
Discovery and Validation of a Compound to Target Ewing’s Sarcoma

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Primary Antibodies
2.3. Cell Lines and Cell Culture
2.4. High-Throughput Screening
2.5. Imaging Caspase 3/7 Activation
2.6. Annexin V Staining
2.7. Z-VAD-FMK Rescue Experiments
2.8. Immunoblot Analyses
2.9. Histology, Immunohistochemistry, and Slide Evaluation
2.10. Immunocytochemistry
2.11. Microtubule Polymerization Assay
2.12. Cell Cycle Analysis
2.13. Effect of ML111 on SK-N-MC Cell Proteomics
2.13.1. Sample Preparation
2.13.2. Basic Reverse-Phase Chromatography
2.13.3. Mass Spectrometry
2.13.4. Data Analysis and Protein Quantification
2.14. Preparation of ML111-Based Nanoparticles (ML111-NP)
2.15. Characterization of ML111-Based Nanoparticles
2.15.1. In Vivo Assessment of the Toxicity of ML111-NP
2.15.2. Physical Parameters
2.15.3. In Vitro ML111 Release
2.15.4. Cellular Toxicity Assay of ML111-NP
2.15.5. Cellular Internalization of ML111-NP
2.16. In Vivo Efficacy of ML111-Based Nanoparticles (ML111-NP)
2.17. Statistical Analysis
3. Results
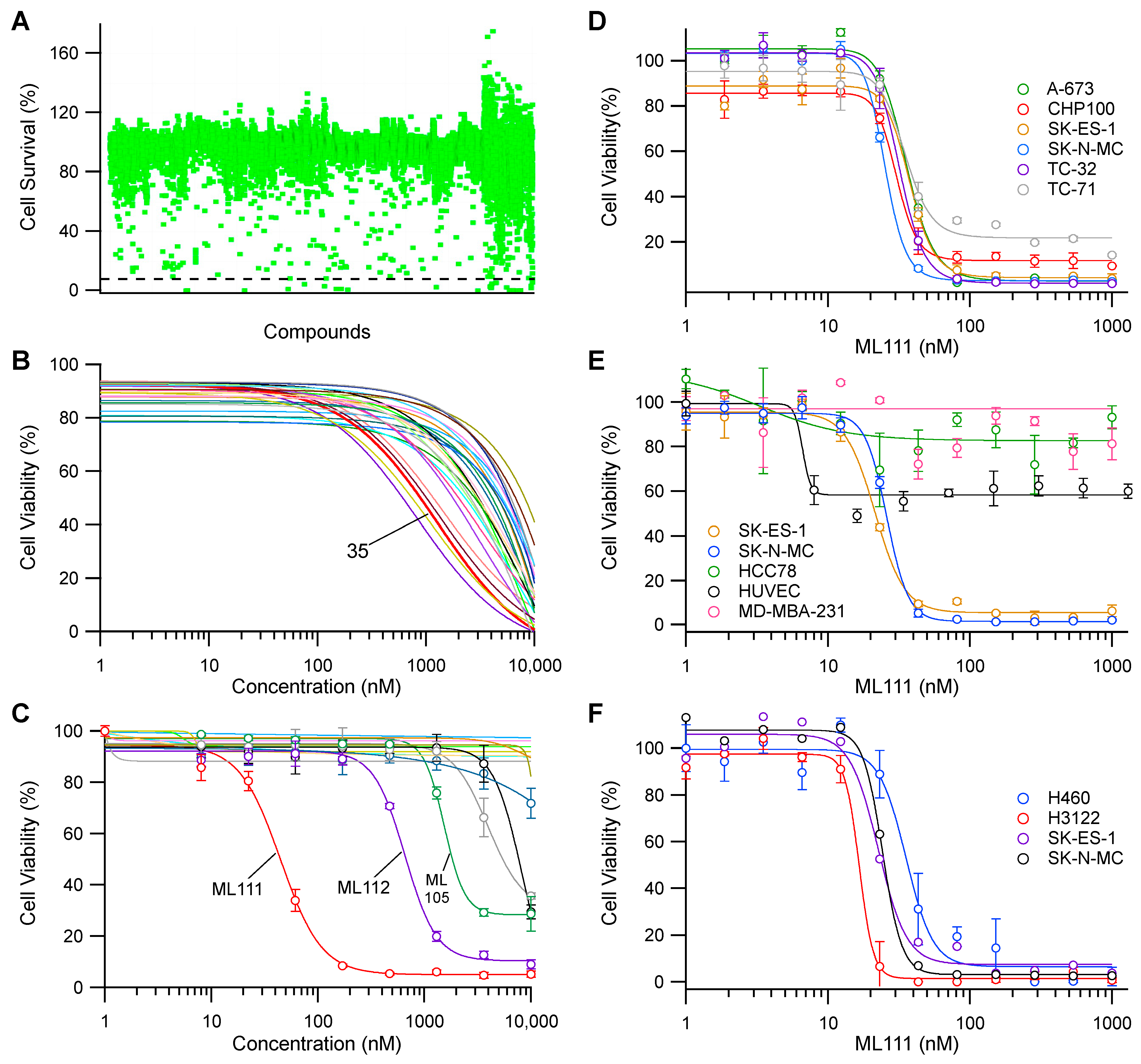
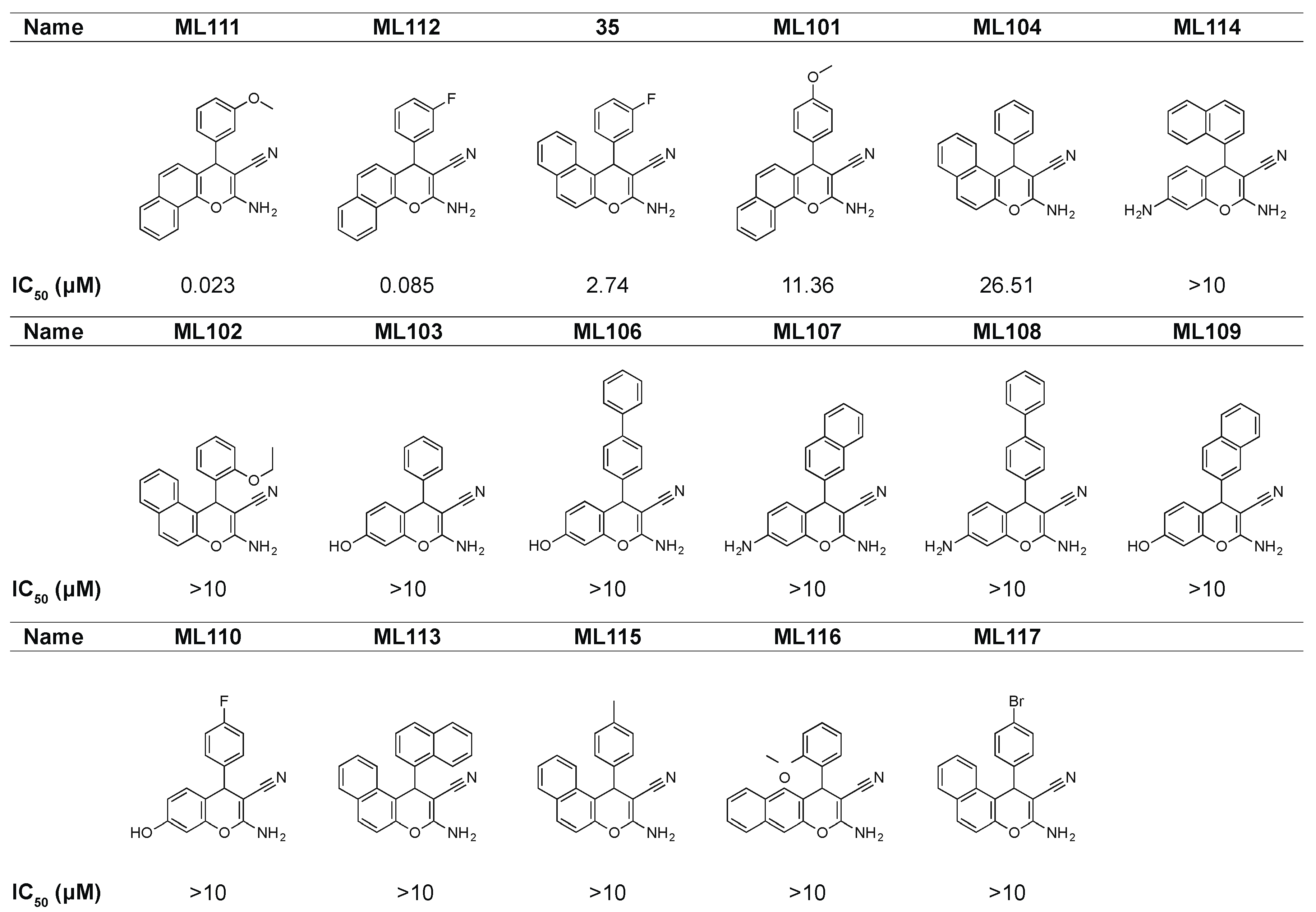
3.1. Discovery of Lead Compound Using High-Throughput Functional Screening
3.2. ML111 Inhibits Viability of Ewing’s Sarcoma Cell Lines Harboring Both Type I and II Fusion Proteins
3.3. Single Enantiomer of ML111 Responsible for Anti-Viability Effect in Ewing’s Sarcoma Cells
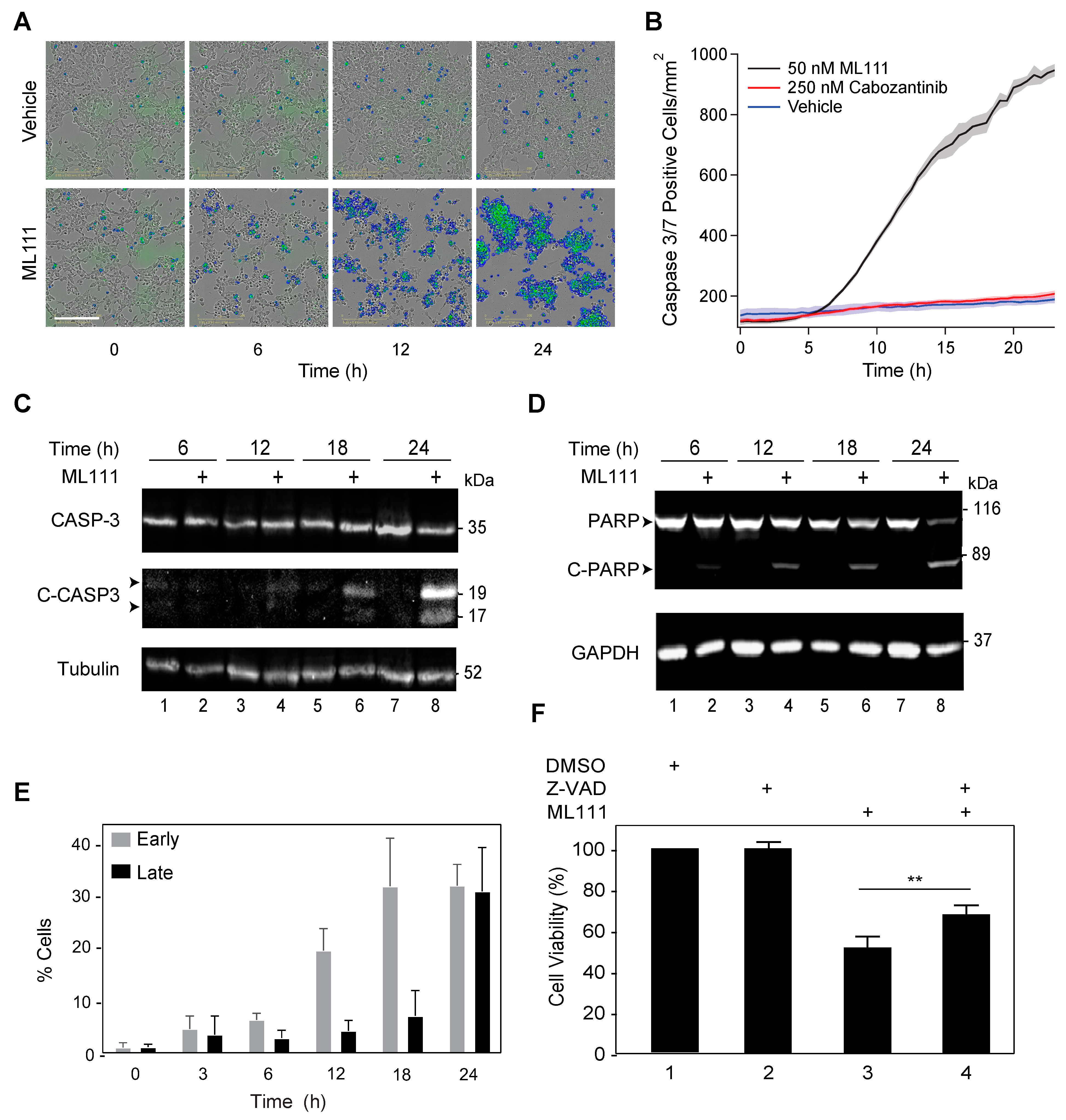
3.4. ML111 Induces Caspase-3/7-Dependent Apoptosis
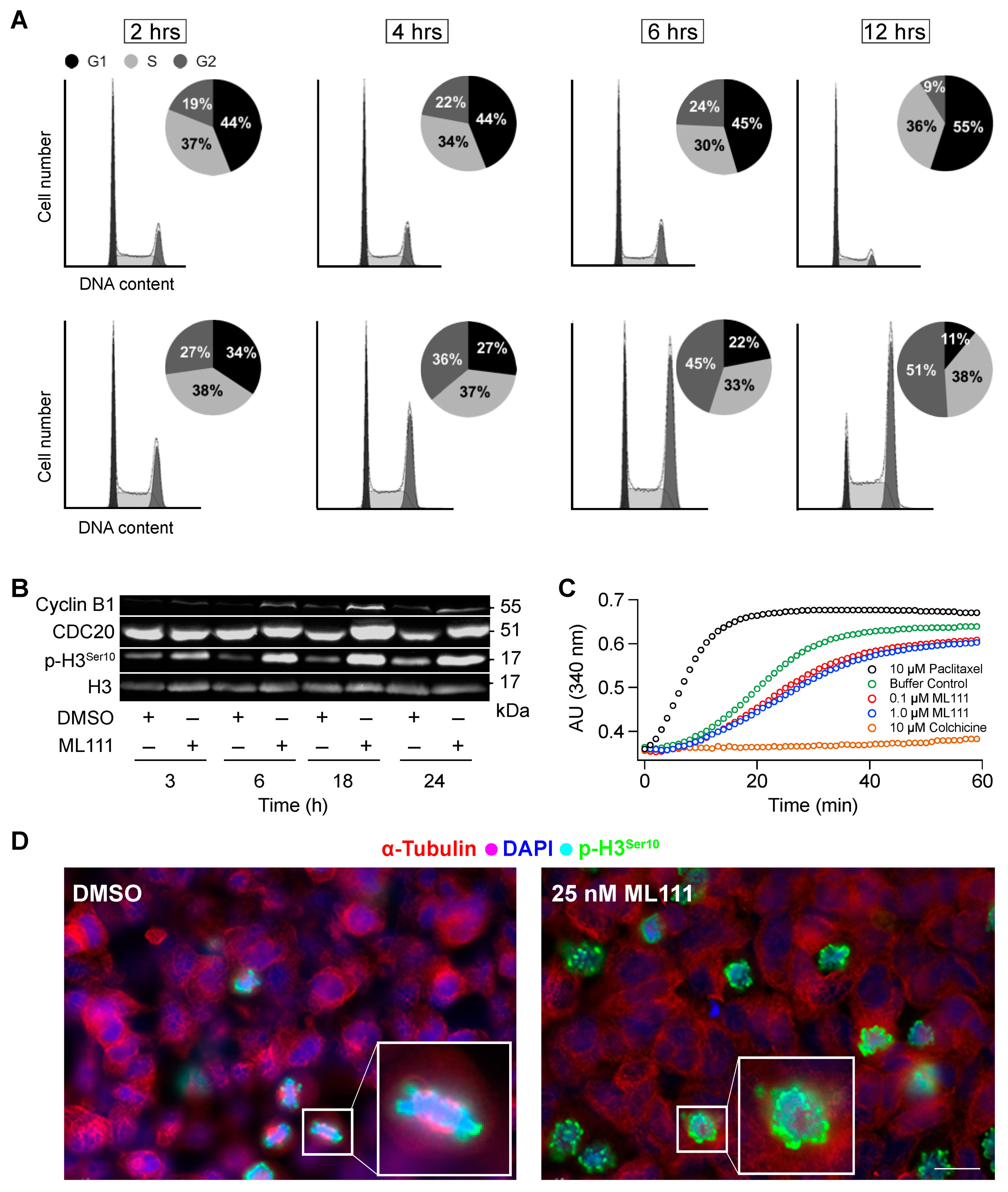
3.5. ML111 Induces Prometaphase Arrest in SK-N-MC Cells Independent of an Effect on Microtubule Dynamics
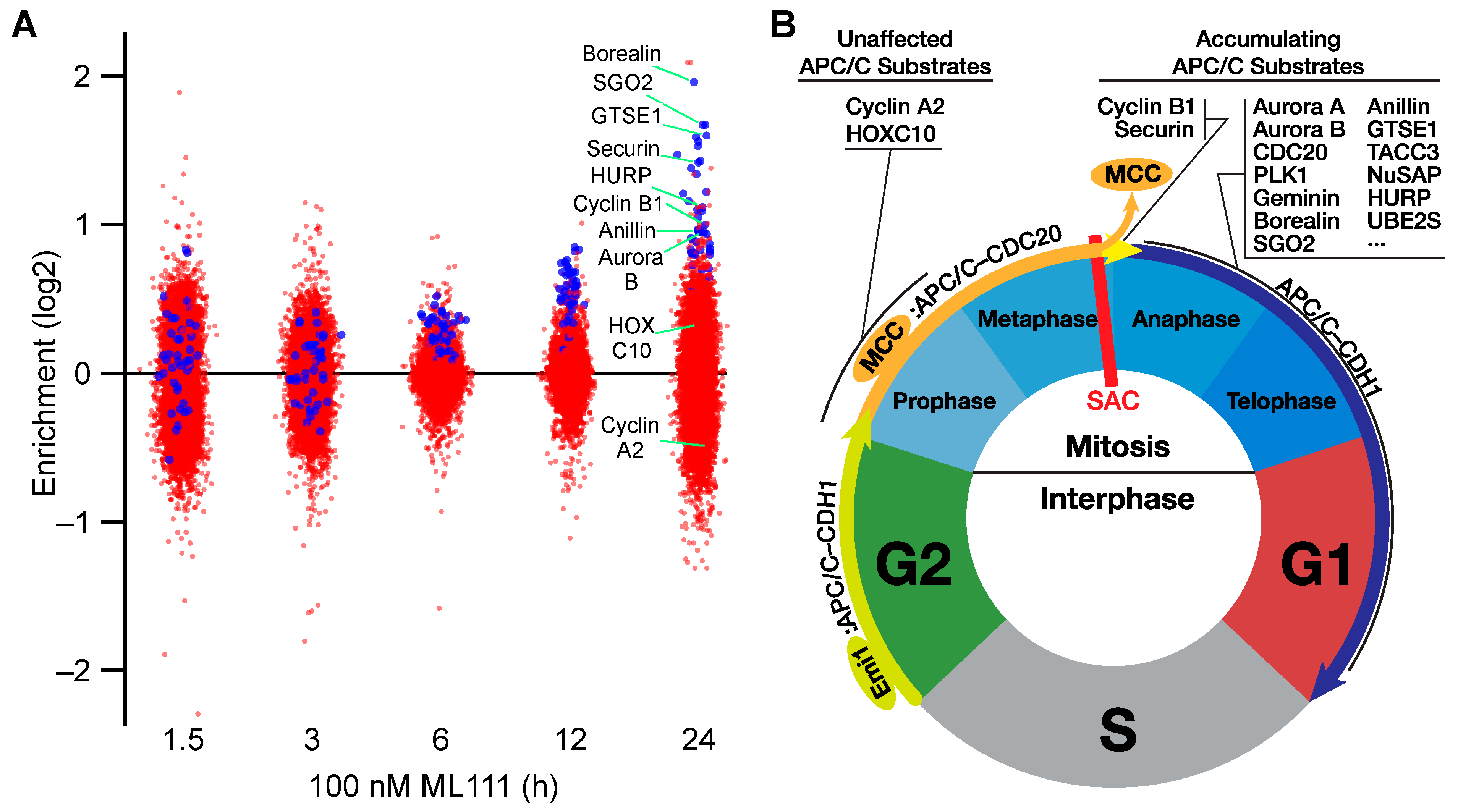
3.6. Quantitative Proteomic Analysis of ML111-Treated Ewing’s Sarcoma Cells
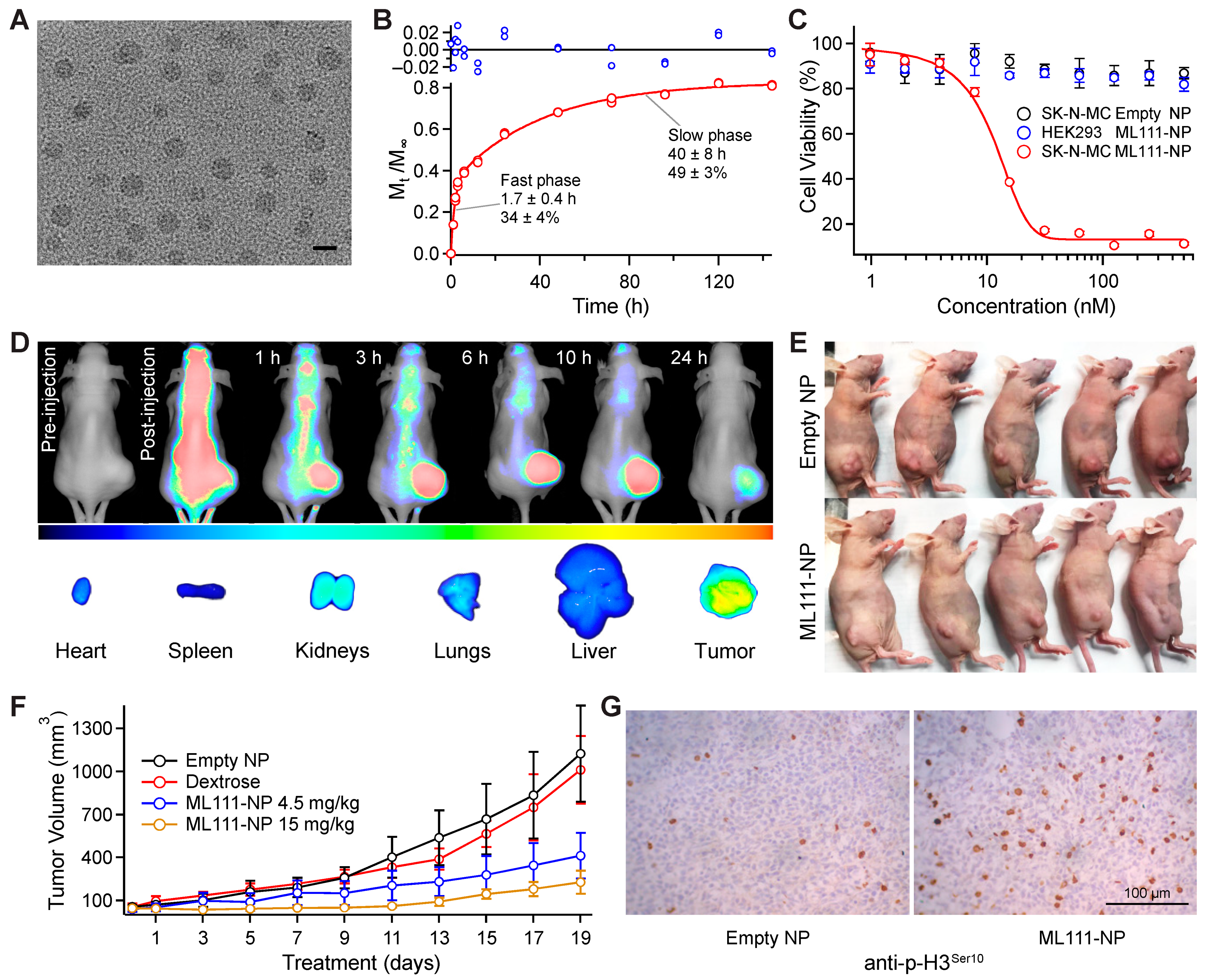
3.7. Formulation of Nanoparticle-Based Drug Delivery System for ML111
3.8. Effect of ML111-NP on Viability of SK-N-MC and HEK293 Cells
3.9. ML111 Decreases Tumor Growth in a Mouse Xenograft Model of Ewing’s Sarcoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1.5 | 3 | 6 | 12 | 24 h | Protein | Uniprot Acc | Gene Symbol | APC/C Substrate References |
|---|---|---|---|---|---|---|---|---|
| Accumulating Cell Cycle Proteins | ||||||||
| 0.29 | −0.21 | 0.38 | 0.74 | 1.96 | Borealin | Q53HL2 | CDCA8 | [59] |
| 0.12 | 0.11 | 0.40 | 0.85 | 1.67 | SGO2 | Q562F6 | SGO2 | [60] |
| 0.83 | 0.19 | 0.16 | 0.82 | 1.67 | Histone H3.2 | Q71DI3 | HIST2H3A | [61] |
| 0.32 | −0.25 | 0.26 | 0.59 | 1.60 | GTSE1 | Q9NYZ3 | GTSE1 | [62] |
| 0.27 | −0.39 | 0.27 | 0.68 | 1.59 | Survivin | O15392 | BIRC5 | [63] |
| 0.34 | 0.02 | 0.33 | 0.59 | 1.56 | TACC3 | Q9Y6A5 | TACC3 | [64] |
| 0.37 | −0.18 | 0.33 | 0.63 | 1.53 | NuSAP | Q9BXS6 | NUSAP1 | [65,66] |
| 0.12 | −0.04 | 0.23 | 0.68 | 1.47 | INCENP | Q9NQS7 | INCENP | |
| 0.12 | −0.04 | 0.26 | 0.47 | 1.43 | UBE2S | Q16763 | UBE2S | [67] |
| 0.23 | −0.20 | 0.42 | 0.46 | 1.42 | Securin | O95997 | PTTG1 | [68,69] |
| 0.13 | −0.03 | 0.41 | 0.71 | 1.38 | TPX2 | Q9ULW0 | TPX2 | [70] |
| 0.09 | 0.26 | 0.46 | 0.76 | 1.34 | BUB1 | O43683 | BUB1 | [71] |
| 0.37 | −0.29 | 0.15 | 0.34 | 1.22 | ZWINT | O95229 | ZWINT | [72] |
| 0.23 | −0.13 | 0.28 | 0.38 | 1.21 | Tome−1 | Q99618 | CDCA3 | [73] |
| 0.81 | 0.34 | 0.36 | 0.75 | 1.16 | LSM14B−var | A0A0C4DFV2 | LSM14B | [60] |
| 0.08 | −0.05 | 0.15 | 0.33 | 1.12 | HURP | Q15398 | DLGAP5 | [66] |
| 0.25 | 0.18 | 0.39 | 0.53 | 1.10 | HMMR | O75330 | HMMR | [66] |
| 0.05 | −0.19 | 0.23 | 0.46 | 1.09 | PIMREG | Q9BSJ6 | PIMREG | [74] |
| 0.08 | 0.00 | 0.13 | 0.43 | 1.05 | Ki−67 | P46013 | MKI67 | [75] |
| −0.26 | 0.41 | 0.52 | 0.83 | 1.00 | Cyclin B1 | P14635 | CCNB1 | [76] |
| 0.06 | 0.14 | 0.31 | 0.51 | 1.00 | PRC1 | O43663 | PRC1 | [77,78,79] |
| 0.02 | 0.15 | 0.35 | 0.57 | 0.97 | CENP−E | P04183 | TK1 | [80] |
| −0.06 | 0.08 | 0.21 | 0.49 | 0.97 | TK1 | Q02224 | CENPE | [81] |
| −0.11 | 0.15 | 0.30 | 0.55 | 0.96 | Anillin | Q9NQW6 | ANLN | [82] |
| −0.27 | 0.26 | 0.39 | 0.72 | 0.95 | Aurora A | O14965 | AURKA | [83] |
| −0.25 | 0.34 | 0.36 | 0.68 | 0.95 | Aurora B | Q96GD4 | AURKB | [84] |
| 0.40 | −0.01 | 0.24 | 0.66 | 0.94 | Sororin | B5MBX0 | CDCA5 | [85,86] |
| 0.00 | 0.15 | 0.32 | 0.51 | 0.91 | CENP−F | P49454 | CENPF | [81] |
| −0.09 | 0.24 | 0.37 | 0.57 | 0.89 | KIFC1 | Q9BW19 | KIFC1 | [87] |
| 0.09 | 0.10 | 0.29 | 0.53 | 0.87 | CKAP2 | Q8WWK9 | CKAP2 | [88] |
| −0.07 | −0.04 | 0.04 | 0.48 | 0.85 | Repo−Man | Q69YH5 | CDCA2 | [89] |
| 0.52 | −0.33 | 0.38 | 0.50 | 0.84 | Geminin | O75496 | GMNN | [90] |
| 0.16 | −0.04 | 0.34 | 0.67 | 0.83 | CDC20 | Q12834 | CDC20 | [62] |
| −0.02 | −0.04 | 0.22 | 0.35 | 0.83 | Spindly | Q96EA4 | SPDL1 | |
| 0.05 | −0.24 | 0.14 | 0.24 | 0.82 | UBE2C | O00762 | UBE2C | [91,92] |
| 0.04 | 0.19 | 0.32 | 0.57 | 0.81 | KIF18A | Q99661 | KIF2C | [87] |
| −0.20 | 0.13 | 0.24 | 0.46 | 0.81 | KIF2C | Q8NI77 | KIF18A | [93] |
| 0.07 | 0.12 | 0.33 | 0.62 | 0.80 | KIF22 | Q14807 | KIF22 | [94] |
| −0.35 | 0.19 | 0.22 | 0.50 | 0.80 | KIF20A | O95235 | KIF20A | [74] |
| −0.24 | 0.35 | 0.34 | 0.60 | 0.73 | PLK1 | P53350 | PLK1 | [95] |
| −0.58 | 0.26 | 0.12 | 0.49 | 0.71 | Top2α | P11388 | TOP2A | [96] |
| 0.45 | −0.25 | 0.09 | 0.16 | 0.70 | LSM14B | Q9BX40 | LSM14B | [60] |
| 0.49 | −0.21 | 0.11 | 0.15 | 0.65 | UPF3B | Q9BZI7 | UPF3B | [60] |
| −0.38 | 0.19 | 0.20 | 0.48 | 0.59 | ECT2 | Q9H8V3 | ECT2 | [88,97] |
| Non−accumulating Prophase APC/C substrates | ||||||||
| 0.28 | 0.06 | 0.29 | −0.01 | 0.33 | HOXC10 | Q9NYD6 | HOXC10 | [36] |
| −0.18 | −0.04 | 0.37 | 0.30 | −0.49 | Cyclin A2 | P20248 | CCNA2 | [35] |
References
- Smith, M.A.; Altekruse, S.F.; Adamson, P.C.; Reaman, G.H.; Seibel, N.L. Declining Childhood and Adolescent Cancer Mortality. Cancer 2014, 120, 2497–2506. [Google Scholar] [CrossRef]
- Riggi, N.; Suvà, M.L.; Stamenkovic, I. Ewing’s Sarcoma. N. Engl. J. Med. 2021, 384, 154–164. [Google Scholar] [CrossRef]
- Bailey, K.; Cost, C.; Davis, I.; Glade-Bender, J.; Grohar, P.; Houghton, P.; Isakoff, M.; Stewart, E.; Laack, N.; Yustein, J.; et al. Emerging Novel Agents for Patients with Advanced Ewing Sarcoma: A Report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force. F1000Research 2019, 8, 493. [Google Scholar] [CrossRef] [Green Version]
- Womer, R.B.; West, D.C.; Krailo, M.D.; Dickman, P.S.; Pawel, B.R.; Grier, H.E.; Marcus, K.; Sailer, S.; Healey, J.H.; Dormans, J.P.; et al. Randomized Controlled Trial of Interval-Compressed Chemotherapy for the Treatment of Localized Ewing Sarcoma: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 4148–4154. [Google Scholar] [CrossRef] [Green Version]
- Lessnick, S.L.; Braun, B.S.; Denny, C.T.; May, W.A. Multiple Domains Mediate Transformation by the Ewing’s Sarcoma EWS/FLI-1 Fusion Gene. Oncogene 1995, 10, 423–431. [Google Scholar]
- Toomey, E.C.; Schiffman, J.D.; Lessnick, S.L. Recent Advances in the Molecular Pathogenesis of Ewing’s Sarcoma. Oncogene 2010, 29, 4504–4516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, W.A.; Lessnick, S.L.; Braun, B.S.; Klemsz, M.; Lewis, B.C.; Lunsford, L.B.; Hromas, R.; Denny, C.T. The Ewing’s Sarcoma EWS/FLI-1 Fusion Gene Encodes a More Potent Transcriptional Activator and Is a More Powerful Transforming Gene Than FLI-1. Mol. Cell. Biol. 1993, 13, 7393–7398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessnick, S.L.; Ladanyi, M. Molecular Pathogenesis of Ewing Sarcoma: New Therapeutic and Transcriptional Targets. Annu. Rev. Pathol. 2012, 7, 145–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.; Owen, L.A.; Trem, D.J.; Wong, J.S.; Whangbo, J.S.; Golub, T.R.; Lessnick, S.L. Expression Profiling of EWS/FLI Identifies NKX2.2 as a Critical Target Gene in Ewing’s Sarcoma. Cancer Cell 2006, 9, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Prieur, A.; Tirode, F.; Cohen, P.; Delattre, O. EWS/FLI-1 Silencing and Gene Profiling of Ewing Cells Reveal Downstream Oncogenic Pathways and a Crucial Role for Repression of Insulin-Like Growth Factor Binding Protein 3. Mol. Cell. Biol. 2004, 24, 7275–7283. [Google Scholar] [CrossRef] [Green Version]
- Minas, T.Z.; Surdez, D.; Javaheri, T.; Tanaka, M.; Howarth, M.; Kang, H.-J.; Han, J.; Han, Z.-Y.; Sax, B.; Kream, B.E.; et al. Combined Experience of Six Independent Laboratories Attempting to Create an Ewing Sarcoma Mouse Model. Oncotarget 2017, 8, 34141–34163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggi, N.; Cironi, L.; Provero, P.; Suvà, M.-L.; Kaloulis, K.; Garcia-Echeverria, C.; Hoffmann, F.; Trumpp, A.; Stamenkovic, I. Development of Ewing’s Sarcoma from Primary Bone Marrow-Derived Mesenchymal Progenitor Cells. Cancer Res. 2005, 65, 11459–11468. [Google Scholar] [CrossRef] [Green Version]
- Üren, A.; Toretsky, J.A. Ewing’s Sarcoma Oncoprotein EWS-FLI1: The Perfect Target without a Therapeutic Agent. Future Oncol. 2005, 1, 521–528. [Google Scholar] [CrossRef]
- Dgachi, Y.; Bautista-Aguilera, O.M.; Benchekroun, M.; Martin, H.; Bonet, A.; Knez, D.; Godyń, J.; Malawska, B.; Gobec, S.; Chioua, M.; et al. Synthesis and Biological Evaluation of Benzochromenopyrimidinones as Cholinesterase Inhibitors and Potent Antioxidant, Non-Hepatotoxic Agents for Alzheimer’s Disease. Molecules 2016, 21, 634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chung, T.; Oldenburg, K. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Albarqi, H.A.; Wong, L.H.; Schumann, C.; Sabei, F.Y.; Korzun, T.; Li, X.; Hansen, M.N.; Dhagat, P.; Moses, A.S.; Taratula, O.; et al. Biocompatible Nanoclusters with High Heating Efficiency for Systemically Delivered Magnetic Hyperthermia. ACS Nano 2019, 13, 6383–6395. [Google Scholar] [CrossRef] [PubMed]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A Contaminant Repository for Affinity Purification-Mass Spectrometry Data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [Green Version]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [Green Version]
- Käll, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-Supervised Learning for Peptide Identification from Shotgun Proteomics Datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef]
- Li, X.; Schumann, C.; Albarqi, H.A.; Lee, C.J.; Alani, A.W.G.; Bracha, S.; Milovancev, M.; Taratula, O.; Taratula, O. A Tumor-Activatable Theranostic Nanomedicine Platform for NIR Fluorescence-Guided Surgery and Combinatorial Phototherapy. Theranostics 2018, 8, 767–784. [Google Scholar] [CrossRef]
- Shin, H.-C.; Alani, A.W.G.; Cho, H.; Bae, Y.; Kolesar, J.M.; Kwon, G.S. A 3-in-1 Polymeric Micelle Nanocontainer for Poorly Water-Soluble Drugs. Mol. Pharm. 2011, 8, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, D.W. An Algorithm for Least-Squares Estimation of Nonlinear Parameters. J. Soc. Ind. Appl. Math. 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Taratula, O.; Dani, R.K.; Schumann, C.; Xu, H.; Wang, A.; Song, H.; Dhagat, P.; Taratula, O. Multifunctional Nanomedicine Platform for Concurrent Delivery of Chemotherapeutic Drugs and Mild Hyperthermia to Ovarian Cancer Cells. Int. J. Pharm. 2013, 458, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.; Biegel, J.A.; Serra, M.; Wang, J.Y.; Wei, Y.H.; Nycum, L.; Emanuel, B.S.; Evans, G.A. Ews-Erg and EWS-Fli1 Fusion Transcripts in Ewing’s Sarcoma and Primitive Neuroectodermal Tumors with Variant Translocations. J. Clin. Investig. 1994, 94, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.P.; Brody, R.I.; Hamelin, A.C.; Bradner, J.E.; Healey, J.H.; Ladanyi, M. Differential Transactivation by Alternative EWS-FLI1 Fusion Proteins Correlates with Clinical Heterogeneity in Ewing’s Sarcoma. Cancer Res. 1999, 59, 1428–1432. [Google Scholar] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Li, J.; Yuan, J. Caspases in Apoptosis and Beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, U.; Jänicke, R.U.; Schulze-Osthoff, K. Many Cuts to Ruin: A Comprehensive Update of Caspase Substrates. Cell Death Differ. 2003, 10, 76–100. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific Proteolytic Cleavage of Poly (ADP-Ribose) Polymerase: An Early Marker of Chemotherapy-Induced Apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Murray, A.W. Recycling the Cell Cycle: Cyclins Revisited. Cell 2004, 116, 221–234. [Google Scholar] [CrossRef] [Green Version]
- Glotzer, M.; Murray, A.W.; Kirschner, M.W. Cyclin Is Degraded by the Ubiquitin Pathway. Nature 1991, 349, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Prinz, S.; Hwang, E.S.; Visintin, R.; Amon, A. The Regulation of Cdc20 Proteolysis Reveals a Role for APC Components Cdc23 and Cdc27 During S Phase and Early Mitosis. Curr. Biol. 1998, 8, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Prigent, C.; Dimitrov, S. Phosphorylation of Serine 10 in Histone H3, What for? J. Cell Sci. 2003, 116, 3677–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.-M.; Lin, X.-T.; Wu, D.; Tan, Y.; Cheng, C.H.K.; Zhang, J. Cardiac Glycoside Bufalin Blocks Cancer Cell Growth by Inhibition of Aurora a and Aurora B Activation via PI3K-Akt Pathway. Oncotarget 2018, 9, 13783–13795. [Google Scholar] [CrossRef] [Green Version]
- Den Elzen, N.; Pines, J. Cyclin a Is Destroyed in Prometaphase and Can Delay Chromosome Alignment and Anaphase. J. Cell Biol. 2001, 153, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Gabellini, D.; Colaluca, I.N.; Vodermaier, H.C.; Biamonti, G.; Giacca, M.; Falaschi, A.; Riva, S.; Peverali, F.A. Early Mitotic Degradation of the Homeoprotein HOXC10 Is Potentially Linked to Cell Cycle Progression. EMBO J. 2003, 22, 3715–3724. [Google Scholar] [CrossRef] [Green Version]
- Hames, R.S.; Wattam, S.L.; Yamano, H.; Bacchieri, R.; Fry, A.M. APC/C-Mediated Destruction of the Centrosomal Kinase Nek2A Occurs in Early Mitosis and Depends upon a Cyclin a-Type D-Box. EMBO J. 2001, 20, 7117–7127. [Google Scholar] [CrossRef] [Green Version]
- Taratula, O.; Doddapaneni, B.S.; Schumann, C.; Li, X.; Bracha, S.; Milovancev, M.; Alani, A.W.G.; Taratula, O. Naphthalocyanine-Based Biodegradable Polymeric Nanoparticles for Image-Guided Combinatorial Phototherapy. Chem. Mater. 2015, 27, 6155–6165. [Google Scholar] [CrossRef]
- Lee, J.H.; Yeo, Y. Controlled Drug Release from Pharmaceutical Nanocarriers. Chem. Eng. Sci. 2015, 125, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Doddapaneni, B.S.; Kyryachenko, S.; Chagani, S.E.; Alany, R.G.; Rao, D.A.; Indra, A.K.; Alani, A.W.G. A Three-Drug Nanoscale Drug Delivery System Designed for Preferential Lymphatic Uptake for the Treatment of Metastatic Melanoma. J. Control. Release 2015, 220, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Pishas, K.I.; Lessnick, S.L. Recent Advances in Targeted Therapy for Ewing Sarcoma. F1000Research 2016, 5, 2077. [Google Scholar] [CrossRef]
- Hans, F.; Dimitrov, S. Histone H3 Phosphorylation and Cell Division. Oncogene 2001, 20, 3021–3027. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.-C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches through Collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Barr, R.; Shulman, L.N.; Forte, G.B.; Magrini, N. Essential Medicines for Cancer: WHO Recommendations and National Priorities. Bull. World Health Organ. 2016, 94, 735–742. [Google Scholar] [CrossRef]
- Casey, D.A.; Wexler, L.H.; Merchant, M.S.; Chou, A.J.; Merola, P.R.; Price, A.P.; Meyers, P.A. Irinotecan and Temozolomide for Ewing Sarcoma: The Memorial Sloan-Kettering Experience. Pediatr. Blood Cancer 2009, 53, 1029–1034. [Google Scholar] [CrossRef]
- Hunold, A.; Weddeling, N.; Paulussen, M.; Ranft, A.; Liebscher, C.; Jürgens, H. Topotecan and Cyclophosphamide in Patients with Refractory or Relapsed Ewing Tumors. Pediatr. Blood Cancer 2006, 47, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Grohar, P.J.; Helman, L.J. Prospects and Challenges for the Development of New Therapies for Ewing Sarcoma. Pharmacol. Ther. 2013, 137, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.-H.; Dakshanamurthy, S.; Brown, M.L.; et al. A Small Molecule Blocking Oncogenic Protein EWS-FLI1 Interaction with RNA Helicase a Inhibits Growth of Ewing’s Sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Van Maldegem, A.M.; Bovée, J.V.M.G.; Peterse, E.F.P.; Hogendoorn, P.C.W.; Gelderblom, H. Ewing Sarcoma: The Clinical Relevance of the Insulin-Like Growth Factor 1 and the Poly-ADP-Ribose-Polymerase Pathway. Eur. J. Cancer 2016, 53, 171–180. [Google Scholar] [CrossRef]
- Vormoor, B.; Curtin, N.J. Poly (ADP-Ribose) Polymerase Inhibitors in Ewing Sarcoma. Curr. Opin. Oncol. 2014, 26, 428–433. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Tap, W.D.; Qin, L.-X.; Livingston, M.B.; Undevia, S.D.; Chmielowski, B.; Agulnik, M.; Schuetze, S.M.; Reed, D.R.; Okuno, S.H.; et al. Cixutumumab and Temsirolimus for Patients with Bone and Soft-Tissue Sarcoma: A Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2013, 14, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Stewart, E.; Goshorn, R.; Bradley, C.; Griffiths, L.M.; Benavente, C.; Twarog, N.R.; Miller, G.M.; Caufield, W.; Freeman, B.B.; Bahrami, A.; et al. Targeting the DNA Repair Pathway in Ewing Sarcoma. Cell Rep. 2014, 9, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabei, F.Y.; Taratula, O.; Albarqi, H.A.; Al-Fatease, A.M.; Moses, A.S.; Demessie, A.A.; Park, Y.; Vogel, W.K.; Esfandiari Nazzaro, E.; Davare, M.A.; et al. A Targeted Combinatorial Therapy for Ewing’s Sarcoma. Nanomedicine 2021, 37, 102446. [Google Scholar] [CrossRef]
- Yang, S.; Wallach, M.; Krishna, A.; Kurmasheva, R.; Sridhar, S. Recent Developments in Nanomedicine for Pediatric Cancer. J. Clin. Med. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Su, Z.; Li, X.; Shi, S. Construction of Triblock Copolymer-Gold Nanorod Composites for Fluorescence Resonance Energy Transfer via pH-Sensitive Allosteric. Chin. Chem. Lett. 2020, 31, 3131–3134. [Google Scholar] [CrossRef]
- Xu, J.; Shang, L. Emerging Applications of Near-Infrared Fluorescent Metal Nanoclusters for Biological Imaging. Chin. Chem. Lett. 2018, 29, 1436–1444. [Google Scholar] [CrossRef]
- Grossen, P.; Witzigmann, D.; Sieber, S.; Huwyler, J. PEG-PCL-Based Nanomedicines: A Biodegradable Drug Delivery System and Its Application. J. Control. Release 2017, 260, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Moses, A.S.; Taratula, O.R.; Lee, H.; Luo, F.; Grenz, T.; Korzun, T.; Lorenz, A.S.; Sabei, F.Y.; Bracha, S.; Alani, A.W.G.; et al. Nanoparticle-Based Platform for Activatable Fluorescence Imaging and Photothermal Ablation of Endometriosis. Small 2020, 16, e1906936. [Google Scholar] [CrossRef]
- Tsunematsu, T.; Arakaki, R.; Kawai, H.; Ruppert, J.; Tsuneyama, K.; Ishimaru, N.; Earnshaw, W.C.; Pagano, M.; Kudo, Y. APC/CCdh1 Is Required for the Termination of Chromosomal Passenger Complex Activity Upon Mitotic Exit. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Bakos, G.; Yu, L.; Gak, I.A.; Roumeliotis, T.I.; Liakopoulos, D.; Choudhary, J.S.; Mansfeld, J. An E2-Ubiquitin Thioester-Driven Approach to Identify Substrates Modified with Ubiquitin and Ubiquitin-Like Molecules. Nat. Commun. 2018, 9, 4776. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.; Mark, K.G.; Mocciaro, A.; Watson, E.R.; Prabu, J.R.; Cha, D.D.; Kampmann, M.; Gamarra, N.; Zhou, C.Y.; Rape, M. Gene Expression and Cell Identity Controlled by Anaphase-Promoting Complex. Nature 2020, 579, 136–140. [Google Scholar] [CrossRef]
- Pfleger, C.M.; Kirschner, M.W. The KEN Box: An APC Recognition Signal Distinct from the D Box Targeted by Cdh1. Genes Dev. 2000, 14, 655–665. [Google Scholar] [PubMed]
- Pines, J. Mitosis: A Matter of Getting Rid of the Right Protein at the Right Time. Trends Cell Biol. 2006, 16, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Jeng, J.-C.; Lin, Y.-M.; Lin, C.-H.; Shih, H.-M. Cdh1 Controls the Stability of TACC3. Cell Cycle 2009, 8, 3537–3544. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhou, Y.; Sun, L.; Xing, G.; Tian, C.; Sun, J.; Zhang, L.; He, F. NuSAP Is Degraded by APC/C-Cdh1 and Its Overexpression Results in Mitotic Arrest Dependent of Its Microtubules’ Affinity. Cell. Signal. 2007, 19, 2046–2055. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Rape, M. Regulated Degradation of Spindle Assembly Factors by the Anaphase-Promoting Complex. Mol. Cell 2010, 38, 369–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, A.; Wickliffe, K.E.; Mellone, B.G.; Song, L.; Karpen, G.H.; Rape, M. Identification of a Physiological E2 Module for the Human Anaphase-Promoting Complex. Proc. Natl. Acad. Sci. USA 2009, 106, 18213–18218. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; McGarry, T.J.; Bernal, T.; Kirschner, M.W. Identification of a Vertebrate Sister-Chromatid Separation Inhibitor Involved in Transformation and Tumorigenesis. Science 1999, 285, 418–422. [Google Scholar] [CrossRef]
- Hagting, A.; Den Elzen, N.; Vodermaier, H.C.; Waizenegger, I.C.; Peters, J.-M.; Pines, J. Human Securin Proteolysis Is Controlled by the Spindle Checkpoint and Reveals When the APC/C Switches from Activation by Cdc20 to Cdh1. J. Cell Biol. 2002, 157, 1125–1137. [Google Scholar] [CrossRef] [Green Version]
- Stewart, S.; Fang, G. Anaphase-Promoting Complex/Cyclosome Controls the Stability of TPX2 During Mitotic Exit. Mol. Cell. Biol. 2005, 25, 10516–10527. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Yu, H. KEN-Box-Dependent Degradation of the Bub1 Spindle Checkpoint Kinase by the Anaphase-Promoting Complex/Cyclosome. J. Biol. Chem. 2007, 282, 3672–3679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Li, R.; Gu, L.; Deng, H.; Zhao, Y.; Guo, Y.; Yu, S.; Wang, G. Anaphase-Promoting Complex/Cyclosome-Cdc-20 Promotes Zwint-1 Degradation. Cell Biochem. Funct. 2020, 38, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Ayad, N.G.; Rankin, S.; Murakami, M.; Jebanathirajah, J.; Gygi, S.; Kirschner, M.W. Tome-1, a Trigger of Mitotic Entry, Is Degraded during G1 via the APC. Cell 2003, 113, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.-M.; Coppinger, J.A.; Seki, A.; Cheng, X.-L.; Yates, J.R.; Fang, G. RCS1, a Substrate of APC/C, Controls the Metaphase to Anaphase Transition. Proc. Natl. Acad. Sci. USA 2008, 105, 13415–13420. [Google Scholar] [CrossRef] [Green Version]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clute, P.; Pines, J. Temporal and Spatial Control of Cyclin B1 Destruction in Metaphase. Nat. Cell Biol. 1999, 1, 82–87. [Google Scholar] [CrossRef]
- Juang, Y.L.; Huang, J.; Peters, J.M.; McLaughlin, M.E.; Tai, C.Y.; Pellman, D. APC-Mediated Proteolysis of Ase1 and the Morphogenesis of the Mitotic Spindle. Science 1997, 275, 1311–1314. [Google Scholar] [CrossRef]
- Visintin, R.; Prinz, S.; Amon, A. CDC20 and CDH1: A Family of Substrate-Specific Activators of APC-Dependent Proteolysis. Science 1997, 278, 460–463. [Google Scholar] [CrossRef]
- Castro, A.; Bernis, C.; Vigneron, S.; Labbé, J.-C.; Lorca, T. The Anaphase-Promoting Complex: A Key Factor in the Regulation of Cell Cycle. Oncogene 2005, 24, 314–325. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.-Y.; Chang, Z.-F. Mitotic Degradation of Human Thymidine Kinase 1 Is Dependent on the Anaphase-Promoting Complex/Cyclosome-CDH1-Mediated Pathway. Mol. Cell. Biol. 2004, 24, 514–526. [Google Scholar] [CrossRef] [Green Version]
- Gurden, M.D.J.; Holland, A.J.; van Zon, W.; Tighe, A.; Vergnolle, M.A.; Andres, D.A.; Spielmann, H.P.; Malumbres, M.; Wolthuis, R.M.F.; Cleveland, D.W.; et al. Cdc20 Is Required for the Post-Anaphase, KEN-Dependent Degradation of Centromere Protein F. J. Cell Sci. 2010, 123, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.-M.; Fang, G. Anillin Is a Substrate of Anaphase-Promoting Complex/Cyclosome (APC/C) That Controls Spatial Contractility of Myosin During Late Cytokinesis. J. Biol. Chem. 2005, 280, 33516–33524. [Google Scholar] [CrossRef] [Green Version]
- Floyd, S.; Pines, J.; Lindon, C. APC/C Cdh1 Targets Aurora Kinase to Control Reorganization of the Mitotic Spindle at Anaphase. Curr. Biol. 2008, 18, 1649–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, S.; Fang, G. Destruction Box-Dependent Degradation of Aurora B Is Mediated by the Anaphase-Promoting Complex/Cyclosome and Cdh1. Cancer Res. 2005, 65, 8730–8735. [Google Scholar] [CrossRef] [Green Version]
- Rankin, S.; Ayad, N.G.; Kirschner, M.W. Sororin, a Substrate of the Anaphase-Promoting Complex, Is Required for Sister Chromatid Cohesion in Vertebrates. Mol. Cell 2005, 18, 185–200. [Google Scholar] [CrossRef]
- Zhang, N.; Pati, D. Sororin Is a Master Regulator of Sister Chromatid Cohesion and Separation. Cell Cycle 2012, 11, 2073–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.A.; Winter, D.; Kirchner, M.; Chauhan, R.; Ahmed, S.; Ozlu, N.; Tzur, A.; Steen, J.A.; Steen, H. Co-Regulation Proteomics Reveals Substrates and Mechanisms of APC/C-Dependent Degradation. EMBO J. 2014, 33, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Fang, G. CKAP2 Is a Spindle-Associated Protein Degraded by APC/C-Cdh1 During Mitotic Exit. J. Biol. Chem. 2007, 282, 15103–15113. [Google Scholar] [CrossRef] [Green Version]
- Manzione, M.G.; Rombouts, J.; Steklov, M.; Pasquali, L.; Sablina, A.; Gelens, L.; Qian, J.; Bollen, M. Co-Regulation of the Antagonistic RepoMan:Aurora-B Pair in Proliferating Cells. Mol. Biol. Cell 2020, 31, 419–438. [Google Scholar] [CrossRef] [PubMed]
- McGarry, T.J.; Kirschner, M.W. Geminin, an Inhibitor of DNA Replication, Is Degraded During Mitosis. Cell 1998, 93, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Rape, M.; Kirschner, M.W. Autonomous Regulation of the Anaphase-Promoting Complex Couples Mitosis to S-Phase Entry. Nature 2004, 432, 588–595. [Google Scholar] [CrossRef]
- Rape, M.; Reddy, S.K.; Kirschner, M.W. The Processivity of Multiubiquitination by the APC Determines the Order of Substrate Degradation. Cell 2006, 124, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Sedgwick, G.G.; Hayward, D.G.; Di Fiore, B.; Pardo, M.; Yu, L.; Pines, J.; Nilsson, J. Mechanisms Controlling the Temporal Degradation of Nek2A and Kif18A by the APC/C-Cdc20 Complex. EMBO J. 2013, 32, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feine, O.; Zur, A.; Mahbubani, H.; Brandeis, M. Human Kid Is Degraded by the APC/C(Cdh1) but Not by the APC/C(Cdc20). Cell Cycle 2007, 6, 2516–2523. [Google Scholar] [CrossRef] [Green Version]
- Lindon, C.; Pines, J. Ordered Proteolysis in Anaphase Inactivates Plk1 to Contribute to Proper Mitotic Exit in Human Cells. J. Cell Biol. 2004, 164, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguren, M.; Álvarez-Fernández, M.; García, F.; López-Contreras, A.J.; Fujimitsu, K.; Yaguchi, H.; Luque-García, J.L.; Fernández-Capetillo, O.; Munoz, J.; Yamano, H.; et al. A Synthetic Lethal Interaction Between APC/C and Topoisomerase Poisons Uncovered by Proteomic Screens. Cell Rep. 2014, 6, 670–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liot, C.; Seguin, L.; Siret, A.; Crouin, C.; Schmidt, S.; Bertoglio, J. APC(Cdh1) Mediates Degradation of the Oncogenic Rho-GEF Ect2 After Mitosis. PLoS ONE 2011, 6, e23676. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esfandiari Nazzaro, E.; Sabei, F.Y.; Vogel, W.K.; Nazari, M.; Nicholson, K.S.; Gafken, P.R.; Taratula, O.; Taratula, O.; Davare, M.A.; Leid, M. Discovery and Validation of a Compound to Target Ewing’s Sarcoma. Pharmaceutics 2021, 13, 1553. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13101553
Esfandiari Nazzaro E, Sabei FY, Vogel WK, Nazari M, Nicholson KS, Gafken PR, Taratula O, Taratula O, Davare MA, Leid M. Discovery and Validation of a Compound to Target Ewing’s Sarcoma. Pharmaceutics. 2021; 13(10):1553. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13101553
Chicago/Turabian StyleEsfandiari Nazzaro, Ellie, Fahad Y. Sabei, Walter K. Vogel, Mohamad Nazari, Katelyn S. Nicholson, Philip R. Gafken, Olena Taratula, Oleh Taratula, Monika A. Davare, and Mark Leid. 2021. "Discovery and Validation of a Compound to Target Ewing’s Sarcoma" Pharmaceutics 13, no. 10: 1553. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13101553