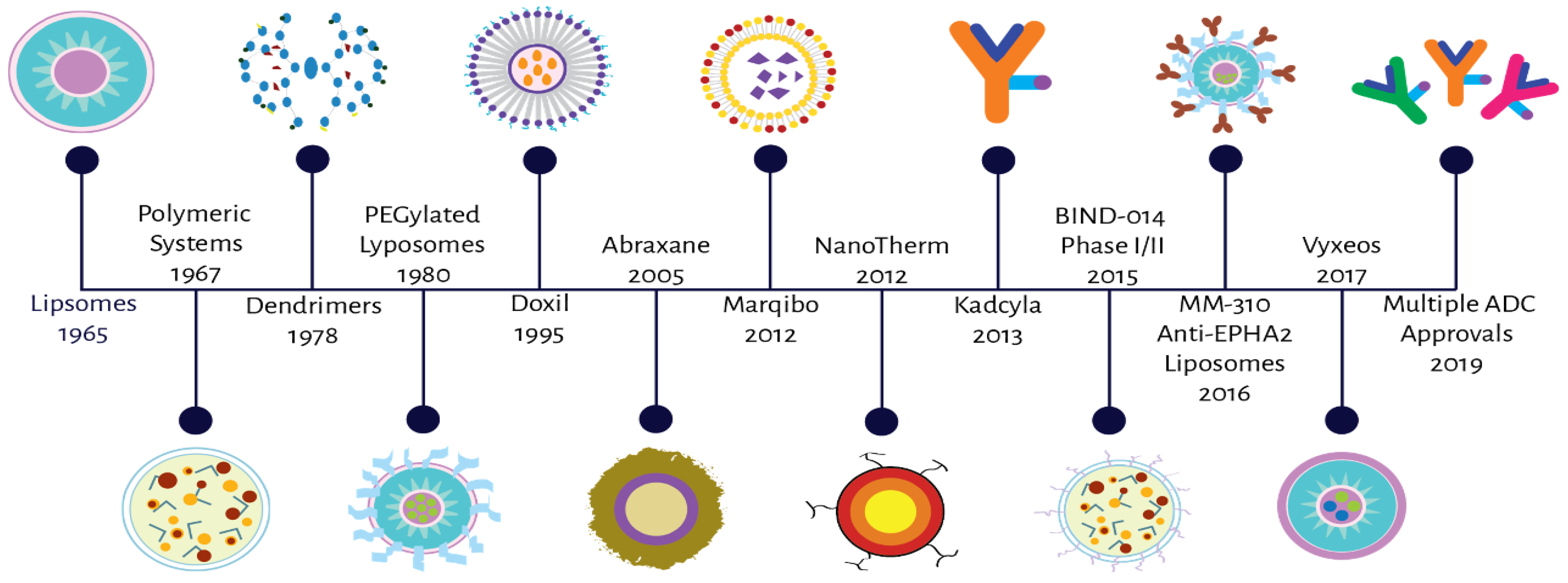
Complex Factors and Challenges that Affect the Pharmacology, Safety and Efficacy of Nanocarrier Drug Delivery Systems


Abstract
:1. Introduction
2. Basic Pharmacokinetic & Analytical Considerations
2.1. Principal Differences in ADME Between Small-Molecule & NPs
2.2. Analytical Characterization to Evaluate the Plasma, Tissue, & Tumor Disposition of NPs
3. Biological Concepts of Nanoparticle Delivery
3.1. Enhanced Permeability and Retention (EPR) Effect & Passive Targeting
3.2. Mechanisms & Factors Involved in Nanoparticle Uptake, Distribution, and Clearance
3.3. Is the EPR Effect Real? Active Pathways as the Primary Mechanism of Nanoparticle Uptake
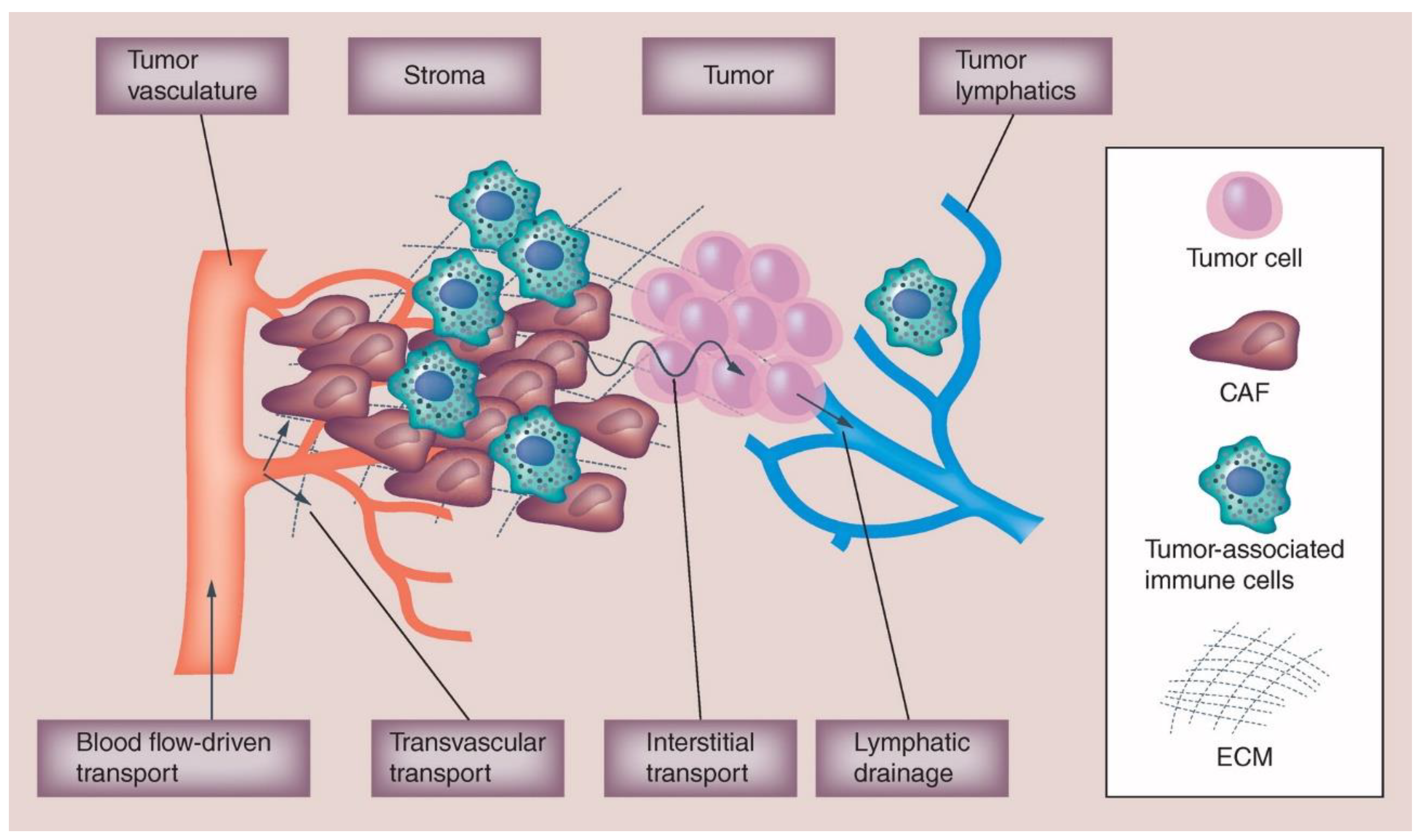
4. Tumor Microenvironment Factors that Affect Disposition of Nanoparticle Agents
4.1. Vascularity: Perfusion & Permeability
4.2. Stroma
4.3. Interstitial Fluid Pressure
4.4. Mononuclear Phagocyte System
5. Considerations in the Safety & Efficacy of Nanoparticle Agents
5.1. Alteration of Pharmacodynamic Toxicity in Nanoparticles
5.2. Payload Release versus Nanoparticle Delivery to Target Organs
5.3. Drug-Drug Interactions
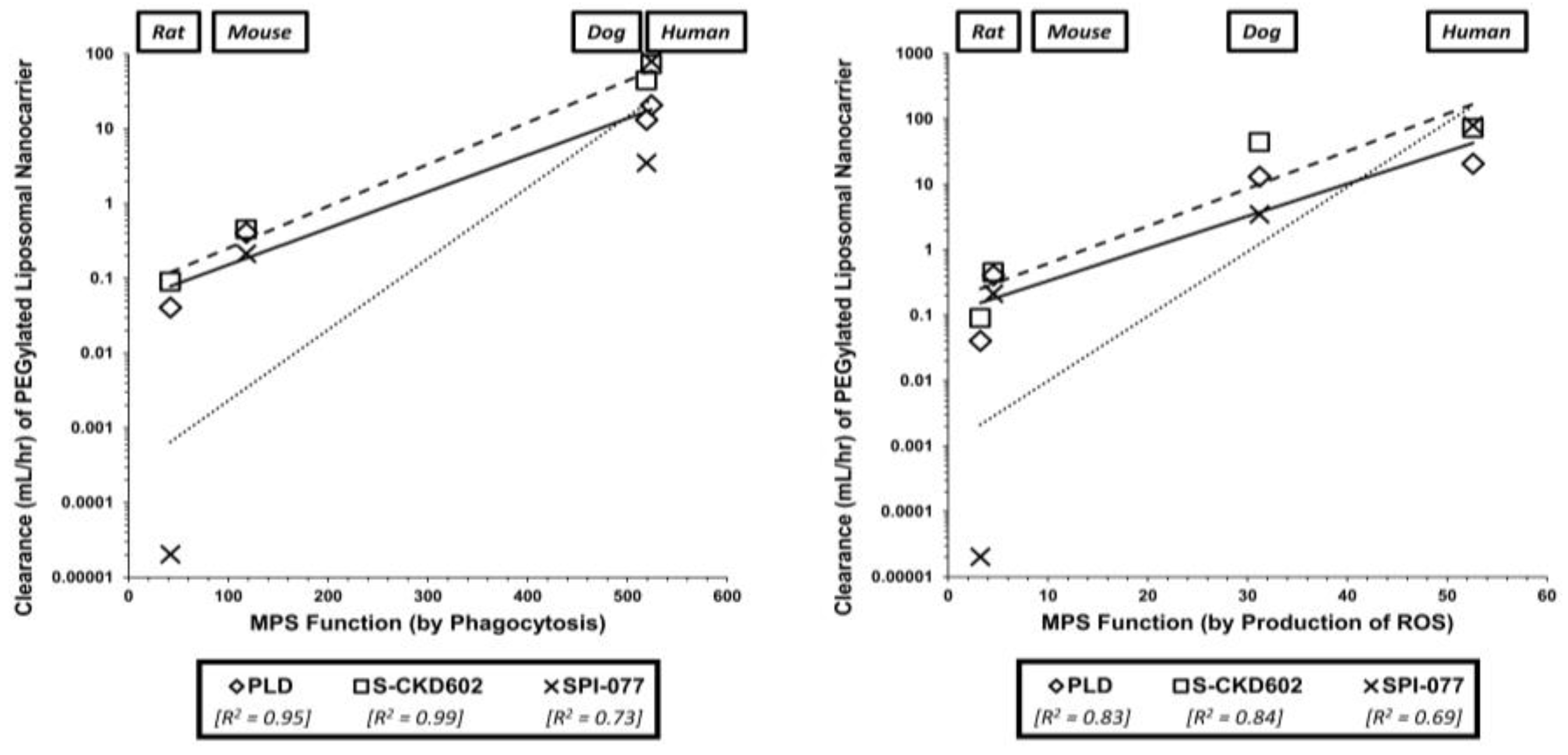
5.4. Preclinical Model Selection: Variability in Patients & Animals
5.5. Which Model is Most Appropriate for Allometric Scaling?
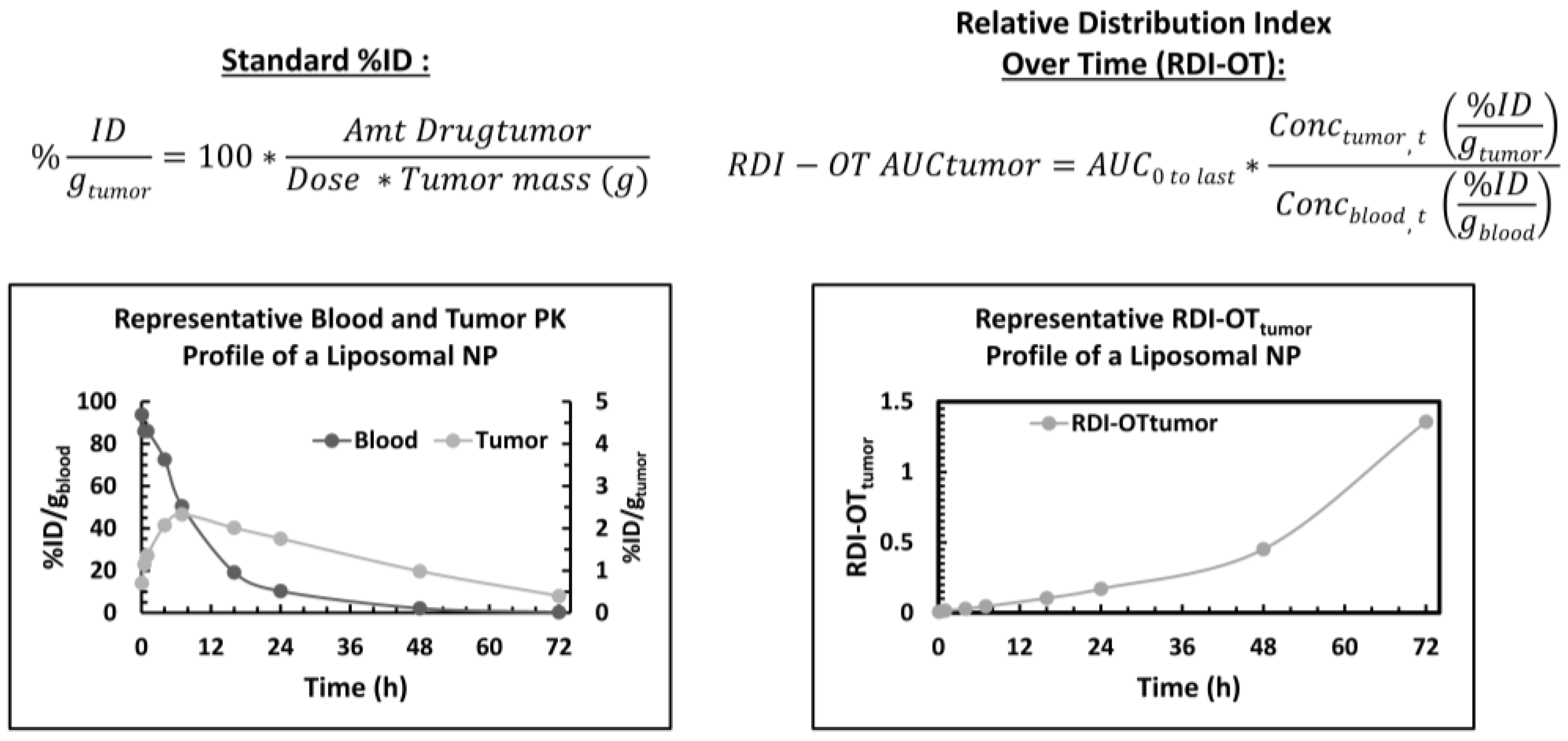
5.6. Pharmacokinetic Parameters used to Describe Nanoparticle Tumor Disposition
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Z.; Tan, S.; Li, S.; Shen, Q.; Wang, K. Cancer drug delivery in the nano era: An overview and perspectives (Review). Oncol. Rep. 2017, 38, 611–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClements, D.J. Encapsulation, protection, and delivery of bioactive proteins and peptides using nanoparticle and microparticle systems: A review. Adv. Colloid Interface Sci. 2018, 253, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; Wang, C.; Ding, J.; Tao, W. Editorial: Applications of Nanobiotechnology in Pharmacology. Front. Pharmacol. 2019, 10, 1451. [Google Scholar] [CrossRef] [PubMed]
- Pitt, G.G.; Gratzl, M.M.; Kimmel, G.L.; Surles, J.; Sohindler, A. Aliphatic polyesters II. The degradation of poly (dl-lactide), poly (ε-caprolactone), and their copolymers In Vivo. Biomateials 1981, 2, 215–220. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and Key Considerations of the Enhanced Permeability and Retention Effect for Nanomedicine Drug Delivery in Oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef] [Green Version]
- Zamboni, W.C.; Torchilin, V.; Patri, A.K.; Hrkach, J.; Stern, S.; Lee, R.; Nel, A.; Panaro, N.J.; Grodzinski, P. Best Practices in Cancer Nanotechnology: Perspective from NCI Nanotechnology Alliance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3229–3241. [Google Scholar] [CrossRef] [Green Version]
- Caron, W.P.; Song, G.; Kumar, P.; Rawal, S.; Zamboni, W.C. Interpatient Pharmacokinetic and Pharmacodynamic Variability of Carrier-Mediated Anticancer Agents. Clin. Pharmacol. Ther. 2012, 91, 802–812. [Google Scholar] [CrossRef]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef] [Green Version]
- Starling, B.R.; Kumar, P.; Lucas, A.T.; Barrow, D.; Farnan, L.; Hendrix, L.; Giovinazzo, H.; Song, G.; Gehrig, P.; Bensen, J.T.; et al. Mononuclear phagocyte system function and nanoparticle pharmacology in obese and normal weight ovarian and endometrial cancer patients. Cancer Chemother. Pharmacol. 2019, 83, 61–70. [Google Scholar] [CrossRef]
- Hamidi, M.; Azadi, A.; Rafiei, P.; Ashrafi, H. A pharmacokinetic overview of nanotechnology-based drug delivery systems: An ADME-oriented approach. Crit. Rev. Ther. Drug 2013, 30, 435–467. [Google Scholar] [CrossRef] [PubMed]
- Malaviya, P.; Shukal, D.; Vasavada, A.R. Nanotechnology-based Drug Delivery, Metabolism and Toxicity. Curr. Drug Metab. 2019, 20, 1167–1190. [Google Scholar] [CrossRef]
- Moss, D.M.; Siccardi, M. Optimizing nanomedicine pharmacokinetics using physiologically based pharmacokinetics modelling. Br. J. Pharmacol. 2014, 171, 3963–3979. [Google Scholar] [CrossRef] [PubMed]
- Zolnik, B.S.; Sadrieh, N. Regulatory perspective on the importance of ADME assessment of nanoscale material containing drugs. Adv. Drug Deliv. Rev. 2009, 61, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.T.; Madden, A.J.; Zamboni, W.C. Formulation and physiologic factors affecting the pharmacology of carrier-mediated anticancer agents. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1419–1433. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zou, P.; Tyner, K.; Lee, S. Physiologically Based Pharmacokinetic (PBPK) Modeling of Pharmaceutical Nanoparticles. AAPS J. 2017, 19, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Rajoli, R.K.R. Pharmacokinetic Modelling to Study the Biodistribution of Nanoparticles. In Mucosal Delivery of Drugs and Biologics in Nanoparticles; Muttil, P., Kunda, N., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 247–267. [Google Scholar]
- Missaoui, W.N.; Arnold, R.D.; Cummings, B.S. Toxicological status of nanoparticles: What we know and what we don’t know. Chem. Biol. Interactions 2018, 295, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chalouni, C.; Doll, S. Fate of Antibody-Drug Conjugates in Cancer Cells. J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Ji, P.; Li, Z.; Roy, P.; Sahajwalla, C.G. The Antibody Drug Absorption Following Subcutaneous or Intramuscular Administration and Its Mathematical Description by Coupling Physiologically Based Absorption Process with the Conventional Compartment Pharmacokinetic Model. J. Clin. Pharmacol. 2013, 53, 314–325. [Google Scholar] [CrossRef]
- Zolnik, B.S.; González-Fernández, Á.; Sadrieh, N.; Dobrovolskaia, M.A. Nanoparticles and the Immune System. Endocrinology 2010, 151, 458–465. [Google Scholar] [CrossRef]
- Lucas, A.T.; Madden, A.J.; Zamboni, W.C. Challenges in preclinical to clinical translation for anticancer carrier-mediated agents. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.T.; Price, L.S.; Schorzman, A.; Zamboni, W.C. Complex effects of tumor microenvironment on the tumor disposition of carrier-mediated agents. Nanomedicine 2017, 12, 2021–2042. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, W.C.; Szebeni, J.; Kozlov, S.V.; Lucas, A.T.; Piscitelli, J.A.; Dobrovolskaia, M.A. Animal models for analysis of immunological responses to nanomaterials: Challenges and considerations. Adv. Drug Deliv. Rev. 2018, 137, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Madden, A.J.; Rawal, S.; Sandison, K.; Schell, R.; Schorzman, A.; Deal, A.; Feng, L.; Ma, P.; Mumper, R.; DeSimone, J.; et al. Evaluation of the efficiency of tumor and tissue delivery of carrier-mediated agents (CMA) and small molecule (SM) agents in mice using a novel pharmacokinetic (PK) metric: Relative distribution index over time (RDI-OT). J. Nanoparticle Res. 2014, 16, 2662. [Google Scholar] [CrossRef] [PubMed]
- Schorzman, A.N.; Lucas, A.T.; Kagel, J.R.; Zamboni, W.C. Methods and Study Designs for Characterizing the Pharmacokinetics and Pharmacodynamics of Carrier-Mediated Agents. In Targeted Drug Delivery; Sirianni, R., Behkam, B., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1831. [Google Scholar]
- Song, G.; Darr, D.B.; Santos, C.M.; Ross, M.; Valdivia, A.; Jordan, J.L.; Midkiff, B.R.; Cohen, S.; Nikolaishvili-Feinberg, N.; Miller, C.R.; et al. Effects of Tumor Microenvironment Heterogeneity on Nanoparticle Disposition and Efficacy in Breast Cancer Tumor Models. Clin. Cancer Res. 2014, 20, 6083–6095. [Google Scholar] [CrossRef] [Green Version]
- Golombek, S.K.; May, J.-N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Wu, H.; Ramanathan, R.K.; Zamboni, B.A.; Strychor, S.; Ramalingam, S.; Edwards, R.P.; Friedland, D.M.; Stoller, R.G.; Belani, C.P.; Maruca, L.J.; et al. Population Pharmacokinetics of Pegylated Liposomal CKD-602 (S-CKD602) in Patients With Advanced Malignancies. J. Clin. Pharmacol. 2012, 52, 180–194. [Google Scholar] [CrossRef]
- Zamboni, W.C.; Ramalingam, S.; Friedland, D.M.; Edwards, R.P.; Stoller, R.G.; Strychor, S.; Maruca, L.; Zamboni, B.A.; Belani, C.P.; Ramanathan, R.K. Phase I and Pharmacokinetic Study of Pegylated Liposomal CKD-602 in Patients with Advanced Malignancies. Clin. Cancer Res. 2009, 15, 1466–1472. [Google Scholar] [CrossRef] [Green Version]
- Salmaso, S.; Caliceti, P. Stealth Properties to Improve Therapeutic Efficacy of Drug Nanocarriers. J. Drug Deliv. 2013, 2013, 374252. [Google Scholar] [CrossRef]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Ruoslahti, E. Specialization of tumour vasculature. Nat. Rev. Cancer 2002, 2, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Greish, K. Enhanced permeability and retention of macromolecular drugs in solid tumors: A royal gate for targeted anticancer nanomedicines. J. Drug Target. 2007, 15, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol. Biol. 2010, 624, 25–37. [Google Scholar]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 98, 1252–1276. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Charrois, G.J.; Allen, T.M. Drug release rate influences the pharmacokinetics, biodistribution, therapeutic activity, and toxicity of pegylated liposomal doxorubicin formulations in murine breast cancer. Biochim. Biophys. Acta 2004, 1663, 167–177. [Google Scholar] [CrossRef]
- Maeda, H. Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 53–71. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; Patri, A.K.; Zheng, J.; Clogston, J.D.; Ayub, N.; Aggarwal, P.; Neun, B.W.; Hall, J.B.; McNeil, S.E. Interaction of colloidal gold nanoparticles with human blood: Effects on particle size and analysis of plasma protein binding profiles. Nanomed. NBM 2009, 5, 106–117. [Google Scholar] [CrossRef] [Green Version]
- França, A.; Aggarwal, P.; Barsov, E.V.; Kozlov, S.V.; Dobrovolskaia, M.A.; González-Fernández, Á. Macrophage scavenger receptor A mediates the uptake of gold colloids by macrophages in vitro. Nanomedicine 2011, 6, 1175–1188. [Google Scholar] [CrossRef] [Green Version]
- Fam, S.Y.; Chee, C.F.; Yong, C.Y.; Ho, K.L.; Mariatulqabtiah, A.R.; Tan, W.S. Stealth Coating of Nanoparticles in Drug-Delivery Systems. Nanomaterials 2020, 10, 787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonarbourg, A.; Passirani, C.; Saulnier, P.; Benoit, J.-P. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials 2006, 27, 4356–4373. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Karmali, P.P.; Simberg, D. Role of Carbohydrate Receptors in the Macrophage Uptake of Dextran-Coated Iron Oxide Nanoparticles. Adv. Exp. Med. Biol. 2012, 733, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Geiser, M. Update on Macrophage Clearance of Inhaled Micro- and Nanoparticles. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Platt, N.; Haworth, R.; Darley, L.; Gordon, S. The Many Roles of the Class A Macrophage Scavenger Receptor. Int. Rev. Cytol. 2002, 212, 1–40. [Google Scholar] [CrossRef]
- Gough, P.J.; Gordon, S. The role of scavenger receptors in the innate immune system. Microbes Infect. 2002, 2, 305–311. [Google Scholar] [CrossRef]
- Guo, C.; Yi, H.; Yu, X.; Hu, F.; Zuo, D.; Subjeck, J.R.; Wang, X.-Y. Absence of scavenger receptor A promotes dendritic cell-mediated cross-presentation of cell-associated antigen and antitumor immune response. Immunol. Cell Biol. 2012, 90, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Pagare, P.P.; Zaidi, S.A.; Zhang, X.; Li, X.; Yu, X.; Wang, X.-Y.; Zhang, Y. Understanding molecular interactions between scavenger receptor A and its natural product inhibitors through molecular modeling studies. J. Mol. Graph. Model. 2017, 77, 189–199. [Google Scholar] [CrossRef]
- Patten, D.; Shetty, S. More Than Just a Removal Service: Scavenger Receptors in Leukocyte Trafficking. Front. Immunol. 2018, 9, 2904. [Google Scholar] [CrossRef] [Green Version]
- Allen, L.-A.H.; Aderem, A. Mechanisms of phagocytosis. Curr. Opin. Immunol. 1996, 8, 36–40. [Google Scholar] [CrossRef]
- Gray, M.; Botelho, R.J. Phagocytosis: Hungry, Hungry Cells. Methods Mol. Biol. 2017, 1519, 1–16. [Google Scholar] [PubMed]
- Swanson, J.A. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 2008, 9, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjelle, T.E.; Lovdal, T.; Berg, T. Phagosome dynamics and function. BioEssays 2000, 22, 255–263. [Google Scholar] [CrossRef]
- Chen, H.; Langer, R.; Edwards, D.A. A Film Tension Theory of Phagocytosis. J. Colloid Interface Sci. 1997, 190, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Davda, J.; Labhasetwar, V. Characterization of nanoparticle uptake by endothelial cells. Int. J. Pharm. 2002, 233, 51–59. [Google Scholar] [CrossRef]
- Panyam, J.; Labhasetwar, V. Dynamics of Endocytosis and Exocytosis of Poly(D,L-Lactide-co-Glycolide) Nanoparticles in Vascular Smooth Muscle Cells. Pharm. Res. 2003, 20, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Kamps, J.A.; Scherphof, G.L. Receptor versus non-receptor mediated clearance of liposomes. Adv. Drug Deliv. Rev. 1998, 32, 81–97. [Google Scholar] [CrossRef]
- Patel, H.M. Serum opsonins and liposomes: Their interaction and opsonophagocytosis. Crit. Rev. Ther. Drug Carr. Syst. 1992, 9, 39–90. [Google Scholar]
- Patel, H.; Moghimi, S.M. Serum-mediated recognition of liposomes by phagocytic cells of the reticuloendothelial system—The concept of tissue specificity. Adv. Drug Deliv. Rev. 1998, 32, 45–60. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Lipoprotein Metabolism in the Macrophage: Implications for Cholesterol Deposition in Atherosclerosis. Annu. Rev. Biochem. 1983, 52, 223–261. [Google Scholar] [CrossRef] [Green Version]
- Linehan, S.A.; Martinez-Pomares, L.; Gordon, S. Mannose Receptor and Scavenger Receptor: Two Macrophage Pattern Recognition Receptors with Diverse Functions in Tissue Homeostasis and Host Defense. Adv. Exp. Med. Biol. 2000, 479, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chnari, E.; Nikitczuk, J.S.; Wang, J.; Uhrich, K.E.; Moghe, P.V. Engineered Polymeric Nanoparticles for Receptor-Targeted Blockage of Oxidized Low Density Lipoprotein Uptake and Atherogenesis in Macrophages. Biomacromolecules 2006, 7, 1796–1805. [Google Scholar] [CrossRef]
- Kamps, J.A.; Morselt, H.W.; Scherphof, G.L. Uptake of Liposomes Containing Phosphatidylserine by Liver Cells In Vivo and by Sinusoidal Liver Cells in Primary Culture: In Vivo–In Vitro Differences. Biochem. Biophys. Res. Commun. 1999, 256, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C. Recognition by macrophages and liver cells of opsonized phospholipid vesicles and phospholipid headgroups. Pharm. Res. 2001, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hu, Q.; Song, Y.K. Liposome clearance from blood: Different animal species have different mechanisms. Biochim. Biophys. Acta 1995, 1240, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Liu, D. Serum independent liposome uptake by mouse liver. Biochim. Biophys. Acta 1996, 1278, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [Green Version]
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Szebeni, J. Complement Activation-Related Pseudoallergy Caused by Liposomes, Micellar Carriers of Intravenous Drugs, and Radiocontrast Agents. Crit. Rev. Ther. Drug Carr. Syst. 2001, 18, 567–606. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy: A stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 2014, 61, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Bedőcs, P.; Rozsnyay, Z.; Weiszhár, Z.; Urbanics, R.; Rosivall, L.; Cohen, R.; Garbuzenko, O.; Báthori, G.; Tóth, M.; et al. Liposome-induced complement activation and related cardiopulmonary distress in pigs: Factors promoting reactogenicity of Doxil and AmBisome. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haney, M.S.; Bohlen, C.J.; Morgens, D.W.; Ousey, J.A.; Barkal, A.A.; Tsui, C.K.; Ego, B.K.; Levin, R.; Kamber, R.A.; Collins, H.; et al. Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens. Nat. Genet. 2018, 50, 1716–1727. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between Defective Endothelial Cells Explain Tumor Vessel Leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612. [Google Scholar] [CrossRef] [Green Version]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; Macmillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef]
- Khawar, I.A.; Kim, J.H.; Kuh, H.-J. Improving drug delivery to solid tumors: Priming the tumor microenvironment. J. Control. Release 2015, 201, 78–89. [Google Scholar] [CrossRef]
- Miao, L.; Lin, C.M.; Huang, L. Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors. J. Control. Release 2015, 219, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Overchuk, M.; Zheng, G. Overcoming obstacles in the tumor microenvironment: Recent advancements in nanoparticle delivery for cancer theranostics. Biomaterials 2018, 156, 217–237. [Google Scholar] [CrossRef]
- Curtis, L.T.; Frieboes, H.B. The Tumor Microenvironment as a Barrier to Cancer Nanotherapy. Adv. Exp. Med. Biol. 2016, 936, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Wong, A.H.-K.; Jain, R.K. Vascular Normalization as a Therapeutic Strategy for Malignant and Nonmalignant Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Z.; Gao, Y.; Wientjes, M.G.; Au, J.L.-S. Improving delivery and efficacy of nanomedicines in solid tumors: Role of tumor priming. Nanomedicine 2011, 6, 1605–1620. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar]
- Tong, R.T.; Boucher, Y.; Kozin, S.V.; Winkler, F.; Hicklin, D.J.; Jain, R.K. Vascular Normalization by Vascular Endothelial Growth Factor Receptor 2 Blockade Induces a Pressure Gradient Across the Vasculature and Improves Drug Penetration in Tumors. Cancer Res. 2004, 64, 3731–3736. [Google Scholar] [CrossRef] [Green Version]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol. Rev. 2001, 53, 283–318. [Google Scholar]
- Hendry, S.A.; Farnsworth, R.H.; Solomon, B.; Achen, M.G.; Stacker, S.A.; Fox, S.B. The Role of the Tumor Vasculature in the Host Immune Response: Implications for Therapeutic Strategies Targeting the Tumor Microenvironment. Front. Immunol. 2016, 7, 621. [Google Scholar] [CrossRef]
- Kim, J.H.; Suh, J.-Y.; Woo, D.-C.; Sung, Y.S.; Son, W.-C.; Choi, Y.S.; Pae, S.J.; Kim, J.K. Difference in the intratumoral distributions of extracellular-fluid and intravascular MR contrast agents in glioblastoma growth. NMR Biomed. 2016, 29, 1688–1699. [Google Scholar] [CrossRef]
- Sarin, H.; Kanevsky, A.S.; Wu, H.; Sousa, A.A.; Wilson, C.M.; Aronova, M.A.; Griffiths, G.L.; Leapman, R.D.; Vo, H.Q. Physiologic upper limit of pore size in the blood-tumor barrier of malignant solid tumors. J. Transl. Med. 2009, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.; Wang, L.; Lin, J. Interaction Pathways between Plasma Membrane and Block Copolymer Micelles. Biomacromolecules 2017, 18, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hoang, B.; Fonge, H.; Reilly, R.M.; Allen, C. In Vivo Distribution of Polymeric Nanoparticles at the Whole-Body, Tumor, and Cellular Levels. Pharm. Res. 2010, 27, 2343–2355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Zhao, Y.; Wang, G.; Zhou, F. Plasma and cellular pharmacokinetic considerations for the development and optimization of antitumor block copolymer micelles. Expert Opin. Drug Deliv. 2015, 12, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Guo, S.; Deng, C. Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Jain, R.K.; Langer, R. Engineering and physical sciences in oncology: Challenges and opportunities. Nat. Rev. Cancer 2017, 17, 659–675. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Brown, E.B.; Boucher, Y.; Nasser, S.; Jain, R.K. Measurement of macromolecular diffusion coefficients in human tumors. Microvasc. Res. 2004, 67, 231–236. [Google Scholar] [CrossRef]
- Buescher, C.; Hoo, K.A.; Janssen, H. An Experimental Approach to Measure Mass Diffusion in Rat Tumor Tissue. IEEE Trans. Biomed. Eng. 2008, 55, 1831–1839. [Google Scholar] [CrossRef]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar]
- Goodman, T.T.; Olive, P.L.; Pun, S.H. Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int. J. Nanomed. 2007, 2, 265–274. [Google Scholar]
- Goins, B.; Phillips, W.T.; Bao, A. Strategies for improving the intratumoral distribution of liposomal drugs in cancer therapy. Expert Opin. Drug Deliv. 2016, 13, 873–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, L.; Huang, L. Exploring the Tumor Microenvironment with Nanoparticles. Cancer Treat. Res. 2015, 166, 193–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogsteen, I.J.; Marres, H.A.M.; Bussink, J.; Van Der Kogel, A.J.; Kaanders, J.H.A.M. Tumor microenvironment in head and neck squamous cell carcinomas: Predictive value and clinical relevance of hypoxic markers. A review. Head Neck 2007, 29, 591–604. [Google Scholar] [CrossRef]
- Ljungkvist, A.S.E.; Bussink, J.; Kaanders, J.H.A.M.; Van Der Kogel, A.J. Dynamics of Tumor Hypoxia Measured with Bioreductive Hypoxic Cell Markers. Radiat. Res. 2007, 167, 127–145. [Google Scholar] [CrossRef] [Green Version]
- Ljungkvist, A.S.E.; Bussink, J.; Rijken, P.F.J.W.; Kaanders, J.H.A.M.; Van Der Kogel, A.J.; Denekamp, J. Vascular architecture, hypoxia, and proliferation in first-generation xenografts of human head-and-neck squamous cell carcinomas. Int. J. Radiat. Oncol. 2002, 54, 215–228. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. Cancer-associated fibroblasts: Secretions, interactions, and therapy. J. Cell. Biochem. 2019, 120, 2791–2800. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Song, X.-T.; Gottschalk, S. Cancer-associated fibroblasts as targets for immunotherapy. Immunotherapy 2012, 4, 1129–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Räsänen, K.; Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010, 316, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Truffi, M.; Mazzucchelli, S.; Bonizzi, A.; Sorrentino, L.; Allevi, R.; Vanna, R.; Morasso, C.; Corsi, F. Nano-Strategies to Target Breast Cancer-Associated Fibroblasts: Rearranging the Tumor Microenvironment to Achieve Antitumor Efficacy. Int. J. Mol. Sci. 2019, 20, 1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ho, S.; Li, B.; Nie, G.; Li, S. Modulating the tumor microenvironment with new therapeutic nanoparticles: A promising paradigm for tumor treatment. Med. Res. Rev. 2020, 40, 1084–1102. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ernsting, M.J.; Undzys, E.; Holwell, N.; Foltz, W.D.; Li, S.-D. Docetaxel Conjugate Nanoparticles That Target α-Smooth Muscle Actin–Expressing Stromal Cells Suppress Breast Cancer Metastasis. Cancer Res. 2013, 73, 4862–4871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, L.; Liu, Q.; Lin, C.M.; Luo, C.; Wang, Y.; Liu, L.; Yin, W.; Hu, S.; Kim, W.Y.; Huang, L. Targeting Tumor-Associated Fibroblasts for Therapeutic Delivery in Desmoplastic Tumors. Cancer Res. 2017, 77, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure — an obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Rippe, B.; Haraldsson, B. Transport of macromolecules across microvascular walls: The two-pore theory. Physiol. Rev. 1994, 74, 163–219. [Google Scholar] [CrossRef] [PubMed]
- Baronzio, G.; Parmar, G.; Baronzio, M. Overview of Methods for Overcoming Hindrance to Drug Delivery to Tumors, with Special Attention to Tumor Interstitial Fluid. Front. Oncol. 2015, 5, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stapleton, S.; Milosevic, M.; Tannock, I.F.; Allen, C.; Jaffray, D.A. The intra-tumoral relationship between microcirculation, interstitial fluid pressure and liposome accumulation. J. Control. Release 2015, 211, 163–170. [Google Scholar] [CrossRef]
- Zhao, Y.; Cao, J.; Melamed, A.; Worley, M.; Gockley, A.; Jones, D.; Nia, H.T.; Zhang, Y.; Stylianopoulos, T.; Kumar, A.S.; et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2019, 116, 2210–2219. [Google Scholar]
- Zamboni, W.C.; Strychor, S.; Joseph, E.; Walsh, D.R.; Zamboni, B.A.; Parise, R.A.; Tonda, M.E.; Yu, N.Y.; Engbers, C.; Eiseman, J.L. Plasma, Tumor, and Tissue Disposition of STEALTH Liposomal CKD-602 (S-CKD602) and Nonliposomal CKD-602 in Mice Bearing A375 Human Melanoma Xenografts. Clin. Cancer Res. 2007, 13, 7217–7223. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, S.K.; Lucas, A.T.; Caron, W.P.; Song, G.; Lay, J.C.; Zamboni, W.C. Bidirectional Interaction between Nanoparticles and Carrier-Mediated Agents and Cells of the Mononuclear Phagocytic System. In Handbook of Immunological Properties of Engineered Nanomaterials, 2nd ed.; Dobrovolskaia, M.A., McNeil, S.E., Eds.; World Scientific: Singapore, 2016; Volume 5, pp. 1–41. [Google Scholar]
- Lucas, A.T.; White, T.F.; Deal, A.M.; Herity, L.B.; Song, G.; Santos, C.M.; Zamboni, W.C. Profiling the relationship between tumor-associated macrophages and pharmacokinetics of liposomal agents in preclinical murine models. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 471–482. [Google Scholar] [CrossRef]
- Alizadeh, D.; Zhang, L.; Hwang, J.; Schluep, T.; Badie, B. Tumor-associated macrophages are predominant carriers of cyclodextrin-based nanoparticles into gliomas. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 382–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Claasen, R.H.H.; Kluin, P.M.; Fleuren, G.J. Tumor infiltrating cells in human cancer. On the possible role of CD16+ macrophages in antitumor cytotoxicity. Lab. Investig. 1992, 67, 166–174. [Google Scholar]
- Al-Sarireh, B.; Eremin, O. Tumour-associated macrophages (TAMS): Disordered function, immune suppression and progressive tumour growth. J. R. Coll. Surg. Edinb. 2000, 45, 1–16. [Google Scholar] [PubMed]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.A.; Baker-Lee, C.; Kennedy, J.; Lai, M.S.; De Vries, P.; Buhler, K.; Singer, J.W. In Vitro and In Vivo metabolism of paclitaxel poliglumex: Identification of metabolites and active proteases. Cancer Chemother. Pharmacol. 2007, 59, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.F.; Esparza-Coss, E.; Wen, X.; Ng, C.S.; Daniel, S.L.; Price, R.E.; Rivera, B.; Charnsangavej, C.; Gelovani, J.G.; Li, C. Magnetic Resonance Imaging of Therapy-Induced Necrosis Using Gadolinium-Chelated Polyglutamic Acids. Int. J. Radiat. Oncol. 2007, 68, 830–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiyath, S.M.; Rasul, M.; Lee, B.; Wei, G.; Lamba, G.; Liu, D. Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: A meta-analysis. Exp. Hematol. Oncol. 2012, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Li, D.; Han, J.; Zhuang, X.; Ding, J. Schiff base bond-linked polysaccharide–doxorubicin conjugate for upregulated cancer therapy. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 76, 1121–1128. [Google Scholar] [CrossRef]
- Li, D.; Han, J.; Ding, J.; Chen, L.; Chen, X. Acid-sensitive dextran prodrug: A higher molecular weight makes a better efficacy. Carbohydr. Polym. 2017, 161, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.S.; Colbern, G.T.; Working, P.K.; Engbers, C.; Amantea, M.A. Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, pegylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemother. Pharmacol. 1999, 43, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Lewanski, C.R.; Northcote, A.D.; Whittaker, J.; Wellbank, H.; Vile, R.G.; Peters, A.M.; Stewart, J.S.W. Phase I–II study of pegylated liposomal cisplatin (SPI-077) in patients with inoperable head and neck cancer. Ann. Oncol: Off. J. Eur. Soc. Med. Oncol. 2001, 12, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Seetharamu, N.; Kim, E.; Hochster, H.; Martin, F.; Muggia, F. Phase II study of liposomal cisplatin (SPI-77) in platinum-sensitive recurrences of ovarian cancer. Anticancer Res. 2010, 30, 541–545. [Google Scholar] [PubMed]
- White, S.C.; Lorigan, P.; Margison, G.P.; Margison, J.M.; Martin, F.; Thatcher, N.; Anderson, H.; Ranson, M. Phase II study of SPI-77 (sterically stabilised liposomal cisplatin) in advanced non-small-cell lung cancer. Br. J. Cancer 2006, 95, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Terwogt, J.M.M.; Groenewegen, G.; Pluim, D.; Maliepaard, M.; Tibben, M.M.; Huisman, A.; Huinink, W.W.T.B.; Schot, M.; Welbank, H.; Voest, E.E.; et al. Phase I and pharmacokinetic study of SPI-77, a liposomal encapsulated dosage form of cisplatin. Cancer Chemother. Pharmacol. 2002, 49, 201–210. [Google Scholar] [CrossRef]
- Zamboni, W.C.; Gervais, A.C.; Egorin, M.J.; Schellens, J.H.M.; Zuhowski, E.G.; Pluim, D.; Joseph, E.; Hamburger, D.R.; Working, P.K.; Colbern, G.; et al. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother. Pharmacol. 2004, 53, 329–336. [Google Scholar] [CrossRef]
- Brown, E.; McKee, T.; diTomaso, E.; Pluen, A.; Seed, B.; Boucher, Y.; Jain, R.K. Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation. Nat. Med. 2003, 9, 796–800. [Google Scholar] [CrossRef]
- Choi, J.; Credit, K.; Henderson, K.; Deverkadra, R.; He, Z.; Wiig, H.; Vanpelt, H.; Flessner, M.F. Intraperitoneal Immunotherapy for Metastatic Ovarian Carcinoma: Resistance of Intratumoral Collagen to Antibody Penetration. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1906–1912. [Google Scholar] [CrossRef] [Green Version]
- Perentes, J.Y.; McKee, T.D.; Ley, C.D.; Mathiew, H.; Dawson, M.; Padera, T.P.; Munn, L.L.; Jain, R.K.; Boucher, Y. In Vivo imaging of extracellular matrix remodeling by tumor-associated fibroblasts. Nat. Methods 2009, 6, 143–145. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, C.D.L.; Lundstrøm, L.M.; Frengen, J.; Eikenes, L.; Bruland, Ø.S.; Kaalhus, O.; Hjelstuen, M.H.B.; Brekken, C. Radiation Improves the Distribution and Uptake of Liposomal Doxorubicin (Caelyx) in Human Osteosarcoma Xenografts. Cancer Res. 2004, 64, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzoor, A.A.; Lindner, L.H.; Landon, C.D.; Park, J.-Y.; Simnick, A.J.; Dreher, M.R.; Das, S.; Hanna, G.; Park, W.; Chilkoti, A.; et al. Overcoming Limitations in Nanoparticle Drug Delivery: Triggered, Intravascular Release to Improve Drug Penetration into Tumors. Cancer Res. 2012, 72, 5566–5575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needham, D.; Anyarambhatla, G.; Kong, G.; Dewhirst, M.W. A new temperature-sensitive liposome for use with mild hyperthermia: Characterization and testing in a human tumor xenograft model. Cancer Res. 2000, 60, 1197–1201. [Google Scholar] [PubMed]
- Zhao, Y.; Alakhova, D.Y.; Kim, J.O.; Bronich, T.K.; Kabanov, A.V. A simple way to enhance Doxil® therapy: Drug release from liposomes at the tumor site by amphiphilic block copolymer. J. Control. Release 2013, 168, 61–69. [Google Scholar] [CrossRef] [Green Version]
- La-Beck, N.M.; Liu, X.; Wood, L.M. Harnessing Liposome Interactions With the Immune System for the Next Breakthrough in Cancer Drug Delivery. Front. Pharmacol. 2019, 10, 220. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Rajan, R.; Sabnani, M.K.; Mavinkurve, V.; Shmeeda, H.; Mansouri, H.; Bonkoungou, S.; Le, A.D.; Wood, L.M.; Gabizon, A.A.; La-Beck, N.M. Liposome-induced immunosuppression and tumor growth is mediated by macrophages and mitigated by liposome-encapsulated alendronate. J. Control. Release 2018, 271, 139–148. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Tsuchiya, S.; Aramaki, Y. Involvement of ERK, a MAP kinase, in the production of TGF-beta by macrophages treated with liposomes composed of phosphatidylserine. Biochem. Biophys. Res. Commun. 2004, 324, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.T.; Herity, L.B.; Kornblum, Z.A.; Madden, A.J.; Gabizon, A.; Kabanov, A.V.; Ajamie, R.T.; Bender, D.M.; Kulanthaivel, P.; Sanchez-Felix, M.; et al. Pharmacokinetic and screening studies of the interaction between mononuclear phagocyte system and nanoparticle formulations and colloid forming drugs. Int. J. Pharm. 2017, 526, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Caron, W.P.; Lay, J.C.; Fong, A.M.; La-Beck, N.M.; Kumar, P.; Newman, S.E.; Zhou, H.; Monaco, J.H.; Clarke-Pearson, D.L.; Brewster, W.R.; et al. Translational Studies of Phenotypic Probes for the Mononuclear Phagocyte System and Liposomal Pharmacology. J. Pharmacol. Exp. Ther. 2013, 347, 599–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falandry, C.; Brain, E.; Bonnefoy, M.; Mefti, F.; Jovenin, N.; Rigal, O.; Guillem, O.; El Kouri, C.; Uwer, L.; Abadie-Lacourtoisie, S.; et al. Impact of geriatric risk factors on pegylated liposomal doxorubicin tolerance and efficacy in elderly metastatic breast cancer patients: Final results of the DOGMES multicentre GINECO trial. Eur. J. Cancer 2013, 49, 2806–2814. [Google Scholar] [CrossRef]
- Sostelly, A.; Henin, E.; Chauvenet, L.; Hardy-Bessard, A.-C.; Tallec, V.J.-L.; Kirsher, S.; Leyronnas, C.; Ligeza-Poisson, C.; Ramdane, S.; Salavt, J.; et al. Can we predict chemo-induced hematotoxicity in elderly patients treated with pegylated liposomal doxorubicin? Results of a population-based model derived from the DOGMES phase II trial of the GINECO. J. Geriatr. Oncol. 2013, 4, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Grobner, T. Gadolinium—A specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol. Dial. Transplant. 2006, 21, 1104–1108. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.-D.; Paudel, R.; Liu, J.; Ma, C.; Zhang, Z.-S.; Zhou, S.-K. MRI contrast agents: Classification and application (Review). Int. J. Mol. Med. 2016, 38, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, S. Gadolinium toxicity: Iron and ferroportin as central targets. Magn. Reson. Imaging 2016, 34, 1373–1376. [Google Scholar] [CrossRef]
- Miyamoto, J.; Tanikawa, A.; Igarashi, A.; Hataya, H.; Kobayashi, K.; Ikegami, M.; Sotome, A.; Nagai, Y.; Kameyama, K.; Ishiko, A. Detection of Iron Deposition in Dermal Fibrocytes Is a Useful Tool for Histologic Diagnosis of Nephrogenic Systemic Fibrosis. Am. J. Dermatopathol. 2011, 33, 271–276. [Google Scholar] [CrossRef]
- Song, G.; Tarrant, T.K.; White, T.F.; Barrow, D.A.; Santos, C.M.; Timoshchenko, R.G.; Hanna, S.K.; Ramanathan, R.K.; Lee, C.R.; Bae-Jump, V.; et al. Roles of chemokines CCL2 and CCL5 in the pharmacokinetics of PEGylated liposomal doxorubicin in vivo and in patients with recurrent epithelial ovarian cancer. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Felici, A.; Verweij, J.; Sparreboom, A. Dosing strategies for anticancer drugs: The good, the bad and body-surface area. Eur. J. Cancer 2002, 38, 1677–1684. [Google Scholar] [CrossRef]
- Mahmood, I. Interspecies scaling: Predicting clearance of anticancer drugs in humans. A comparative study of three different approaches using body weight or body surface area. Eur. J. Drug Metab. Pharmacokinet. 1996, 21, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Sahneh, F.D.; Scoglio, C.M.; Monteiro-Riviere, N.A.; Riviere, J.E. Predicting the impact of biocorona formation kinetics on interspecies extrapolations of nanoparticle biodistribution modeling. Nanomedicine 2015, 10, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Caron, W.P.; Clewell, H.; Dedrick, R.; Ramanathan, R.K.; Davis, W.L.; Yu, N.; Tonda, M.; Schellens, J.H.; Beijnen, J.H.; Zamboni, W.C. Allometric scaling of pegylated liposomal anticancer drugs. J. Pharmacokinet. Pharmacodyn. 2011, 38, 653–669. [Google Scholar] [CrossRef]
- Caron, W.P.; Morgan, K.P.; Zamboni, B.A.; Zamboni, W.C. A Review of Study Designs and Outcomes of Phase I Clinical Studies of Nanoparticle Agents Compared with Small-Molecule Anticancer Agents. Clin. Cancer Res. 2013, 19, 3309–3315. [Google Scholar] [CrossRef] [Green Version]
- Price, L.S.L.; Stern, S.T.; Deal, A.M.; Kabanov, A.V.; Zamboni, W.C. A reanalysis of nanoparticle tumor delivery using classical pharmacokinetic metrics. Sci. Adv. 2020, 6, eaay9249. [Google Scholar] [CrossRef]
- Schell, R.F.; Sidone, B.J.; Caron, W.P.; Walsh, M.D.; White, T.F.; Zamboni, B.A.; Ramanathan, R.K.; Zamboni, W.C. Meta-analysis of inter-patient pharmacokinetic variability of liposomal and non-liposomal anticancer agents. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traditional Nanomedicines | ||
| Generic Name | Brand Name | Approval Date |
| Liposomal doxorubicin | Doxil | 1995-11-17 |
| Liposomal daunorubicin (discontinued) | DaunoXome | 1996-04-08 |
| Liposomal cytarabine (discontinued) | DepoCyt | 1999-08-01 |
| Nab-paclitaxel | Abraxane | 2005-01-07 |
| Liposomal vincristine sulfate | Marqibo | 2012-08-09 |
| Liposomal irinotecan | Onivyde | 2015-10-22 |
| Liposomal cytarabine & daunorubicin | Vyxeos | 2017-08-23 |
| Therapeutic Conjugates | ||
| Generic Name | Brand Name | Approval Date |
| Pegfilgrastim | Neulasta | 2002-01-31 |
| Pegaspariginase | Oncaspar | 2006-07-24 |
| Antibody-Drug Conjugates | ||
| Generic Name | Brand Name | Approval Date |
| Brentuximab vedotin | Adcetris | 2011-08-19 |
| Ado-trastuzumab emtansine | Kadcyla | 2013-02-22 |
| Inotuzumab ozogamicin | Besponsa | 2017-08-17 |
| Gemtuzumab ozogamicin | Mylotarg | 2017-09-01 |
| Moxetumomab pasudotox-tdfk | Lumoxiti | 2018-09-23 |
| Polatuzumab vedotin | Polivy | 2019-06-10 |
| Enfortumab vedotin | Padcev | 2019-12-18 |
| Trastuzumab deruxtecan | Enhertu | 2019-12-20 |
| Sacituzumab govitecan | Trodelvy | 2020-04-22 |
| Belantamab mafodotin-blmf | Blenrep | 2020-08-05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piscatelli, J.A.; Ban, J.; Lucas, A.T.; Zamboni, W.C. Complex Factors and Challenges that Affect the Pharmacology, Safety and Efficacy of Nanocarrier Drug Delivery Systems. Pharmaceutics 2021, 13, 114. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13010114
Piscatelli JA, Ban J, Lucas AT, Zamboni WC. Complex Factors and Challenges that Affect the Pharmacology, Safety and Efficacy of Nanocarrier Drug Delivery Systems. Pharmaceutics. 2021; 13(1):114. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13010114
Chicago/Turabian StylePiscatelli, Joseph A., Jisun Ban, Andrew T. Lucas, and William C. Zamboni. 2021. "Complex Factors and Challenges that Affect the Pharmacology, Safety and Efficacy of Nanocarrier Drug Delivery Systems" Pharmaceutics 13, no. 1: 114. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13010114