1. Introduction
Protein therapy has emerged as a promising therapeutic alternative due to its high specificity and reduced off-target effects [
1]. Among protein-based therapies, the use of antibodies have been largely explored and numerous treatments are currently available for a broad spectrum of clinical indications, from transplant rejection to cancer [
2]. One of the most interesting features of antibody-based therapies is their potential to reach a wide range of targets, including those that are usually unattainable by other strategies (i.e., undruggable targets). However, because most antibodies are unable to easily cross the extracellular membrane, their applications are often limited to extracellular targets [
3,
4]. In the last years, several research groups have explored the use of antibodies against intracellular targets. Straightforward techniques for antibody delivery, such as microinjection, electroporation and antibody modification with cell penetration peptides (CPPs), have been tested. Unfortunately, even though experimentally effective in vitro, they have not been efficiently translated to in vivo models or into clinical applications [
5,
6]. In addition, although different nanoparticles, mainly metallic and lipidic, have been developed for intracellular delivery of antibodies, only few of them showed efficient intracellular delivery [
7,
8]. The current drawbacks are related with limited cargo for hydrophilic substances, un-adequate endosomal escape, in vivo instability and a short half-life circulation time. Thus, intracellular delivery of antibodies is still a considerable challenge [
9,
10].
On the other hand, although extensively investigated and besides constant development of new therapies, cancer often remains as an incurable disease. Insights into cancer physiology have shown the complexity of cancer regulatory networks in which a large variety of different cell types are involved and largely interconnected to determine disease progression. In this regard, particular attention should be addressed to metastatic spreading, resistance to treatment and tumor recurrence. These features are related with the presence of a cancer cell subpopulation known as cancer stem cells (CSC). CSC display stemness properties, such as self-renewal, quiescence and the capability to remain in an undifferentiated state [
11,
12]. Besides, CSC have been shown to exhibit high expression levels of drug efflux transporters and unregulated DNA repair machinery, conferring them drug resistance potential. In this context, CSC have become a highly appealing target in order to improve current anti-cancer therapies [
13,
14]. Although the number of new drugs with therapeutic potential against CSC is rising and several products are already in clinical trials, there is still strong need to broaden the catalogue of therapeutic options.
In this sense, the Structural Maintenance of Chromosomes 2 (SMC2) protein could be a good therapeutic target candidate. SMC2 forms part of the condensin complex that has a central role in many aspects of chromosome biology, including the segregation of sister chromatids and compaction of chromosomes during cell division, as well as the regulation of gene expression during the interphase [
15,
16,
17,
18]. Importantly, SMC2 protein has been mostly located at the cell cytoplasm during cell interphase with only a minor amount remaining associated to chromatin in the nucleus. The condensin complex is formed by five subunits and requires the initial arrangement of a SMC2/SMC4 heterodimer. The condensin complex is completed by the union of a subcomplex, formed by the three non-SMC subunits (Cnd1, Cnd2 and Cnd3), to the heterodimer of SMC2/SMC4 [
15,
16,
17]. Of note, SMC2 has been found to be over-expressed in a significant number of patients with colorectal cancer, gastric cancer, lymphoma and some types of neuroblastoma [
19,
20], and has been suggested as a risk biomarker in pancreatic cancers [
21]. Moreover, when SMC2 expression was knocked down it drastically reduced tumor growth in colorectal cancer mice models [
18]. This data and the central role of the condensin complex in cell division suggested that an effective inhibition of the activity of this complex would prevent cancer cells from dividing. Under this premise, directing SMC2-targeted inhibitory molecules into the cell cytoplasm should ensure SMC2 protein blockade and serve as a novel therapeutic strategy.
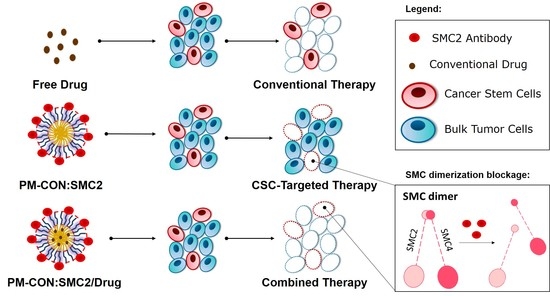
Hereby we propose a polymeric micelle delivery system (PM) based on amphiphilic polymers, namely the Pluronic
® F127 as a nanoplatform to cluster the antibodies directed against the SMC2 protein. Because of its small size, biocompatibility, stealth properties and amphiphilic nature, this is an ideal system to overcome major drawbacks related to intracellular delivery of antibodies. Furthermore, poloxamer polymers like Pluronic
® F127 have shown to interfere with the function of P-glycoprotein, being therefore helpful to further overcome drug resistance and reduce cancer recurrence [
22,
23,
24]. Moreover, taking advantage of the plasticity provided by amphiphilic PM we have also explored potential combinatorial therapeutic strategies with 5-FU and PTX, drugs commonly used in colon and breast cancer, respectively.
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
The HCT116 (ATCC® CCL-247™) colon cancer cell line, MDA-MB-231 (ATCC® HTB-26™) breast cancer cell line and PANC-1 (ATCC® CRL-1469™) pancreas cancer cell line were obtained from American Type Culture Collection (ATTC®, LGC Standards, Barcelona, Spain). Cells were cultured in RPMI medium (ThermoFisher Scientific, Madrid, Spain supplemented with 10% Fetal Bovine Serum (FBS) (ThermoFisher Scientific, Madrid, Spain), a 1% penicillin–streptomycin mixture (ThermoFisher Scientific, Madrid, Spain), 1% L-glutamine (Lonza, Basel, Switzerland) and 1% Non-Essential Amino Acids (NEAA) (ThermoFisher Scientific, Madrid, Spain). All cell lines were maintained at 37 °C under a 5% CO2-saturated atmosphere. The medium was changed every two days, and upon sub-confluence, cells were harvested from plates with 0.25% trypsin-EDTA and subcultured (ThermoFisher Scientific, Madrid, Spain).
Cells were also cultured in low attachment conditions as tumorspheres in serum-free media and seeded in ultra-low attachment surface plates (Corning Life Sciences, Chorges, France). The medium for breast and pancreas sphere growth was supplemented with glucose, 60 mg/mL (Merck Life Science S.L.U., Madrid, Spain), as well as 10 µL/mL L-Glutamax (Merck Life Science S.L.U., Madrid, Spain), 10 µL/mL antibiotic–antimitotic mixture (Merck Life Science S.L.U., Madrid, Spain), 4 µg/mL heparin (Merck Life Science S.L.U., Madrid, Spain), 2 mg/mL BSA (Merck Life Science S.L.U., Madrid, Spain), 0.02 µg/mL of human recombinant EGF (Merck Life Science S.L.U., Madrid, Spain), 0.01 µg/mL of human recombinant bFGF (ThermoFisher Scientific, Madrid, Spain) and 10% of Hormone Mix: 10 µg/mL putrescin, 20 µM progesterone (both from Merck Life Science S.L.U., Madrid, Spain), 0.1 mg/mL apo-transferrin, 25 µg/mL insulin and 30 µM selen (ThermoFisher Scientific, Madrid, Spain). Tumorspheres from the HCT116 cell line were also cultured in serum-free media supplemented with a 1% antibiotic–antimitotic mixture, 10 ng/mL of human recombinant EGF, 20 ng/mL of human recombinant bFGF and 10 ng/mL of LIF Recombinant Human Protein (TermoFisher Scientific, Madrid, Spain).
2.2. Phase Contrast Microscopy
The morphology and structure of adherent cells and spheres were routinely assessed by phase contrast microscopy imaging after treatment with the distinct compounds. Images were acquired at 10× amplification using a Nikon Eclipse TS100 (Nikon Instruments Europe BV, Amsterdam, Netherlands) inverted microscope and further edited with ImageJ 1.49v software.
2.3. Cell Transfection with siRNA
2.3.1. Gene Expression Determination
10
5 cells were seeded in 6-well plates and transfected with SMC2-siRNA (SI02654260, Qiagen, Hilden, Germany) using Lipofectamine
® 2000 (ThermoFisher Scientific, Madrid, Spain) according to the manufacturer’s instructions. A nonspecific sequence was used as the control (siControl). The culture medium was changed 6 h after transfection and cells harvested after 72 h of incubation. Samples were further processed for RNA extraction as detailed below in
Section 2.4.
2.3.2. Cell Growth Assay
After cell transfection, cells were washed in PBS and harvested by trypsin digestion following standard procedures. Cells were counted in a CountessTM II (ThermoFisher Scientific, Madrid, Spain).
2.3.3. Sphere Formation Assay
SMC2 knock-down was performed following a 2-times siRNA shot strategy in order to guarantee an efficient gene expression inhibition along the assay. Briefly, a first treatment with SMC2-siRNA using Lipofectamine
® 2000 (ThermoFisher Scientific, Madrid, Spain) was performed as described above. After 72 h an identical SMC-siRNA shot was performed. After 6 h upon the second siRNA transfection, cells were harvested and washed 3 times in serum free media. Then, 1000 cells were seeded in ultra-low attachment 96-well plates, adding sphere growth media for assessing sphere formation (as described in the cell viability assay
Section 2.10.1).
2.4. RNA Extraction and Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from cells using RNeasy Micro Kit (Qiagen, Hilden, Germany), and the obtained RNA was reverse transcribed with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Madrid, Spain), according to the manufacturer’s instructions. The cDNA reverse transcription product was amplified with specific primers for SMC2 (hSMC2 F: 5′ AAT GAG CTG CGG GCT CTA GA 3′; hSMC2 R: 5′ TTG TTG CTT GTG ATA TGA GCT TTG 3′); GADPH (hGADPH F: 5′ ACC CAC TCC TCC ACC TTT GAC; hGADPH R: 5′ CAT ACC AGG AAA TGA GCT TGA CAA 3′) and Actin (hActin F: 5′ CAT CCA CGA AAC TAC CTT CAA CTC C 3′; hActin R: 5′GAG CCG CCG ATC CAC AC 3′) by qPCR using SYBR Green (ThermoFisher Scientific, Madrid, Spain) to fluorescently label double stranded DNA. The reaction was performed on a 7500 Real time PCR system (Applied Biosystems, Madrid, Spain). At least three biological replicates, each comprising two technical replicates, were performed. Relative normalized quantities (NRQ) of mRNA expression were calculated using the comparative Ct method (2e-ΔΔCt) with two reference genes (hGAPDH and hActin) used as endogenous controls through Qbase™ software v3.2.
2.5. Protein Extraction and Western Blotting
Cell pellets were lysed with the Cell Lytic M reagent (Merck Life Science S.L.U., Madrid, Spain) containing a protease inhibitor cocktail (Roche cOmplete™, Merck Life Science S.L.U., Madrid, Spain). Proteins in the crude lysates were quantified using the PierceTM BCA protein quantification kit (ThermoFisher Scientific, Madrid, Spain) following the manufacturer’s instructions. A total of 20 μg of whole-cell lysates were separated by SDS-PAGE and transferred onto PVDF membranes. Blots were probed using primary antibodies anti-SMC2 (Merck Life Science S.L.U., Madrid, Spain) and β-Tubulin (Invitrogen, ThermoFisher Scientific, Madrid, Spain). Proteins were detected using HRP-conjugated secondary antibodies (Dako, Palex Medical SA, Barcelona, Spain) and developed after appropriate incubation in an Immobilion® Western reagent (Merck Life Science S.L.U., Madrid, Spain) using an Odissey FC imaging system (LI-COR Biotechnology GmbH, Bad Homburg, Germany) for chemiluminescence detection.
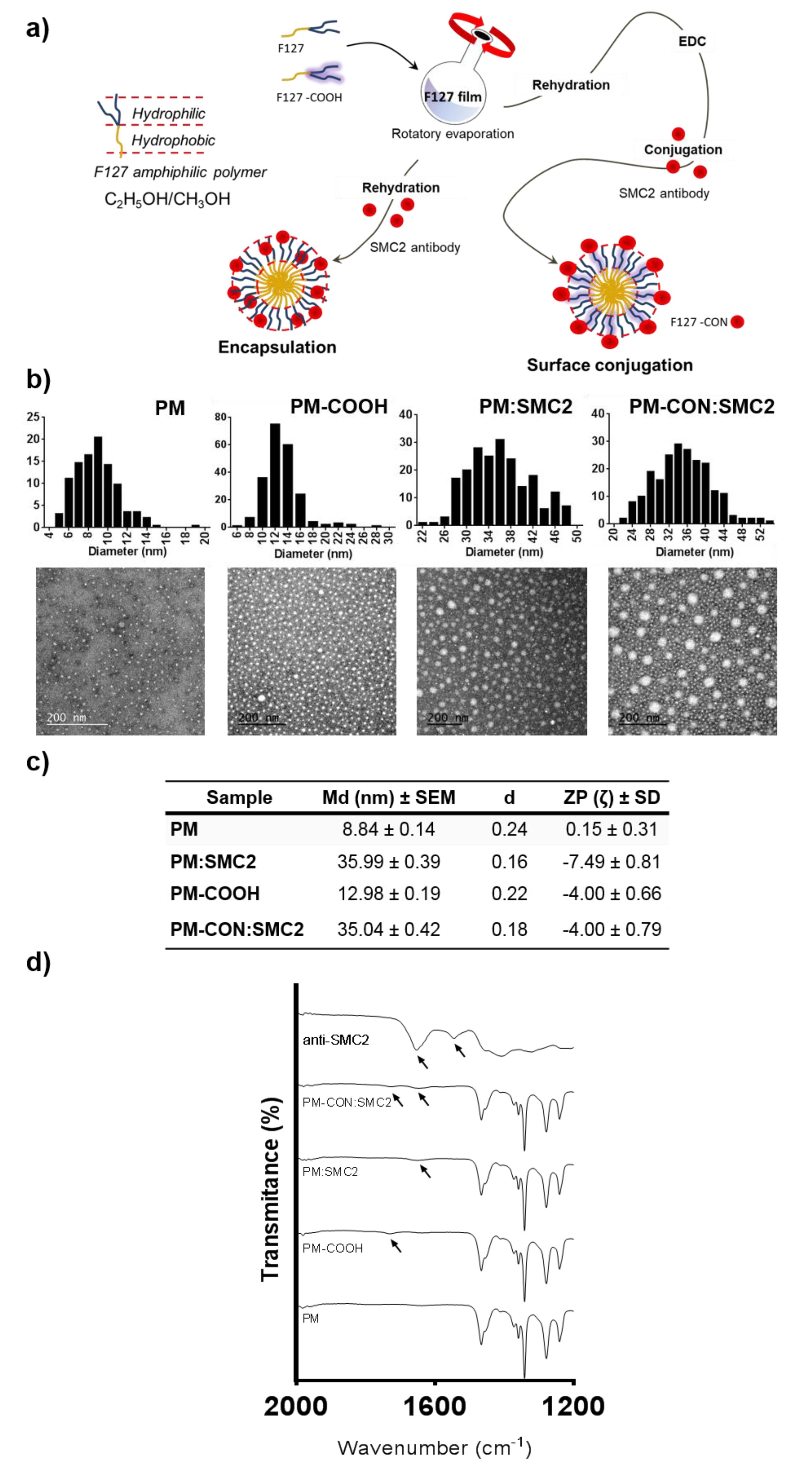
2.6. Production of Polymeric Micelles
Pluronic
® F127 was kindly provided by BASF (Ludwigshafen, Germany). PM were prepared using the thin-film hydration technique [
22]. Pluronic
® F127 carboxylation was performed as previously reported [
25]. Briefly, F127 and F127:COOH polymers were individually weighed in an 8:2 (
w/
w) ratio and dissolved in a mixture of methanol:ethanol (1:1) (Merck Life Science S.L.U., Madrid, Spain). For the formation of loaded micelles, the drug, 5-FU or PTX (both from Merck Life Science S.L.U., Madrid, Spain) were added to the organic solution at the desired concentration. Then, the solvent was removed under vacuum in a rotary evaporator (bath temperature 60 °C, 200 rpm), and the resulting film left to dry overnight at room temperature to eliminate any remaining solvent. The film was then hydrated with PBS and vortexed for 5 min at full speed. For the PM encapsulating SMC2 antibodies (PM:SMC2), the antibody (Anti-SMC2/hCAP-E Antibody, Merck Life Science S.L.U., Madrid, Spain) was added at the aqueous phase during the rehydration step. For PM functionalized with SMC2 antibodies (PM-CON:SMC2), an adequate amount of EDC (polymer:EDC ratio 1:1.5) (Merck Life Science S.L.U., Madrid, Spain) was incubated with the formulation during 30 min at RT. Afterwards, the SMC2 antibody solution was added and incubated under stirring during 2 h at RT. Samples were freeze-dried for long-term storage using a VirTisBenchTop Freeze-Dryer (SP Scientific, Ipswich, UK) when required.
2.7. Physicochemical Characterization of the Polymeric Micelles
2.7.1. Zeta Potential Measurements
Zeta potential was assessed by laser Doppler microelectrophoresis using a NanoZS (Malvern Instruments, Malvern, UK) with an angle of 173°. Samples were diluted 1:5 in Milli-Q® water (18.2 MΩ·cm at 25 °C) in order to obtain an adequate nanoparticle concentration. Data obtained from each formulation were represented as mean values and measured at least in triplicate.
2.7.2. Transmission Electron Microscopy (TEM)
Particle shape and morphology were observed by TEM, using a JEM-1400 Electron Microscope (JEOL Ltd., Croissy-sur-Seine, France) with an applied voltage of 120 kV. For that, a drop of sample (previously diluted 1:20 in Milli-Q
® water) was placed on a carbon 400-mesh copper grid, the liquid excess removed, and the sample contrasted with uranyl acetate before visualization. Image J 1.49v software was used to process information and determine diameter measures (
n > 200) from TEM images, while histogram plots from nanoparticles size distribution were generated by GraphPad Prism 6. The dispersion index (d) was determined by Equation (1).
2.7.3. Loading/Association Efficiency Determination
The efficacy of SMC2 loading in the case of PM:SMC2 and association efficiency in the case of PM-CON:SMC2 was assessed by BCA protein assay. Briefly, the amount of free SMC2 antibody in the aqueous phase of the PM was separated by centrifugation with filtration (10,000
g, 10 min at RT) using 300k membrane centrifugal devices (Nanosep
® Centrifugal Devices, Pall España, Madrid, Spain). Then, the protein level was measured from the flow using a Pierce
TM BCA protein assay kit (ThermoFisher Scientific, Madrid, Spain). The Loading/Association efficiency percentage was determined according to Equation (2):
2.7.4. Fourier-Transform Infrared Spectroscopy (FTIR)
The conjugation of the SMC2 antibody at the PM surface was also confirmed by FTIR analysis. FTIR was carried out using a spectrometer Perkin-Elmer Spectrum One (energy range: 450–4000 cm−1) equipped with a Universal Attenuated Total Reflectance Accessory (U-ATR, Perkin-Elmer, Madrid, Spain). Prior to analysis, samples were freeze-dried using a VirTisBenchTop Freeze-Dryer (SP Scientific, Ipswich, UK).
2.8. Micelles Internalization: Flow Cytometry and Confocal Microscopy
Cell internalization of the different formulations was assessed in HCT116 and MDA-MB-231 using 5-DTAF (Merck Life Science S.L.U., Madrid, Spain) fluorescently labeled PM [
25,
26].
2.8.1. Flow Cytometry
Briefly, 20,000 cells were seeded in complete RPMI medium in 96-well plates and left to attach for 24 h. PM were added to cells (10 mg/mL) and incubated for 15, 30, 60, 180, 360 and 1440 min, respectively. Then, cells were washed with 1× PBS, trypzinized and neutralized with PBS supplemented with 10% FBS and 1 μg/mL DAPI (Merck Life Science S.L.U., Madrid, Spain) used for vital staining. The plate was read in a cytometer Fortessa (BD Biosciences, Madrid, Spain) and data was analyzed with FCS Express 4 Flow Research Edition software (De Novo Software, Los Angeles, USA, Version 4). Three biological replicated were performed for each condition and only DAPI negative cells were admitted for the analysis, while cell debris or possible aggregates were removed by forward and side scatter gating. For each sample, at least 10,000 individual cells were collected, and the percentage of fluorescent cells evaluated.
2.8.2. Confocal Microscopy
A total of 50,000 cells were seeded in complete RPMI medium in 8-well chambered coverglass (ThermoFisher Scientific, Madrid, Spain) and incubated overnight at 37 °C and 5% CO2 to allow cell adhesion. Cells were incubated with 5-DTAF-fluorescently labelled PM (10 mg/mL) for 6 h. Lysosomes were stained with 1 µM Lysotracker® Red DND-99 (ThermoFisher Scientific Madrid, Spain) for 30 min at 37 °C while the cell membrane was stained with 5 µg/mL CellMask™ (Invitrogen, ThermoFisher Scientific, Madrid, Spain) for 15 min at 37 °C. Subsequently, cells were fixed in 4% PFA (Merck Life Science S.L.U., Madrid, Spain) at 4 °C for 20 min followed by nuclei staining with DAPI (1 μg/mL) for 5 min at RT in the dark. Cells were viewed under a Spectral Confocal Microscope MFV1000 Olympus (Olympus Iberia, S.A.U., L’Hospitalet de Llobregat, Spain). The 561 nm excitation wavelength of the green laser (10 mW) was used for selective detection of the red fluorochromes (Lysotracker® Red and CellMask™). The 488 nm excitation wavelength of Argon multiline laser (40 mW) was used for selective detection of the green fluorochrome (5-DTAF). The nuclear staining DAPI was excited at 405 nm with a violet laser (6 mW). Minimal single optical sections were collected for each fluorochrome sequentially. Images were merged and analyzed with Image J 1.49v software.
2.9. Cell Cycle Assay by Flow Cytometry
A flow cytometric analysis of cell cycle with propidium iodide DNA staining was carried out. Briefly, 40,000 cells of HCT116 cells and 30,000 cells of MDA-MB-231 cells were seeded in 24-well plates and left to adhere during 24 h at 37 °C and a 5% CO2-saturated atmosphere. Then, cells were treated and incubated with empty PM (control PM) (5 mg/mL) and PM-CON:SMC2 (5 mg/mL polymer and 32.9 µg/mL of SMC2 antibody) for 48 h. Untreated cells were used as the negative control. Afterwards, cells were harvested, centrifuged (8000 rpm, 5 min at 4 °C) and fixed in cold 70% ethanol for 30 min at 4 °C. After centrifugation, cells were washed two times with PBS and the cell pellets resuspended in 250 µL of the staining solution containing 100 µg/mL of ribonuclease PureLink™ RNase A (ThermoFisher Scientific, Madrid, Spain) and 50 µg/mL of propidium iodide (Merck Life Science S.L.U., Madrid, Spain) in order to ensure that only DNA, not RNA, was stained.
Cell cycle evaluation was performed using an FACS Calibur (BD Biosciences, Madrid, Spain) flow cytometer and the resulting histograms analysed with FCS Express 4 Flow Research Edition software (De Novo Software, Version 4). Three biological replicates were performed and at least 5000 individual cells were collected for the analysis once debris were removed by forward and side scatter gating. The multicycle option in autofit mode was performed in order to automatically quantify the percentage of cells in each cell cycle phase. By this method a Gaussian curve was fitted to each phase allowing the precise determination of the area under the curve. In addition, the G2/G1 ratio was also represented, scoring around 2 in all the cases, in compliance with the accepted quality standards for cell-cycle assays.
2.10. Tumor Cell Viability Assays
2.10.1. Viability Assay in Adherent Cell Cultures
3000 cells of the HCT116 cell line and 5000 cells of the MDA-MB-231 cell line were seeded in 96-well plates and incubated in overnight to allow adhesion. Then, cells were treated and incubated during 72 h with crescent concentrations of the different formulations (PM, PM:SMC2 and PM-CON:SMC2) at 5 mg/mL of polymer and 32.9 µg/mL of SMC2 antibody. To determine the cytotoxicity of free 5-FU and free PTX (Merck Life Science S.L.U., Madrid, Spain), cells were incubated during 48 h with a range concentration of 5-FU and PTX of 384.40 µM to 0.023 µM and 1 µM to 0.0078 µM, respectively. Complete medium was used as negative control and 10% DMSO as positive control of toxicity. Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Merck Life Science S.L.U., Madrid, Spain). The absorbance of each well was read on an absorbance microplate reader ELx800 (BioTek, Colmar, France), at 590 and 630 nm for 5-FU and PTX, respectively. The half-maximal inhibitory concentration (IC50) was determined by nonlinear regression of the concentration-effect curve fit using Prism 6.02 software (GraphPad Software, Inc.).
2.10.2. Tumorsphere Viability Assays
Studies were performed using ultra-low attachment surface plates (Corning Life Sciences, Chorges, France). Cells were cultured in serum free media supplemented differently for HCT116 colon cell line and MDA-MB-231 breast cancer cell line, as mentioned above. HCT116, MDA-MB-231 and Panc-1 cells were transfected with siRNA anti-SMC2, as previously described. A total of 1000 cells were seeded and cultured for 6 days in ultra-low attachment plates.
For PM and drug efficacy assays, 1000 cells from the HCT116 cell line and 2000 cells from the MDA-MB-231 cell line were seeded in 96-well ultra-low attachment plates in serum-free media, supplemented as described above. After overnight incubation, cells were treated with different PM formulations (PM, PM-CON:SMC2, PM/5-FU, PM-PTX, PM-CON:SMC2/5-FU, PM-CON:SMC2/PTX) (1 mg/mL of polymer), the free drugs (5-FU, 3.84 µM and PTX, 0.1 µM) and free SMC2 antibody (6.58 µg/mL) for 7 days. The IC50 of the free tested drugs were also determined in low attachment conditions. Cells were treated with serial 1:2 dilutions of 5-FU (from 768.80 µM to 0.047 µM) and PTX (from 10 µM to 0.078 µM) for 7 days. Free serum medium was used as negative control and 10% DMSO as positive control of toxicity. In all cases, sphere formation was monitored with a Nikon Eclipse TS100 inverted microscope (Nikon Instruments Europe BV, Amsterdam, Netherlands) and quantified by Presto Blue® Reagent assay (ThermoFisher Scientifics, Madrid, Spain). The absorbance of each well was read on a MultiskanTM FC Microplate Photomer (ThermoFisher Scientific, Madrid, Spain), at 570 and 620 nm, and the data were processed by GraphPad Prism 6 software.
2.11. Statistical Analysis
At least three batches of each PM were produced and characterized. The results were expressed as the mean ± standard deviation. For biological studies, at least 3 replicates, each involving at least two technical replicates, were involved in the final results expressed as the mean ± standard deviation. Statistical analysis was performed in GraphPad Prism 6 software using a non-parametric Dunn test for multiple comparisons and Mann–Whitney U test for simple comparisons. Differences were regarded as statistically significant when the p-value was smaller than 0.05.
4. Discussion
SMC2 forms part of the condensin I and II complexes, thus being a crucial player in several cellular biological processes related to mitotic and meiotic chromosome condensation and rigidity, interphase ribosomal DNA compactation, as well as removal of cohesion during mitosis and meiosis [
15,
16,
17]. Moreover, SMC2 overexpression and mutations in some of the condensing subunits had been reported in cancer genomes, suggesting that functional alterations affecting condensin complexes are common in tumorigenesis [
19,
20]. Given the essential role of SMC2 in the survival of embryonic stem cells, it is reasonable to speculate that SMC2 could play an important function also in the homeostasis of tumoral CSC. This is a relevant issue since finding an efficient target against this subpopulation is of major importance in order to avoid cancer resistance and tumor recurrence [
11,
13]. Ideally, a therapy should be able to eradicate not only the primary tumor but also CSC that often survive after most conventional treatments. In order to investigate CSC response to new therapeutic strategies, several methods have been described to generate CSC models in vitro [
31]. A simple and rapid approach is based on culturing tumor cell lines in non-adherent conditions, exploiting CSC ability to form pseudo-spherical colonies. This method was firstly reported in 1992 by Reynolds and colleagues [
32] and lately improved by Tatianna Herheliuk et al., in 2019 [
33], among others.
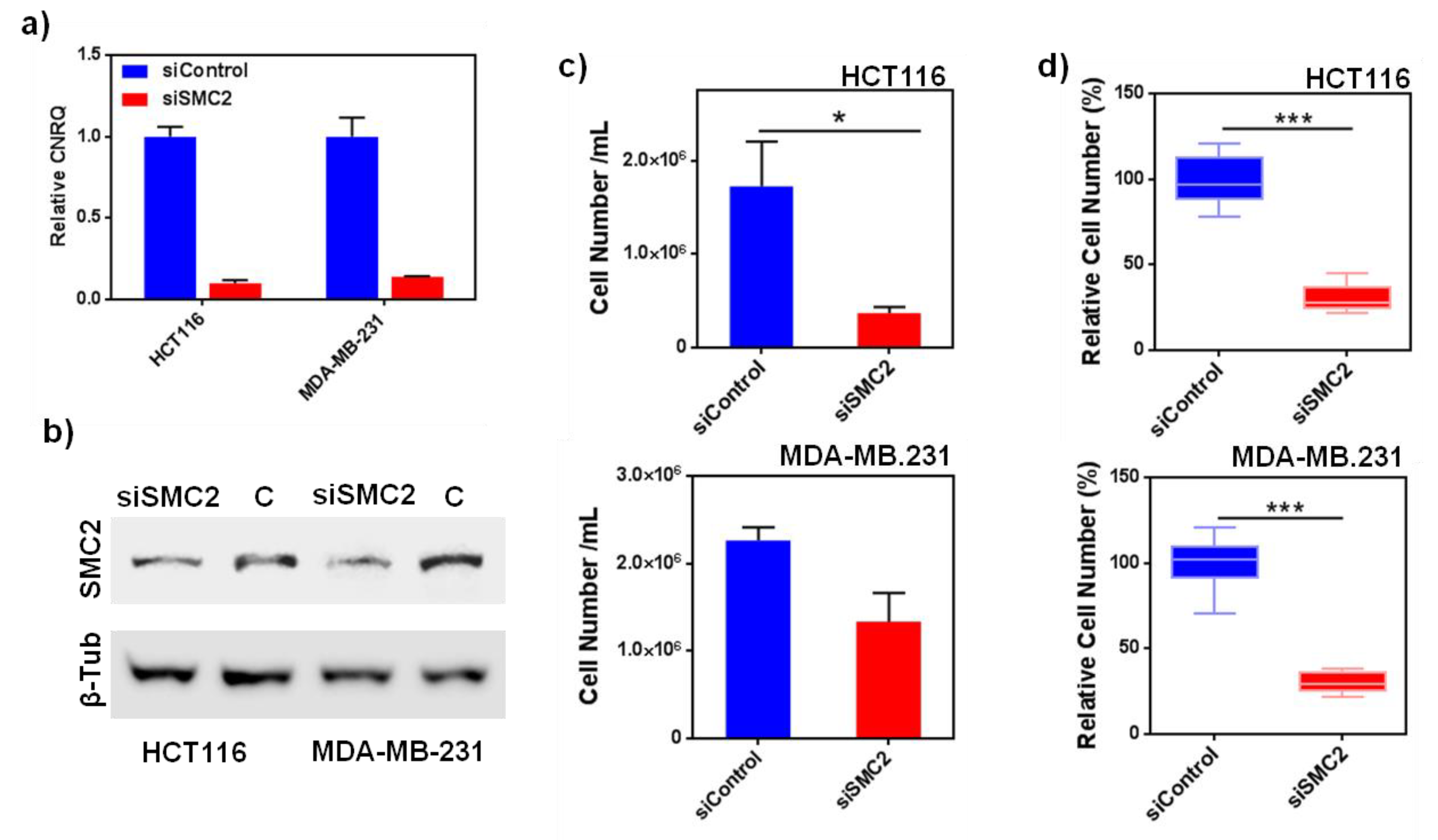
Aiming to validate SMC2 as a CSC target, SMC2 was silenced in different cancer cell lines (HT116, MDA-MB-231 and PANC-1, from colorectal, breast and pancreatic origin) using RNA interference technology. Its effect was assessed in terms of cell viability and colony formation impairment. As predicted, the silencing of SMC2 was able to reduce cell viability in adherent conditions and the formation of spheres for HCT116 cells in non-adherent cultures. In the case of MDA-MB-231 and PANC-1 the effect of SMC2 silencing was only observed in low attachment conditions. This result revealed that SMC2 could be an important target within stem cell subpopulations (
Figure 1 and
Figure S1). After SMC2 validation as a promising target, different therapeutic silencing strategies were considered. SMC2 silencing by siRNA arose as the most straightforward approach since a previous report by our group showed effective in vivo tumor growth inhibition after ex vivo silencing of the SMC2 gene in tumor cells [
18]. However, unforeseen challenges related with siRNA stability and the need to reach high doses to render biological efficacy after i.v. administration, prompted us to choose an alternative strategy [
34]. Thus, we attempted to block SMC2 activity by specific interaction with an antibody against the protein. In order to protect the antibody integrity and improve its intracellular delivery, Ab-SMC2 was conjugated with Pluronic
® F127-based PM. The use of nanocarriers allows (i) the systemic delivery of high amount of Ab, (ii) to decrease off-target related toxicity in other organs, (iii) to protect the cargo from enzymatic degradation, and (iv) the sustained release of the Ab alone or in combination with other chemo-therapeutic compounds [
9]. In the last years, several formulations have been developed for the intracellular release of antibodies. One of them consists of cationic lipid-based carriers, which are also employed for siRNA delivery, as proven by Courtete et al. (2007) [
35]. Nonetheless, even though some of these formulations are reaching clinical trials, strong concerns about their reported toxicity are delaying its entrance into the clinical practice. Moreover, inorganic and viral carriers have also been developed but with the same toxicity and immunogenicity concerns that reduce their clinical expectations. Some polymeric-based nanoparticles also has been developed for intracellular delivery such as the polymersomes described by Canton et al. (2013) and Tian et al. (2014) [
36,
37], or the self-assembling pyridylthiourea modified polyethylenimine nanoparticles designed by Postupalenko et al. (2014) [
8]. Although great efforts have been invested until today, the proposed formulations still present important limitations, such as (i) difficult and expensive production methods, (ii) high mean diameter and immunogenicity issues, (iii) lack of endosomal escape capacity, and (iv) toxicity, among others [
9]. In this regard, the proposed delivery system solves some of these drawbacks. Accordingly, they are biocompatible and non-immunogenic, present a small size, and can be produced in compliance to a simple and easily scalable production method. In previous studies, we have demonstrated that these PM can be repeatedly administered in vivo without causing toxicities and can accumulate into tumors upon i.v. administration [
38]. Furthermore, our PM were previously used to efficiently conjugate Cetuximab, showing an effective active targeting against the epidermal growth factor receptor (EGFR) in overexpressing breast cancer cells [
25].
In the present work, we focused into the intracellular delivery of Ab-SMC2 to silence the activity of SMC2 in CSC. For that, Ab-SMC2 was conjugated to PM through two different approaches. The first one consisted in the encapsulation of the antibody into the micelles hydrophilic shield (PM:SMC2). The second one was based on the covalent bonding of the amine groups of the antibody with the carboxylic terminals of the modified polymer (PM-CON:SMC2). As expected, the formulations with Ab-SMC2 presented larger mean diameter. The conjugation was also confirmed by the FTIR where was possible to see the appearance of a peak at 1646 cm
−1 (
Figure 2 and
Figure S3). Since the two encapsulation techniques presented adequate physicochemical features and similar association efficiency, both formulations were tested for cell toxicity in HCT116 and MDA-MB-231 colon and breast cancer cells, to evaluate whether the level of exposure to Ab-SMC2 might affect its in vitro efficacy. As expected, control PM and free antibody did not show cell toxicity in any tested cell lines. However, when conjugated, Ab-SMC2 showed higher efficacy in terms of cell toxicity and impairment of colony formation, suggesting specific positive activity against CSC. In addition, the way Ab-SMC2 was loaded into the PM might affect the efficacy of the formulation, since a slight higher effect was displayed by cells treated with PM-CON:SMC2 in comparison with the ones treated with PM:SMC2 in both tested cell lines (
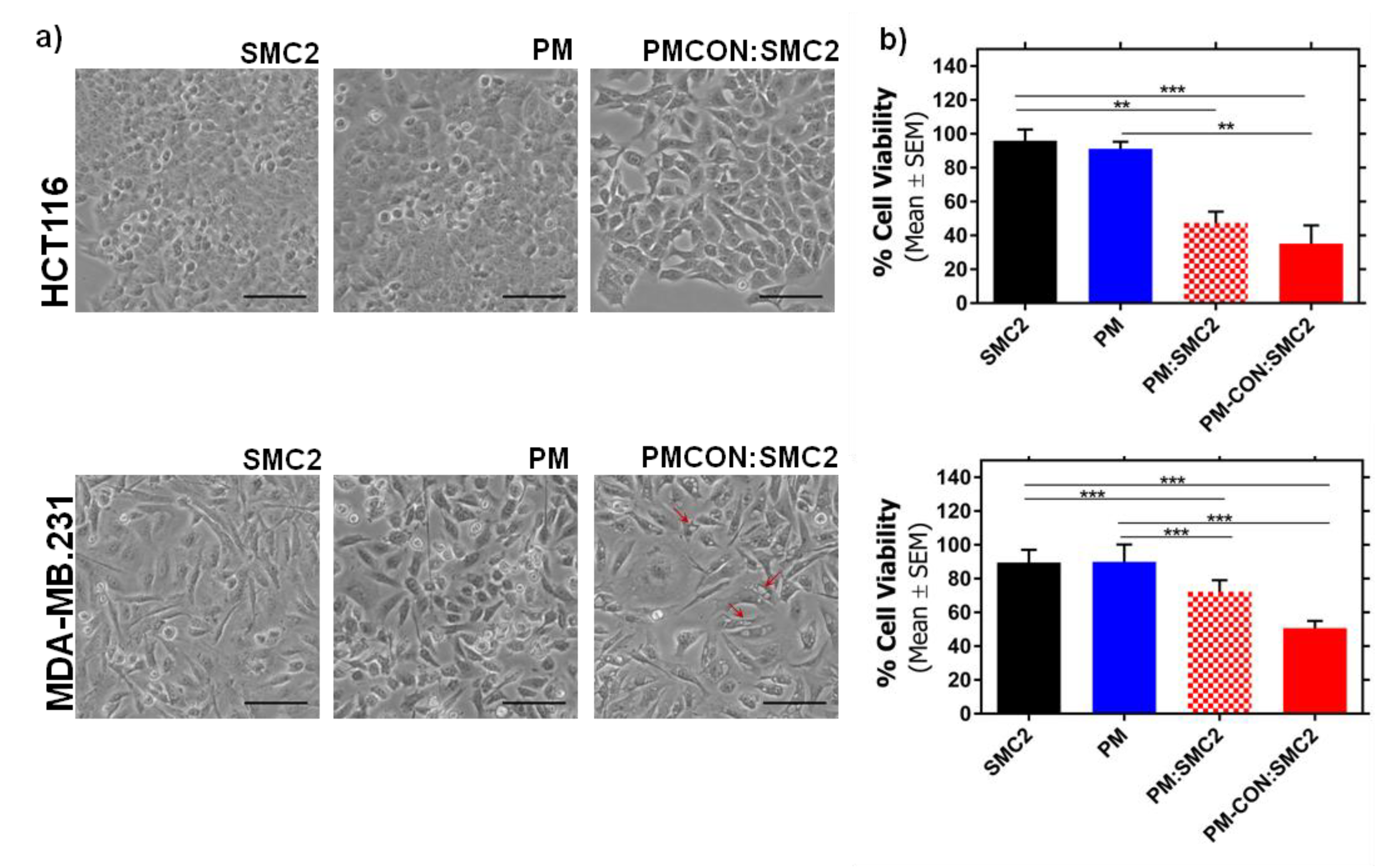
Figure 3). These results and the fact that the covalent conjugation of Ab-SMC2 would be more stable and controlled, made us select PM-CON:SMC2 for further studies. Moreover, the differential effect observed between free antibody and the one conjugated onto PM was clear when analyzing the morphology of treated cells. Thus, it was possible to detect strong morphologic changes in HCT116 cells and the formation of vacuoles in MDA-MB-231 after treatment with PM-CON:SMC2. In contrast, no changes were detected when cells were treated with free antibody or non-conjugated PM. These data indicate that PM-CON:SMC2 were exerting a specific cytotoxic action visibly affecting cellular structures. This was also in accordance with the results seen in the viability assays.
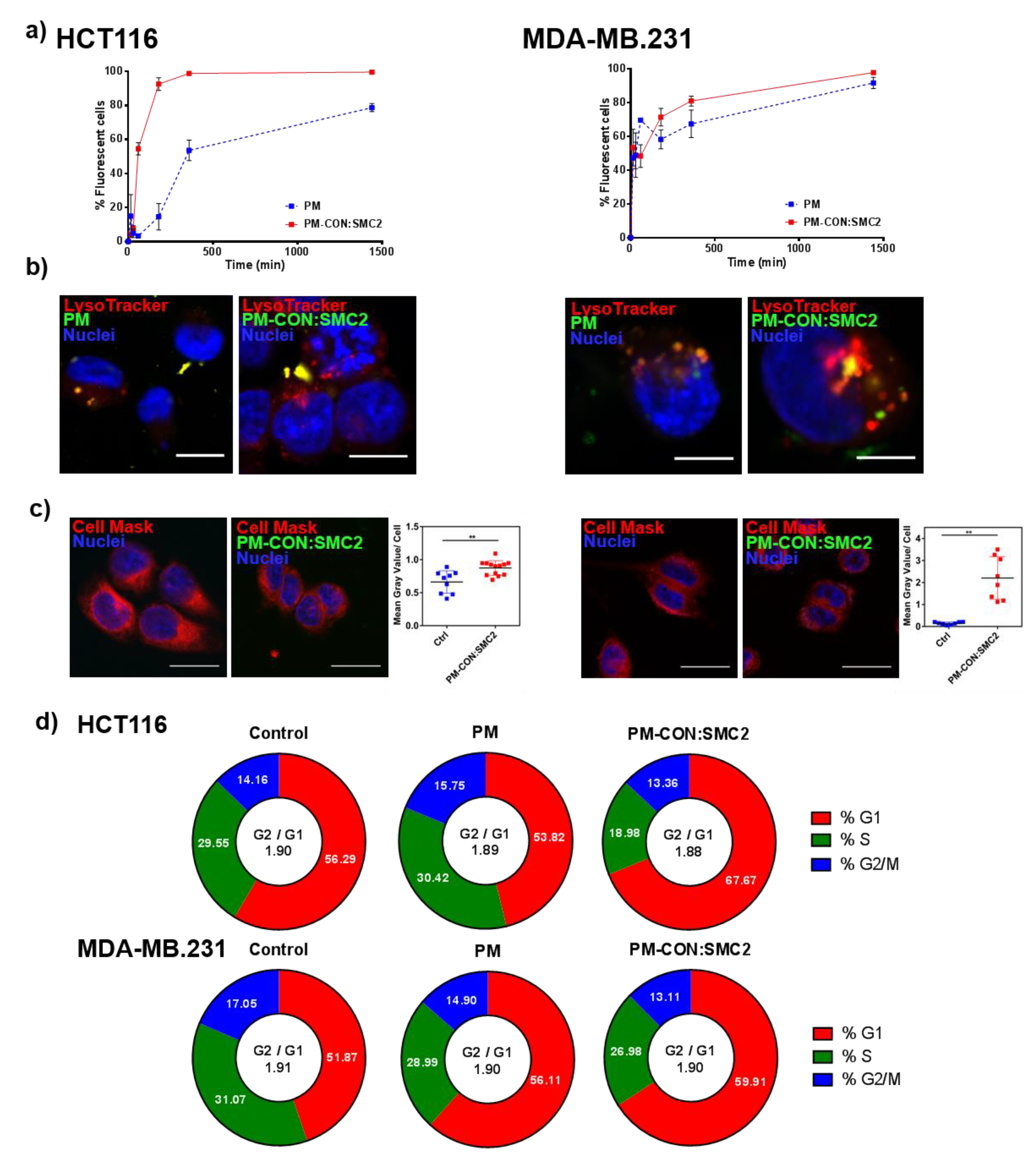
Additionally, in terms of internalization, PM-CON:SMC2 demonstrated to have a faster uptake when compared with the control PM, probably due to its more negatively charged particle surface and Van der Waals interactions between the functional groups of the particle and the cellular membrane. Confocal microscopy images revealed the co-localization of fluorescently labeled PM with endocytic vesicles, suggesting that PM formulations could enter into the cells via endocytosis, at least partially. Other cellular entry routes cannot be discarded, nonetheless. One of the most critical steps regarding the intracellular delivery of antibodies is the need of their endosomal escape. The antibody must be released to the cytosol before reaching the lysosomes, otherwise it might be degraded inside these vehicles. By analyzing the levels of green fluorescence in the whole cytoplasm, it was possible to conclude that a substantial part of Ab-SMC2 PM was able to escape the endosomes and reach the cytoplasm, where the Ab-SMC2 exert its activity. Importantly, these results strongly support the in vitro efficacy outcomes in terms of cell toxicity and colony formation. In order to confirm the biological action of PM-CON:SMC2 inhibiting the SMC2 protein, a cell-cycle assay was performed. Of note, the condensin complex is mostly found in the cell cytoplasm in interphase while during mitosis is found associated to chromatin. Therefore, in order to effectively block SMC2 dimerization and the activity of the condensin complex, the antibody must reach the cell cytosol. Our results demonstrate that at least in the case of HCT116 cells a significant arrest of cell cycle in G1 was driven by the incubation with PM-CON:SMC2, reinforcing our previous results showing a decrease in cell viability. These also suggest the capacity of our system to deliver the antibody to the cell cytoplasm. This pattern, however, was not clear for MDA-MB-231 cells. The effect of the SMC2 inhibition in adherent cultures was restricted to the CSC-like subpopulation in this cell line (
Figure 1).
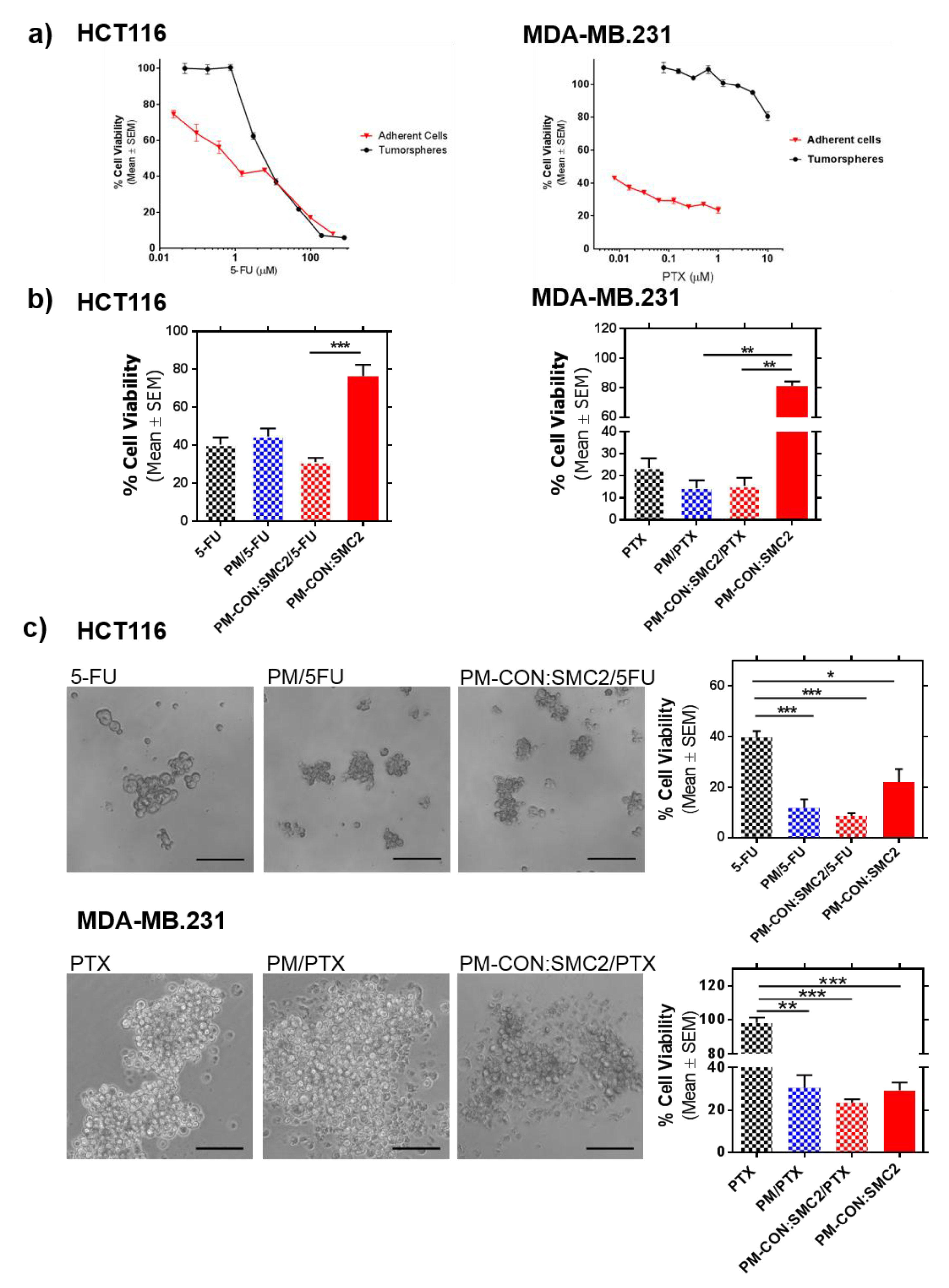
Finally, we further investigated the potential use of PM-CON:SMC2 as adjuvant treatment with Standard-of-Care (SoC) drugs. 5-FU and PTX are the SoC therapy for colon and breast cancer, respectively. Therefore, 5-FU and PTX were loaded into the hydrophobic core of PM-CON:SMC2. In a previous study, we showed that Zileuton™, a drug with reported efficacy in breast CSC, presented higher efficacy when encapsulated in similar micelles than its free form [
38]. In agreement with this, both 5-FU and PTX also presented higher efficacy in terms of inhibition of colony formation when encapsulated into PM-CON:SMC2. Interestingly, both 5-FU and PTX showed increased efficacy when encapsulated into PM, particularly regarding tumorsphere formation. This was remarkable in the case of the MDA-MB-231 cell line, known for its high resistance to most treatments. In this case, no efficacy was detected with free PTX (
Figure 6c). Although, we could not detect a clear additive efficacy when treating cells with PM-CON:SMC2 loaded with the respective chemotherapeutic drugs, a significant effect was observed in CSC-like in vitro models in comparison with SoC. Our results suggest that the combination of drugs and Ab-SMC2 delivery might cooperate in the eradication not only of bulk tumor cells but also of cells with stemness properties.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





