In Vivo Fluorescence Imaging of Passive Inflammation Site Accumulation of Liposomes via Intravenous Administration Focused on Their Surface Charge and PEG Modification

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials, Cells, and Mice
2.2. Liposome Preparation and Physical Property Measurement
2.3. Preparation of Collagen-Induced Arthritis Mice
2.4. Evaluation of Inflammatory Site Accumulation Using In Vivo Imaging after Intravenous Liposome Administration in CIA Mice
2.5. Evaluation of Inflammatory Site Accumulation Using In Vivo Imaging after Intravenous Liposome Administration in an Ulcerative Colitis Mouse Model
2.6. Evaluation of the Uptake Efficiency of Each Liposome by RAW264.7 Macrophages
2.7. Statistical Analysis
3. Results
3.1. Physical Properties of Various Liposomes
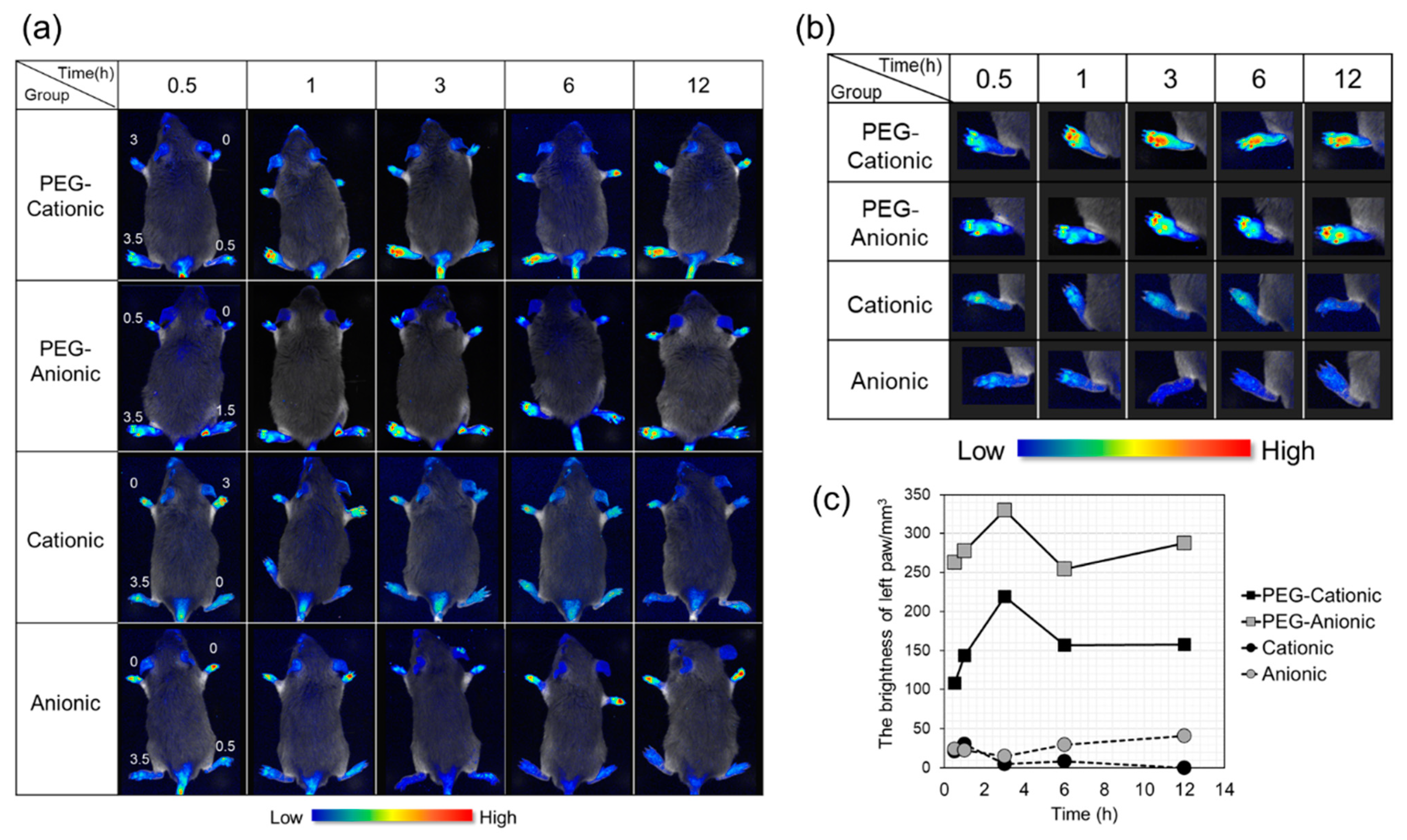
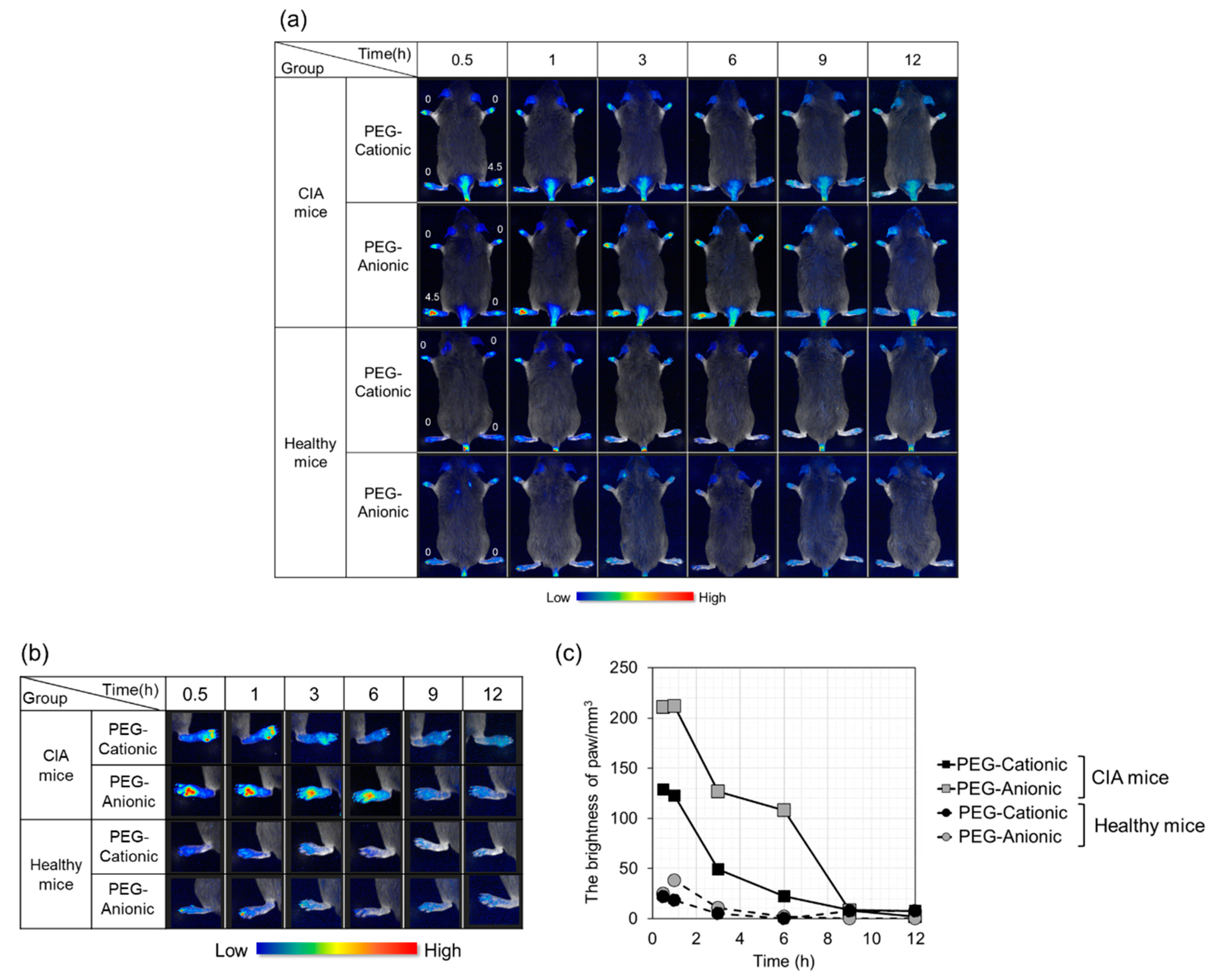
3.2. In Vivo Imaging of Inflammatory Sites after Intravenous Liposome Administration to CIA Mice
3.3. Effects of Liposome Surface Charge on Inflammatory Site Accumulation after Intravenous PEG-Modified Liposome Administration to CIA Mice
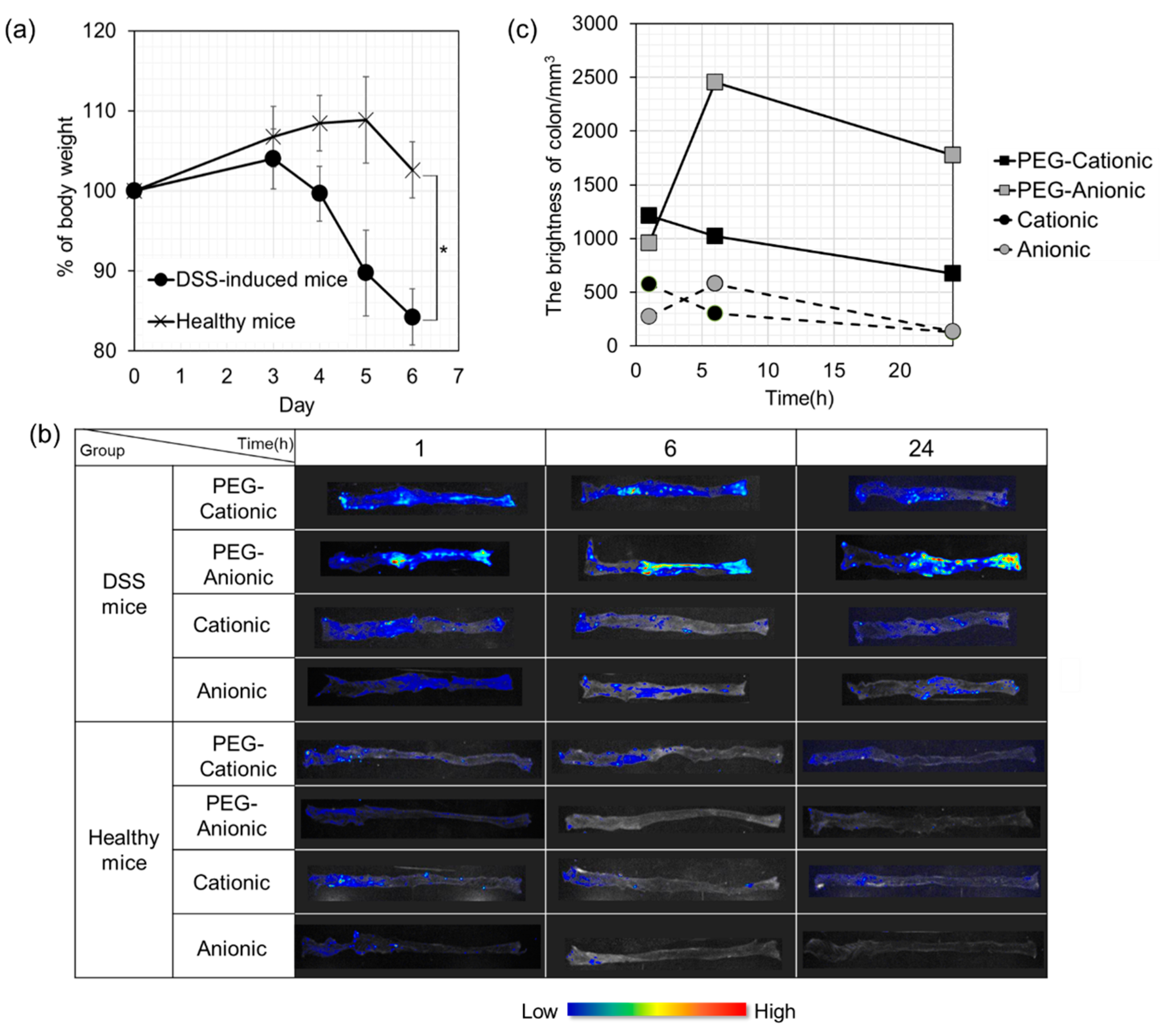
3.4. In Vivo Imaging Evaluation of Inflammatory Sites in DSS Mice after Intravenous Liposome Administration
3.5. Evaluation of PEG Liposome Uptake by RAW264.7 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CFA | Complete Freund’s adjuvant |
| CHEMS | Cholesteryl hemisuccinate |
| CIA | Collagen-induced arthritis |
| CII | Bovine type II collagen |
| DEPE-PEG2000 | 2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DOPE | 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine |
| DOTAP | 1,2-Dioleoyl-3-trimethylammonium-propane |
| DSS | Dextran sulfate sodium |
| EPR | Enhanced permeation and retention |
| FBS | Fetal bovine serum |
| IBD | Inflammatory bowel disease |
| IFA | Incomplete Freund’s adjuvant |
| PBS | Phosphate-buffered saline |
| PEG | Polyethylene glycol |
| RA | Rheumatoid arthritis |
| RAW264.7 | Murine macrophage |
| RES | Reticuloendothelial system |
References
- NIH Autoimmune Diseases Coordinating Committee: Autoimmune Diseases Research Plan. Available online: https://www.niaid.nih.gov/sites/default/files/adccfinal.pdf (accessed on 31 December 2020).
- Bach, J. Infections and autoimmune diseases. J. Autoimmun. 2005, 25, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Ortona, E.; Pierdominici, M.; Maselli, A.; Veroni, C.; Aloisi, F.; Shoenfeld, Y. Sex-based differences in autoimmune diseases. Ann. Ist. Super. Sanità 2016, 52, 205–212. [Google Scholar] [PubMed]
- Moreland, L.W.; O’Dell, J.R. Glucocorticoids and rheumatoid arthritis: Back to the future? Arthritis Rheum. 2002, 46, 2553–2563. [Google Scholar] [CrossRef] [PubMed]
- Magro, F.; Portela, F. Management of Inflammatory Bowel Disease with Infliximab and Other Anti-Tumor Necrosis Factor Alpha Therapies. BioDrugs 2010, 24, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Guo, J.; Gui, S. Orally targeted galactosylated chitosan poly(lactic-co-glycolic acid) nanoparticles loaded with TNF-ɑ siRNA provide a novel strategy for the experimental treatment of ulcerative colitis. Eur. J. Pharm. Sci. 2018, 125, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Catrina, A.I.; Paget, S. Rheumatoid arthritis. Lancet 2009, 373, 21–27. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. mAbs 2019, 11, 219–238. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Sandborn, W.J.; Feagan, B.G.; Reinisch, W.; Olson, A.; Johanns, J.; Travers, S.; Rachmilewitz, D.; Hanauer, S.B.; Lichtenstein, G.R.; et al. Infliximab for Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2005, 353, 2462–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandborn, W.J.; Van Assche, G.; Reinisch, W.; Colombel, J.; D’Haens, G.; Wolf, D.C.; Kron, M.; Tighe, M.B.; Lazar, A.; Thakkar, R.B. Adalimumab Induces and Maintains Clinical Remission in Patients with Moderate-to-Severe Ulcerative Colitis. Gastroenterology 2012, 142, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.V.; Khan, D.A. Adverse Reactions to Biologic Therapy. Immunol. Allergy Clin. N. Am. 2017, 37, 397–412. [Google Scholar] [CrossRef]
- Entyvio®. Available online: https://www.entyvio.com/cost (accessed on 31 December 2020).
- Zahin, N.; Anwar, R.; Tewari, D.; Kabir, T.; Sajid, A.; Mathew, B.; Uddin, S.; Aleya, L.; Abdel-Daim, M.M. Nanoparticles and its biomedical applications in health and diseases: Special focus on drug delivery. Environ. Sci. Pollut. Res. 2019, 27, 19151–19168. [Google Scholar] [CrossRef]
- Kandeil, M.A.; Mohammed, E.T.; Hashem, K.S.; Aleya, L.; Abdel-Daim, M.M. Moringa seed extract alleviates titanium oxide nanoparticles (TiO2-NPs)-induced cerebral oxidative damage, and increases cerebral mitochondrial viability. Environ. Sci. Pollut. Res. 2020, 27, 19169–19184. [Google Scholar] [CrossRef] [PubMed]
- Taurin, S.; Nehoff, H.; Greish, K. Anticancer nanomedicine and tumor vascular permeability; Where is the missing link? J. Control. Release 2012, 164, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Ishihara, T.; Hayashi, E.; Yamamoto, S.; Kobayashi, C.; Tamura, Y.; Sawazaki, R.; Tamura, F.; Tahara, K.; Kasahara, T.; Ishihara, T.; et al. Encapsulation of beraprost sodium in nanoparticles: Analysis of sustained release properties, targeting abilities and pharmacological activities in animal models of pulmonary arterial hypertension. J. Control. Release 2015, 197, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Fang, J.; Liao, L.; Nakamura, H.; Maeda, H. Styrene-maleic acid copolymer-encapsulated CORM2, a water-soluble carbon monoxide (CO) donor with a constant CO-releasing property, exhibits therapeutic potential for inflammatory bowel disease. J. Control. Release 2014, 187, 14–21. [Google Scholar] [CrossRef]
- Fang, J.; Yin, H.; Liao, L.; Qin, H.; Ueda, F.; Uemura, K.; Eguchi, K.; Bharate, G.Y.; Maeda, H. Water soluble PEG-conjugate of xanthine oxidase inhibitor, PEG–AHPP micelles, as a novel therapeutic for ROS related inflammatory bowel diseases. J. Control. Release 2016, 223, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Hayder, M.; Poupot, M.; Baron, M.; Nigon, D.; Turrin, C.-O.; Caminade, A.-M.; Majoral, J.-P.; Eisenberg, R.A.; Fournié, J.-J.; Cantagrel, A.; et al. A Phosphorus-Based Dendrimer Targets Inflammation and Osteoclastogenesis in Experimental Arthritis. Sci. Transl. Med. 2011, 3, 81ra35. [Google Scholar] [CrossRef]
- Kelly, C.; Jefferies, C.; Cryan, S.-A. Targeted Liposomal Drug Delivery to Monocytes and Macrophages. J. Drug Deliv. 2011, 2011, 1–11. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Oku, N.; Namba, Y. Long-circulating liposomes. Crit. Rev. Ther. Drug Carrier Syst. 1994, 11, 231–270. [Google Scholar] [PubMed]
- Yamamoto, Y.; Nagasaki, Y.; Kato, Y.; Sugiyama, Y.; Kataoka, K. Long-circulating poly(ethylene glycol)–poly(d,l-lactide) block copolymer micelles with modulated surface charge. J. Control. Release 2001, 77, 27–38. [Google Scholar] [CrossRef]
- Ibaraki, H.; Kanazawa, T.; Takashima, Y.; Okada, H.; Seta, Y. Development of an Innovative Intradermal siRNA Delivery System Using a Combination of a Functional Stearylated Cytoplasm-Responsive Peptide and a Tight Junction-Opening Peptide. Molecules 2016, 21, 1279. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Shizawa, Y.; Takeuchi, M.; Tamano, K.; Ibaraki, H.; Seta, Y.; Takashima, Y.; Okada, H. Topical Anti-Nuclear Factor-Kappa B Small Interfering RNA with Functional Peptides Containing Sericin-Based Hydrogel for Atopic Dermatitis. Pharmaceutics 2015, 7, 294–304. [Google Scholar] [CrossRef]
- Kanazawa, T.; Hamasaki, T.; Endo, T.; Tamano, K.; Sogabe, K.; Seta, Y.; Ohgi, T.; Okada, H. Functional peptide nanocarriers for delivery of novel anti-RelA RNA interference agents as a topical treatment of atopic dermatitis. Int. J. Pharm. 2015, 489, 261–267. [Google Scholar] [CrossRef]
- Kanazawa, T.; Endo, T.; Arima, N.; Ibaraki, H.; Takashima, Y.; Seta, Y. Systemic delivery of small interfering RNA targeting nuclear factor κB in mice with collagen-induced arthritis using arginine-histidine-cysteine based oligopeptide-modified polymer nanomicelles. Int. J. Pharm. 2016, 515, 315–323. [Google Scholar] [CrossRef]
- Kanazawa, T.; Tamano, K.; Sogabe, K.; Endo, T.; Ibaraki, H.; Takashima, Y.; Seta, Y. Intra-articular retention and anti-arthritic effects in collagen-induced arthritis model mice by injectable small interfering RNA containing hydrogel. Biol. Pharm. Bull. 2017, 40, 1929–1933. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Kanazawa, T.; Ogawa, T.; Takashima, Y.; Fukuda, T.; Okada, H. Disulfide crosslinked stearoyl carrier peptides containing arginine and histidine enhance siRNA uptake and gene silencing. Int. J. Pharm. 2010, 398, 219–224. [Google Scholar] [CrossRef]
- Tanaka, K.; Kanazawa, T.; Ogawa, T.; Suda, Y.; Takashima, Y.; Fukuda, T.; Okada, H. A Novel, Bio-Reducible Gene Vector Containing Arginine and Histidine Enhances Gene Transfection and Expression of Plasmid DNA. Chem. Pharm. Bull. 2011, 59, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Kanazawa, T.; Horiuchi, S.; Ando, T.; Sugawara, K.; Takashima, Y.; Seta, Y.; Okada, H. Cytoplasm-responsive nanocarriers conjugated with a functional cell-penetrating peptide for systemic siRNA delivery. Int. J. Pharm. 2013, 455, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ibaraki, H.; Kanazawa, T.; Oogi, C.; Takashima, Y.; Seta, Y. Effects of surface charge and flexibility of liposomes on dermal drug delivery. J. Drug Deliv. Sci. Technol. 2019, 50, 155–162. [Google Scholar] [CrossRef]
- Ibaraki, H.; Kanazawa, T.; Kurano, T.; Oogi, C.; Takashima, Y.; Seta, Y. Anti-RelA siRNA-Encapsulated Flexible Liposome with Tight Junction-Opening Peptide as a Non-invasive Topical Therapeutic for Atopic Dermatitis. Biol. Pharm. Bull. 2019, 42, 1216–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibaraki, H.; Kanazawa, T.; Chien, W.-Y.; Nakaminami, H.; Aoki, M.; Ozawa, K.; Kaneko, H.; Takashima, Y.; Noguchi, N.; Seta, Y. The effects of surface properties of liposomes on their activity against Pseudomonas aeruginosa PAO-1 biofilm. J. Drug Deliv. Sci. Technol. 2020, 57, 101754. [Google Scholar] [CrossRef]
- Ibaraki, H.; Kanazawa, T.; Owada, M.; Iwaya, K.; Takashima, Y.; Seta, Y. Anti-Metastatic E ects on Melanoma via Intravenous. Administration of Anti-NF-κB siRNA Complexed with Functional Peptide-Modified Nano-Micelles. Pharmaceutics 2019, 12, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangham, A.D.; Hill, M.W.; Miller, N.G.A. Preparation and use of liposomes as models of biological membranes. In Methods in Membrane Biology; Springer: Boston, MA, USA, 1974; pp. 1–68. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghamib, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Brand, D.D.; Latham, K.A.; Rosloniec, E.F. Collagen-induced arthritis. Nat. Protoc. 2007, 2, 1269–1275. [Google Scholar] [CrossRef]
- Suzuki, K.; Sun, X.; Nagata, M.; Kawase, T.; Yamaguchi, H.; Sukumaran, V.; Kawauchi, Y.; Kawachi, H.; Nishino, T.; Watanabe, K.; et al. Analysis of intestinal fibrosis in chronic colitis in mice induced by dextran sulfate sodium. Pathol. Int. 2011, 61, 228–238. [Google Scholar] [CrossRef]
- Wilson, D.S.; Dalmasso, G.; Wang, L.; Sitaraman, S.V.; Merlin, D.; Murthy, N. Orally delivered thioketal nanoparticles loaded with TNF-α–siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater. 2010, 9, 923–928. [Google Scholar] [CrossRef]
- Boshtam, M.; Asgary, S.; Kouhpayeh, S.; Shariati, L.; Khanahmad, H. Aptamers Against Pro- and Anti-Inflammatory Cytokines: A Review. Inflammation 2017, 40, 340–349. [Google Scholar] [CrossRef]
- Nemati, H.; Ghahramani, M.-H.; Faridi-Majidi, R.; Izadi, B.; Bahrami, G.; Madani, S.-H.; Tavoosidana, G. Using siRNA-based spherical nucleic acid nanoparticle conjugates for gene regulation in psoriasis. J. Control. Release 2017, 268, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Champion, J.A.; Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4930–4934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Mori, A.; Huang, L. Large liposomes containing ganglioside GM1 accumulate effectively in spleen. Biochim. Biophys. Acta Biomembr. 1991, 1066, 159–165. [Google Scholar] [CrossRef]
- Ren, H.; He, Y.; Liang, J.; Cheng, Z.; Zhang, M.; Zhu, Y.; Hong, C.; Qin, J.; Xu, X.; Wang, J. Role of Liposome Size, Surface Charge, and PEGylation on Rheumatoid Arthritis Targeting Therapy. ACS Appl. Mater. Interfaces 2019, 11, 20304–20315. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, N.; Paré, A.; Farndale, R.W.; Schumacher, H.R.; Nigrovic, P.A.; Lacroix, S.; Boilard, E. Platelets can enhance vascular permeability. Blood 2012, 120, 1334–1343. [Google Scholar] [CrossRef]
- Hong, R.L.; Huang, C.J.; Tseng, Y.L.; Pang, V.F.; Chen, S.T.; Liu, J.J.; Chang, F.H. Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in C-26 tumor-bearing mice: Is surface coating with polyethylene glycol beneficial? Clin. Cancer Res. 1999, 5, 3645–3652. [Google Scholar]
- Maldiney, T.; Richard, C.; Seguin, J.; Wattier, N.; Bessodes, M.; Scherman, D. Effect of Core Diameter, Surface Coating, and PEG Chain Length on the Biodistribution of Persistent Luminescence Nanoparticles in Mice. ACS Nano 2011, 5, 854–862. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Koczy-Baron, E.; Kasperska-Zając, A. The role of vascular endothelial growth factor in inflammatory processes. Postępy Hig. Med. Doświadczalnej 2014, 68, 57–65. [Google Scholar] [CrossRef]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Tanaka, H.; Sakurai, Y.; Tange, K.; Nakai, Y.; Ohkawara, T.; Takeda, H.; Harashima, H.; Akita, H. Effect of particle size on their accumulation in an inflammatory lesion in a dextran sulfate sodium (DSS)-induced colitis model. Int. J. Pharm. 2016, 509, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Tang, Y.; Lv, Z.; Lin, Y.; Chen, L. Nanomedicine—Advantages for their use in rheumatoid arthritis theranostics. J. Control. Release 2019, 316, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Mold, C. Effect of membrane phospholipids on activation of the alternative complement pathway. J. Immunol. 1989, 143, 1663–1668. [Google Scholar] [PubMed]
- Sakurai, F.; Nishioka, T.; Saito, H.; Baba, T.; Okuda, A.; Matsumoto, O.; Taga, T.; Yamashita, F.; Takakura, Y.; Hashida, M. Interaction between DNA–cationic liposome complexes and erythrocytes is an important factor in systemic gene transfer via the intravenous route in mice: The role of the neutral helper lipid. Gene Ther. 2001, 8, 677–686. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Yeo, Y. Drug delivery to macrophages: Challenges and opportunities. J. Control. Release 2016, 240, 202–211. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, Y.; Hou, T.; Zeng, H.; Kalambhe, D.; Wang, B.; Shen, X.; Huang, Y. Macrophage-based nanotherapeutic strategies in ulcerative colitis. J. Control. Release 2020, 320, 363–380. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Liposomes | ATTO-DOPE + DOPE (mol%) | DOTAP (mol%) | CHEMS (mol%) | DSPE-PEG2000 (mol%) | Total Lipid (µmol) |
|---|---|---|---|---|---|
| PEG-cationic | 46.15 | 46.15 | - | 7.70 | 6.50 |
| PEG-anionic | 54.54 | - | 36.36 | 9.10 | 5.50 |
| Cationic | 50.00 | 50.00 | - | - | 6.00 |
| Anionic | 60.00 | - | 40.00 | - | 5.00 |
| Liposomes | Particle Size (nm) | Zeta Potential (mV) | PDI |
|---|---|---|---|
| PEG-Cationic | 67.8 ± 7.9 | +18.9 ± 0.9 | 0.201 ± 0.008 |
| PEG-Anionic | 50.6 ± 0.9 | −19.8 ± 1.3 | 0.309 ± 0.051 |
| Cationic | 62.4 ± 2.1 | +51.2 ± 0.8 | 0.231 ± 0.007 |
| Anionic- | 86.8 ± 2.4 | −42.7 ± 3.5 | 0.276 ± 0.015 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibaraki, H.; Takeda, A.; Arima, N.; Hatakeyama, N.; Takashima, Y.; Seta, Y.; Kanazawa, T. In Vivo Fluorescence Imaging of Passive Inflammation Site Accumulation of Liposomes via Intravenous Administration Focused on Their Surface Charge and PEG Modification. Pharmaceutics 2021, 13, 104. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13010104
Ibaraki H, Takeda A, Arima N, Hatakeyama N, Takashima Y, Seta Y, Kanazawa T. In Vivo Fluorescence Imaging of Passive Inflammation Site Accumulation of Liposomes via Intravenous Administration Focused on Their Surface Charge and PEG Modification. Pharmaceutics. 2021; 13(1):104. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13010104
Chicago/Turabian StyleIbaraki, Hisako, Akihiro Takeda, Naoki Arima, Naruhiro Hatakeyama, Yuuki Takashima, Yasuo Seta, and Takanori Kanazawa. 2021. "In Vivo Fluorescence Imaging of Passive Inflammation Site Accumulation of Liposomes via Intravenous Administration Focused on Their Surface Charge and PEG Modification" Pharmaceutics 13, no. 1: 104. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13010104