. Med. 2019, 70, 273–288. [Google Scholar] [CrossRef] [Green Version]
- Áyen, Á.; Jiménez Martínez, Y.; Marchal, J.; Boulaiz, H. Recent Progress in Gene Therapy for Ovarian Cancer. Int. J. Mol. Sci. 2018, 19, 1930. [Google Scholar] [CrossRef] [Green Version]
- Navarro, S.A.; Carrillo, E.; Griñán-Lisón, C.; Martín, A.; Perán, M.; Marchal, J.A.; Boulaiz, H. Cancer Suicide Gene Therapy: A Patent Review. Expert Opin. Ther. Pat. 2016, 26, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Áyen, Á.; Jiménez Martínez, Y.; Boulaiz, H. Targeted Gene Delivery Therapies for Cervical Cancer. Cancers 2020, 12, 1301. [Google Scholar] [CrossRef]
- Ramírez, A.; Conejo-García, A.; Griñán-Lisón, C.; López-Cara, L.C.; Jiménez, G.; Campos, J.M.; Marchal, J.A.; Boulaiz, H. Enhancement of Tumor Cell Death by Combining Gef Gene Mediated Therapy and New 1,4-Benzoxazepin-2,6-Dichloropurine Derivatives in Breast Cancer Cells. Front. Pharmacol. 2018, 9, 798. [Google Scholar] [CrossRef]
- Nobles, C.L.; Sherrill-Mix, S.; Everett, J.K.; Reddy, S.; Fraietta, J.A.; Porter, D.L.; Frey, N.; Gill, S.I.; Grupp, S.A.; Maude, S.L.; et al. CD19-Targeting CAR T Cell Immunotherapy Outcomes Correlate with Genomic Modification by Vector Integration. J. Clin. Investig. 2019, 130, 673–685. [Google Scholar] [CrossRef]
- Chen, Y.H.; Keiser, M.S.; Davidson, B.L. Viral Vectors for Gene Transfer. Curr. Protoc. Mouse Biol. 2018, 8, e58. [Google Scholar] [CrossRef]
- Philpott, N.J.; Thrasher, A.J. Use of Nonintegrating Lentiviral Vectors for Gene Therapy. Hum. Gene Ther. 2007, 18, 483–489. [Google Scholar] [CrossRef]
- Boulaiz, H.; Marchal, J.A.; Prados, J.; Melguizo, C.; Aránega, A. Non-Viral and Viral Vectors for Gene Therapy. Cell. Mol. Biol. (Noisy-le-Grand) 2005, 51, 3–22. [Google Scholar]
- FDA Approves Innovative Gene Therapy to Treat Pediatric Patients with Spinal Muscular Atrophy, a Rare Disease and Leading Genetic Cause of Infant Mortality | FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease (accessed on 9 October 2020).
- Patel, U.; Boucher, M.; de Léséleuc, L.; Visintini, S. Voretigene Neparvovec: An Emerging Gene Therapy for the Treatment of Inherited Blindness. CADTH Issues Emerg. Health Technol. 2016, 1–11. [Google Scholar]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-Associated Virus Vector as a Platform for Gene Therapy Delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Mullard, A. EMA Greenlights Second Gene Therapy. Nat. Rev. Drug Discov. 2016, 15, 299. [Google Scholar] [CrossRef]
- Strimvelis | European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/strimvelis (accessed on 9 October 2020).
- FDA Approves CAR-T Cell Therapy to Treat Adults with Certain Types of Large B-Cell Lymphoma | FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-car-t-cell-therapy-treat-adults-certain-types-large-b-cell-lymphoma (accessed on 8 October 2020).
- Baldo, A.; Galanis, E.; Tangy, F.; Herman, P. Biosafety Considerations for Attenuated Measles Virus Vectors Used in Virotherapy and Vaccination. Hum. Vaccines Immunother. 2016, 12, 1102–1116. [Google Scholar] [CrossRef] [Green Version]
- Frantz, P.N.; Teeravechyan, S.; Tangy, F. Measles-Derived Vaccines to Prevent Emerging Viral Diseases. Microbes Infect. 2018, 20, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, I.R.; Sebastian, S. Novel Viral Vectors in Infectious Diseases. Immunology 2018, 153, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.C.; Guan, X.H.; Li, Y.H.; Huang, J.Y.; Jiang, T.; Hou, L.H.; Li, J.X.; Yang, B.F.; Wang, L.; Wang, W.J.; et al. Immunogenicity and Safety of a Recombinant Adenovirus Type-5-Vectored COVID-19 Vaccine in Healthy Adults Aged 18 Years or Older: A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet 2020, 396, 479–488. [Google Scholar] [CrossRef]
- Haase, A.T. Pathogenesis of Lentivirus Infections. Nature 1986, 322, 130–136. [Google Scholar] [CrossRef]
- Sakuma, T.; Barry, M.A.; Ikeda, Y. Lentiviral Vectors: Basic to Translational. Biochem. J. 2012, 618, 603–618. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; O’Doherty, U. Clinical Use of Lentiviral Vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Ferris, A.L.; Wu, X.; Hughes, C.M.; Stewart, C.; Smith, S.J.; Milne, T.A.; Wang, G.G.; Shun, M.C.; Allis, C.D.; Engelman, A.; et al. Lens Epithelium-Derived Growth Factor Fusion Proteins Redirect HIV-1 DNA Integration. Proc. Natl. Acad. Sci. USA 2010, 107, 3135–3140. [Google Scholar] [CrossRef] [Green Version]
- Craigie, R.; Bushman, F.D. HIV DNA Integration. Cold Spring Harb. Perspect. Med. 2012, 2, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Cesana, D.; Ranzani, M.; Volpin, M.; Bartholomae, C.; Duros, C.; Artus, A.; Merella, S.; Benedicenti, F.; Sergi, L.S.; Sanvito, F.; et al. Uncovering and Dissecting the Genotoxicity of Self-Inactivating Lentiviral Vectors In Vivo. Mol. Ther. 2014, 22, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The Genotoxic Potential of Retroviral Vectors Is Strongly Modulated by Vector Design and Integration Site Selection in a Mouse Model of HSC Gene Therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-Inactivating Lentivirus Vector for Safe and Efficient In Vivo Gene Delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [CrossRef] [Green Version]
- Vink, C.A.; Counsell, J.R.; Perocheau, D.P.; Karda, R.; Buckley, S.M.K.; Brugman, M.H.; Galla, M.; Schambach, A.; McKay, T.R.; Waddington, S.N.; et al. Eliminating HIV-1 Packaging Sequences from Lentiviral Vector Proviruses Enhances Safety and Expedites Gene Transfer for Gene Therapy. Mol. Ther. 2017, 25, 1790–1804. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 4 September 2020).
- FDA Approval Brings First Gene Therapy to the United States | FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approval-brings-first-gene-therapy-united-states (accessed on 19 August 2020).
- New Gene Therapy to Treat Rare Inherited Blood Condition | European Medicines Agency. Available online: https://www.ema.europa.eu/en/news/new-gene-therapy-treat-rare-inherited-blood-condition (accessed on 19 August 2020).
- Ikawa, Y.; Miccio, A.; Magrin, E.; Kwiatkowski, J.L.; Rivella, S.; Cavazzana, M. Gene Therapy of Hemoglobinopathies: Progress and Future Challenges. Hum. Mol. Genet. 2019, 28, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanber, K.S.; Knight, S.B.; Stephen, S.L.; Bailey, R.; Escors, D.; Minshull, J.; Santilli, G.; Thrasher, A.J.; Collins, M.K.; Takeuchi, Y. Construction of Stable Packaging Cell Lines for Clinical Lentiviral Vector Production. Sci. Rep. 2015, 5, 9021. [Google Scholar] [CrossRef]
- Magrin, E.; Miccio, A.; Cavazzana, M. Lentiviral and Genome-Editing Strategies for the Treatment of β-Hemoglobinopathies. Blood 2019, 134, 1203–1213. [Google Scholar] [CrossRef]
- Urbinati, F.; Campo Fernandez, B.; Masiuk, K.E.; Poletti, V.; Hollis, R.P.; Koziol, C.; Kaufman, M.L.; Brown, D.; Mavilio, F.; Kohn, D.B. Gene Therapy for Sickle Cell Disease: A Lentiviral Vector Comparison Study. Hum. Gene Ther. 2018, 29, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy in Patients with Wiskott-Aldrich Syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef] [Green Version]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen–Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef] [Green Version]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T Cells: The Long and Winding Road to Solid Tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef]
- Crawford, K.H.D.; Eguia, R.; Dingens, A.S.; Loes, A.N.; Malone, K.D.; Wolf, C.R.; Chu, H.Y.; Tortorici, M.A.; Veesler, D.; Murphy, M.; et al. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 2020, 12, 513. [Google Scholar] [CrossRef] [PubMed]
- De Ravin, S.S.; Wu, X.; Moir, S.; Kardava, L.; Anaya-O’Brien, S.; Kwatemaa, N.; Littel, P.; Theobald, N.; Choi, U.; Su, L.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy for X-Linked Severe Combined Immunodeficiency. Sci. Transl. Med. 2016, 8, 335ra57. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, A.; Thalheimer, F.B.; Hartmann, S.; Frank, A.M.; Bender, R.R.; Danisch, S.; Costa, C.; Wels, W.S.; Modlich, U.; Stripecke, R.; et al. In Vivo Generation of Human CD 19-CAR T Cells Results in B-cell Depletion and Signs of Cytokine Release Syndrome. EMBO Mol. Med. 2018, 10, e9158. [Google Scholar] [CrossRef]
- Dumont, J.; Euwart, D.; Mei, B.; Estes, S.; Kshirsagar, R. Human Cell Lines for Biopharmaceutical Manufacturing: History, Status, and Future Perspectives. Crit. Rev. Biotechnol. 2016, 36, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Yan, R.; Li, A.; Zhang, Y.; Li, J.; Du, H.; Chen, B.; Wei, W.; Zhang, Y.; Sumners, C.; et al. Lentiviral Vectors Mediate Long-Term and High Efficiency Transgene Expression in HEK 293T Cells. Int. J. Med. Sci. 2015, 12, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Baekelandt, V.; Eggermont, K.; Michiels, M.; Nuttin, B.; Debyser, Z. Optimized Lentiviral Vector Production and Purification Procedure Prevents Immune Response after Transduction of Mouse Brain. Gene Ther. 2003, 10, 1933–1940. [Google Scholar] [CrossRef] [Green Version]
- Matasci, M.; Hacker, D.L.; Baldi, L.; Wurm, F.M. Recombinant Therapeutic Protein Production in Cultivated Mammalian Cells: Current Status and Future Prospects. Drug Discov. Today Technol. 2008, 5, 37–42. [Google Scholar] [CrossRef]
- Ikeda, Y.; Takeuchi, Y.; Martin, F.; Cosset, F.-L.; Mitrophanous, K.; Collins, M. Continuous High-Titer HIV-1 Vector Production. Nat. Biotechnol. 2003, 21, 569–572. [Google Scholar] [CrossRef]
- Merten, O.W.; Hebben, M.; Bovolenta, C. Production of Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2016, 3, 16017. [Google Scholar] [CrossRef]
- Stornaiuolo, A.; Piovani, B.M.; Bossi, S.; Zucchelli, E.; Corna, S.; Salvatori, F.; Mavilio, F.; Bordignon, C.; Rizzardi, G.P.; Bovolenta, C. RD2-MolPack-Chim3, a Packaging Cell Line for Stable Production of Lentiviral Vectors for Anti-HIV Gene Therapy. Hum. Gene Ther. Methods 2013, 240, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Marin, V.; Stornaiuolo, A.; Piovan, C.; Corna, S.; Bossi, S.; Pema, M.; Giuliani, E.; Scavullo, C.; Zucchelli, E.; Bordignon, C.; et al. RD-MolPack Technology for the Constitutive Production of Self-Inactivating Lentiviral Vectors Pseudotyped with the Nontoxic RD114-TR Envelope. Mol. Ther. Methods Clin. Dev. 2016, 3, 16033. [Google Scholar] [CrossRef]
- Tomás, H.A.; Rodrigues, A.F.; Carrondo, M.J.T.; Coroadinha, A.S. LentiPro26: Novel Stable Cell Lines for Constitutive Lentiviral Vector Production. Sci. Rep. 2018, 8, 5271. [Google Scholar] [CrossRef] [Green Version]
- Stewart, H.J.; Leroux-Carlucci, M.A.; Sion, C.J.M.; Mitrophanous, K.A.; Radcliffe, P.A. Development of Inducible EIAV-Based Lentiviral Vector Packaging and Producer Cell Lines. Gene Ther. 2009, 16, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Throm, R.E.; Ouma, A.A.; Zhou, S.; Chandrasekaran, A.; Lockey, T.; Greene, M.; De Ravin, S.S.; Moayeri, M.; Malech, H.L.; Sorrentino, B.P.; et al. Efficient Construction of Producer Cell Lines for a SIN Lentiviral Vector for SCID-X1 Gene Therapy by Concatemeric Array Transfection. Blood 2009, 113, 5104–5110. [Google Scholar] [CrossRef] [Green Version]
- Broussau, S.; Jabbour, N.; Lachapelle, G.; Durocher, Y.; Tom, R.; Transfiguracion, J.; Gilbert, R.; Massie, B. Inducible Packaging Cells for Large-Scale Production of Lentiviral Vectors in Serum-Free Suspension Culture. Mol. Ther. 2008, 16, 500–507. [Google Scholar] [CrossRef]
- Stewart, H.J.; Fong-Wong, L.; Strickland, I.; Chipchase, D.; Kelleher, M.; Stevenson, L.; Thoree, V.; McCarthy, J.; Ralph, G.S.; Mitrophanous, K.A.; et al. A Stable Producer Cell Line for the Manufacture of a Lentiviral Vector for Gene Therapy of Parkinson’s Disease. Hum. Gene Ther. 2011, 22, 357–369. [Google Scholar] [CrossRef]
- Milani, M.; Annoni, A.; Bartolaccini, S.; Biffi, M.; Russo, F.; Di Tomaso, T.; Raimondi, A.; Lengler, J.; Holmes, M.C.; Scheiflinger, F.; et al. Genome Editing for Scalable Production of Alloantigen-free Lentiviral Vectors for in Vivo Gene Therapy. EMBO Mol. Med. 2017, 9, 1558–1573. [Google Scholar] [CrossRef]
- Manceur, A.P.; Kim, H.; Misic, V.; Andreev, N.; Dorion-Thibaudeau, J.; Lanthier, S.; Bernier, A.; Tremblay, S.; Gélinas, A.-M.; Broussau, S.; et al. Scalable Lentiviral Vector Production Using Stable HEK293SF Producer Cell Lines. Hum. Gene Ther. Methods 2017, 28, 330–339. [Google Scholar] [CrossRef]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Tiscornia, G.; Singer, O.; Verma, I.M. Production and Purification of Lentiviral Vectors. Nat. Protoc. 2006, 1, 241–245. [Google Scholar] [CrossRef]
- Segura, M.M.; Mangion, M.; Gaillet, B.; Garnier, A. New Developments in Lentiviral Vector Design, Production and Purification. Expert Opin. Biol. Ther. 2013, 13, 987–1011. [Google Scholar] [CrossRef]
- Toledo, J.R.; Prieto, Y.; Oramas, N.; Sánchez, O. Polyethylenimine-Based Transfection Method as a Simple and Effective Way to Produce Recombinant Lentiviral Vectors. Appl. Biochem. Biotechnol. 2009, 157, 538–544. [Google Scholar] [CrossRef]
- Valkama, A.J.; Leinonen, H.M.; Lipponen, E.M.; Turkki, V.; Malinen, J.; Heikura, T.; Ylä-Herttuala, S.; Lesch, H.P. Optimization of Lentiviral Vector Production for Scale-up in Fixed-Bed Bioreactor. Gene Ther. 2018, 25, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Shi, B.; Xue, M.; Wang, Y.; Wang, Y.; Li, D.; Zhao, X.; Li, X. An Improved Method for Increasing the Efficiency of Gene Transfection and Transduction. Int. J. Physiol. Pathophysiol. Pharmacol. 2018, 10, 95–104. [Google Scholar]
- Witting, S.R.; Li, L.-H.; Jasti, A.; Allen, C.; Cornetta, K.; Brady, J.; Shivakumar, R.; Peshwa, M.V. Efficient Large Volume Lentiviral Vector Production Using Flow Electroporation. Hum. Gene Ther. 2012, 23, 243–249. [Google Scholar] [CrossRef]
- Ansorge, S.; Lanthier, S.; Transfiguracion, J.; Durocher, Y.; Henry, O.; Kamen, A. Development of a Scalable Process for High-Yield Lentiviral Vector Production by Transient Transfection of HEK293 Suspension Cultures. J. Gene Med. 2009, 11, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.B.; Sumner, R.P.; Rodriguez-Plata, M.T.; Rasaiyaah, J.; Milne, R.S.; Thrasher, A.J.; Qasim, W.; Towers, G.J. Lentiviral Vector Production Titer Is Not Limited in HEK293T by Induced Intracellular Innate Immunity. Mol. Ther. Methods Clin. Dev. 2020, 17, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Kutner, R.H.; Puthli, S.; Marino, M.P.; Reiser, J. Simplified Production and Concentration of HIV-1-Based Lentiviral Vectors Using HYPERFlask Vessels and Anion Exchange Membrane Chromatography. BMC Biotechnol. 2009, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Merten, O.-W.; Charrier, S.; Laroudie, N.; Fauchille, S.; Dugué, C.; Jenny, C.; Audit, M.; Zanta-Boussif, M.-A.; Chautard, H.; Radrizzani, M.; et al. Large-Scale Manufacture and Characterization of a Lentiviral Vector Produced for Clinical Ex Vivo Gene Therapy Application. Hum. Gene Ther. 2011, 22, 343–356. [Google Scholar] [CrossRef]
- Rout-Pitt, N.; McCarron, A.; McIntyre, C.; Parsons, D.; Donnelley, M. Large-Scale Production of Lentiviral Vectors Using Multilayer Cell Factories. J. Biol. Methods 2018, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Olszewska, M.; Qu, J.; Wasielewska, T.; Bartido, S.; Hermetet, G.; Sadelain, M.; Rivière, I. Large-Scale Clinical-Grade Retroviral Vector Production in a Fixed-Bed Bioreactor. J. Immunother. 2015, 38, 127–135. [Google Scholar] [CrossRef]
- Lesch, H.P.; Heikkilä, K.M.; Lipponen, E.M.; Valonen, P.; Müller, A.; Räsänen, E.; Tuunanen, T.; Hassinen, M.M.; Parker, N.; Karhinen, M.; et al. Process Development of Adenoviral Vector Production in Fixed Bed Bioreactor: From Bench to Commercial Scale. Hum. Gene Ther. 2015, 26, 560–571. [Google Scholar] [CrossRef]
- Powers, A.D.; Piras, B.A.; Clark, R.K.; Lockey, T.D.; Meagher, M.M. Development and Optimization of AAV HFIX Particles by Transient Transfection in an ICELLis® Fixed-Bed Bioreactor. Hum. Gene Ther. Methods 2016, 27, 112–121. [Google Scholar] [CrossRef]
- Moolten, F.L. Tumor Chemosensitivity Conferred by Inserted Herpes Thymidine Kinase Genes: Paradigm for a Prospective Cancer Control Strategy. Cancer Res. 1986, 46, 5276–5281. [Google Scholar]
- Huszthy, P.C.; Giroglou, T.; Tsinkalovsky, O.; Euskirchen, P.; Skaftnesmo, K.O.; Bjerkvig, R.; von Laer, D.; Miletic, H. Remission of Invasive, Cancer Stem-Like Glioblastoma Xenografts Using Lentiviral Vector-Mediated Suicide Gene Therapy. PLoS ONE 2009, 4, e6314. [Google Scholar] [CrossRef]
- Leinonen, H.M.; Lipponen, E.M.; Valkama, A.J.; Hynynen, H.; Oruetxebarria, I.; Turkki, V.; Olsson, V.; Kurkipuro, J.; Samaranayake, H.; Määttä, A.; et al. Preclinical Proof-of-Concept, Analytical Development, and Commercial Scale Production of Lentiviral Vector in Adherent Cells. Mol. Ther. Methods Clin. Dev. 2019, 15, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Valkama, A.J.; Oruetxebarria, I.; Lipponen, E.M.; Leinonen, H.M.; Käyhty, P.; Hynynen, H.; Turkki, V.; Malinen, J.; Miinalainen, T.; Heikura, T.; et al. Development of Large-Scale Downstream Processing for Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2020, 17, 717–730. [Google Scholar] [CrossRef]
- Leinonen, H.M.; Lepola, S.; Lipponen, E.M.; Heikura, T.; Koponen, T.; Parker, N.; Ylä-Herttuala, S.; Lesch, H.P. Benchmarking of Scale-X Bioreactor System in Lentiviral and Adenoviral Vector Production. Hum. Gene Ther. 2020, 31, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Segura, M.M.; Garnier, A.; Durocher, Y.; Coelho, H.; Kamen, A. Production of Lentiviral Vectors by Large-Scale Transient Transfection of Suspension Cultures and Affinity Chromatography Purification. Biotechnol. Bioeng. 2007, 98, 789–799. [Google Scholar] [CrossRef]
- Lesch, H.P.; Laitinen, A.; Peixoto, C.; Vicente, T.; Makkonen, K.-E.; Laitinen, L.; Pikkarainen, J.T.; Samaranayake, H.; Alves, P.M.; Carrondo, M.J.T.; et al. Production and Purification of Lentiviral Vectors Generated in 293T Suspension Cells with Baculoviral Vectors. Gene Ther. 2011, 18, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Cervera, L.; Fuenmayor, J.; González-Domínguez, I.; Gutiérrez-Granados, S.; Segura, M.M.; Gòdia, F. Selection and Optimization of Transfection Enhancer Additives for Increased Virus-like Particle Production in HEK293 Suspension Cell Cultures. Appl. Microbiol. Biotechnol. 2015, 99, 9935–9949. [Google Scholar] [CrossRef]
- Bauler, M.; Roberts, J.K.; Wu, C.; Fan, B.; Ferrara, F.; Yip, B.H.; Diao, S.; Kim, Y.; Moore, J.; Zhou, S.; et al. Production of Lentiviral Vectors Using Suspension Cells Grown in Serum-Free Media. Mol. Ther. Methods Clin. Dev. 2020, 17, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, M.; Merten, O.-W. Large-Scale Production Means for the Manufacturing of Lentiviral Vectors. Curr. Gene Ther. 2010, 10, 474–486. [Google Scholar] [CrossRef]
- Reeves, L.; Cornetta, K. Clinical Retroviral Vector Production: Step Filtration Using Clinically Approved Filters Improves Titers. Gene Ther. 2000, 7, 1993–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura, M.D.L.M.; Kamen, A.; Trudel, P.; Garnier, A. A Novel Purification Strategy for Retrovirus Gene Therapy Vectors Using Heparin Affinity Chromatography. Biotechnol. Bioeng. 2005, 90, 391–404. [Google Scholar] [CrossRef]
- Geraerts, M.; Micheils, M.; Baekelandt, V.; Debyser, Z.; Gijsbers, R. Upscaling of Lentiviral Vector Production by Tangential Flow Filtration. J. Gene Med. 2005, 7, 1299–1310. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, T.; Carrondo, M.J.T.; Alves, P.M.; Cruz, P.E. Purification of Retroviral Vectors for Clinical Application: Biological Implications and Technological Challenges. J. Biotechnol. 2007, 127, 520–541. [Google Scholar] [CrossRef]
- Scherr, M.; Battmer, K.; Eder, M.; Schüle, S.; Hohenberg, H.; Ganser, A.; Grez, M.; Blömer, U. Efficient Gene Transfer into the CNS by Lentiviral Vectors Purified by Anion Exchange Chromatography. Gene Ther. 2002, 9, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; McCarty, D.M.; Madden, V.J.; Walsh, C.E. Lentivirus Vector Purification Using Anion Exchange HPLC Leads to Improved Gene Transfer. Biotechniques 2003, 34, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mccarron, A.; Donnelley, M.; Mcintyre, C. Challenges of Up-Scaling Lentivirus Production and Processing. J. Biotechnol. 2016, 240, 23–30. [Google Scholar] [CrossRef]
- Reiter, K.; Aguilar, P.P.; Wetter, V.; Steppert, P.; Tover, A.; Jungbauer, A. Separation of Virus-like Particles and Extracellular Vesicles by Flow-through and Heparin Affinity Chromatography. J. Chromatogr. A 2019, 1588, 77–84. [Google Scholar] [CrossRef]
- De las Mercedes Segura, M.; Kamen, A.; Lavoie, M.C.; Garnier, A. Exploiting Heparin-Binding Properties of MoMLV-Based Retroviral Vectors for Affinity Chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 846, 124–131. [Google Scholar] [CrossRef]
- Higashikawa, F.; Chang, L.-J. Kinetic Analyses of Stability of Simple and Complex Retroviral Vectors. Virology 2001, 280, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Bandeira, V.; Peixoto, C.; Rodrigues, A.F.; Cruz, P.E.; Alves, P.M.; Coroadinha, A.S.; Carrondo, M.J.T. Downstream Processing of Lentiviral Vectors: Releasing Bottlenecks. Hum. Gene Ther. Methods 2012, 23, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Kumru, O.S.; Wang, Y.; Gombotz, C.W.R.; Kelley-Clarke, B.; Cieplak, W.; Kim, T.; Joshi, S.B.; Volkin, D.B. Physical Characterization and Stabilization of a Lentiviral Vector Against Adsorption and Freeze-Thaw. J. Pharm. Sci. 2018, 107, 2764–2774. [Google Scholar] [CrossRef] [PubMed]
- Poorebrahim, M.; Sadeghi, S.; Fakhr, E.; Abazari, M.F.; Poortahmasebi, V.; Kheirollahi, A.; Askari, H.; Rajabzadeh, A.; Rastegarpanah, M.; Linē, A.; et al. Production of CAR T-Cells by GMP-Grade Lentiviral Vectors: Latest Advances and Future Prospects. Crit. Rev. Clin. Lab. Sci. 2019, 56, 393–419. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Intervention | Reference | Phase |
|---|---|---|---|
| X-linked severe combined immunodeficiency (SCID-X1) | Transfer IL2RG complementary DNA (ex vivo) | NCT04286815 | I |
| Mucopolysaccharidosis type IIIA | CD34+ cells expressing the human SGSH gene (ex vivo) | NCT04201405 | I, II |
| Metastatic breast cancer | CAR T cells specific for a cleaved form of MUC1 (ex vivo) | NCT04020575 | I |
| Fanconi anemia subtype A | CD34+ cells expressing the FANCA gene (ex vivo) | NCT04069533 | II |
| Sickle cell disease/anemia | CD34+ cells expressing the βAS3 globin gene (ex vivo) | NCT03964792 | I, II |
| CD34+ cells expressing human γ-globinG16D and short-hairpin RNA734 (ex vivo) | NCT04091737 | I | |
| CD34+ cells expressing LentiGlobin BB305 Drug Product (ex vivo) | NCT04293185 | III | |
| Non-Hodgkin’s lymphoma | Autologous CD19-directed chimeric antigen receptor (CAR) T cells (ex vivo) | NCT03938987 | I, II |
| Infusion of UCD19 CAR T cells (ex vivo) | NCT04240808 | I | |
| CD22 CAR T cells (ex vivo) | NCT04088890 | I | |
| Hemophilia A | CD34+ cells expressing the B domain deleted from of human coagulation factor VIII (ex vivo) | NCT03818763 | I |
| CD34+ cells expressing human factor VIII gene (ex vivo) | NCT04418414 | I | |
| CD34+ cells expressing a functional FVIII gene to overcome human clotting FVIII gene defect (ex vivo) | NCT03217032 | I | |
| COVID-19 | Use LV to modify artificial antigen-presenting cells to express SARS-CoV-2 proteins and immunomodulatory genes and activate T cells (in vivo) | NCT04299724 | I |
| Use LV to express SARS-CoV-2 proteins and immunomodulatory genes to modify dendritic cells and to activate T cells (in vivo) | NCT04276896 | I, II | |
| Metachromatic leukodystrophy | CD34+ cells expressing the human arylsulfatase A gene (ex vivo) | NCT04283227 | III |
| Pyruvate kinase deficiency | CD34+ cells expressing a correct copy of the deficient PKD gene (ex vivo) | NCT04105166 | I |
| Hemophilia B | LV to deliver a functional FIX gene to overcome human clotting FIX gene defect (ex vivo) | NCT03961243 | I |
| CD19 negative B-cell malignancies | Gene-modified T cells targeting CD19-negative B malignancies (ex vivo) | NCT04430530 | I, II |
| Hematological malignancy | Genetically engineered NK cells (ex vivo) | NCT04093622 | - |
| Hepatitis C | Immunotherapy (HCVax™) (in vivo) | NCT04318379 | I |
| Multiple myeloma | C-CAR088 Drug (ex vivo) | NCT04295018 | I |
| CD4+ chimeric antigen receptor immunotherapy (ex vivo) | NCT04162340 | II, III | |
| CD4+ chimeric antigen receptor immunotherapy (ex vivo) | NCT04162353 | I | |
| B-Cell lymphoma | CD22 CAR T cells (ex vivo) | NCT04088864 | I |
| Device | Surface | Yield | Total Production | |
|---|---|---|---|---|
| T-150 Flask |  | 150 cm2 | 6.9 × 107 TU/mL | 1.2 × 1010 TU |
| HYPERFlask |  | 1720 cm2 | 2.3 × 108 TU/mL | 1.26 × 1011 TU |
| Multilayer Cell Factory |  | 6320 cm2 | 2 × 109 TU/mL | 6 × 1011 TU |
| Device | iCELLis Nano | Scale-X Hydro | iCELLis 500+ | Scale-X Nitro |
|---|---|---|---|---|
 |  |  |  | |
| Surface | 2.67 m2 | 2.4 m2 | 333 m2 | 200–600 m2 |
| Total viral particles (vp) | >1013 | >1013 | 9.27 × 1014 | Not optimized yet |
| Vp/cm2 | >109 | >109 | 9.27 × 108 | Not optimized yet |
| Total transducing units (TU) | 1.3 × 1010 | 2.4 × 1010 | 1.12 × 1012 | Not optimized yet |
| TU/cm2 | 4.7 × 105 | 9.8 × 105 | 2.3 × 105 | Not optimized yet |
| Yield | Not concentrated | Not concentrated | 1.97 × 109 TU/mL | Not optimized yet |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Molina, E.; Chocarro-Wrona, C.; Martínez-Moreno, D.; Marchal, J.A.; Boulaiz, H. Large-Scale Production of Lentiviral Vectors: Current Perspectives and Challenges. Pharmaceutics 2020, 12, 1051. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12111051
Martínez-Molina E, Chocarro-Wrona C, Martínez-Moreno D, Marchal JA, Boulaiz H. Large-Scale Production of Lentiviral Vectors: Current Perspectives and Challenges. Pharmaceutics. 2020; 12(11):1051. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12111051
Chicago/Turabian StyleMartínez-Molina, Eduardo, Carlos Chocarro-Wrona, Daniel Martínez-Moreno, Juan A. Marchal, and Houria Boulaiz. 2020. "Large-Scale Production of Lentiviral Vectors: Current Perspectives and Challenges" Pharmaceutics 12, no. 11: 1051. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12111051