
Current Advances in Lipid and Polymeric Antimicrobial Peptide Delivery Systems and Coatings for the Prevention and Treatment of Bacterial Infections


Abstract
:1. Introduction
2. Literature Strategy
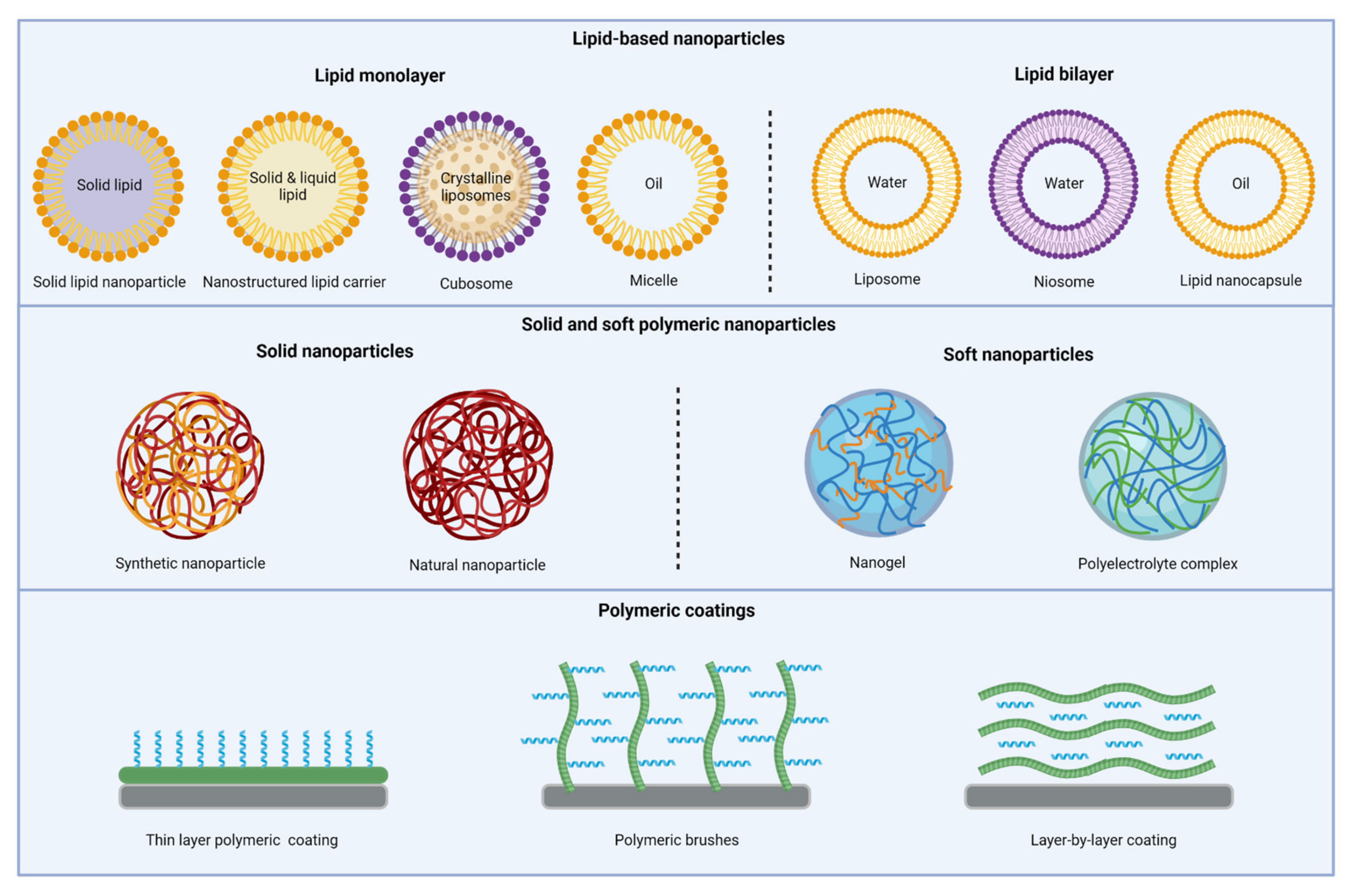
3. Nanoparticles
3.1. Lipid-Based Nanoparticles
3.1.1. Liposomes
3.1.2. Niosomes
3.1.3. Solid Lipid Nanoparticles
3.1.4. Nanostructured Lipid Carriers
3.1.5. Lipid Nanocapsules
3.1.6. Cubosomes
3.1.7. Micelles

{kind=link}
{kind=link}
{kind=link}
| Type of Nanoparticles and Particle Composition | AMP | Physicochemical Properties (Size, Surface Charge, Encapsulation Efficiency, Release) | In Vitro and In Vivo Results | Application | Refs. |
|---|---|---|---|---|---|
| Liposomes | |||||
| SCS-Lipoid® S75 SPC liposomes | Colistin | 113–137 nm, −66 to −53 mV, EE = 84–92%, 15–43% release in 24 h |
| Systemic/pulmonary infections | [41,85] |
| Surface-modified liposomes | |||||
| CHL-DSPC-DSPE-mPEG2000 liposomes | LL-37 Indolicidin | 107 nm, −2.1 mV, EE = 53% (LL-37) 121 nm, −3.1 mV, EE = 35% (indolicidin) |
| Topical/intracellular infections | [20] |
| CHL-DOPE-lecithin liposomes coated with chitosan | Colistin | 485 nm, +5.3 mV |
| Systemic/pulmonary infections | [47] |
| CHL-S60-lecithin liposomes coated with chitosan | Colistin | 156 nm, +16.7 mV, EE = 45–82%, 85% release in 24 h |
| Oral delivery/systemic infections | [48] |
| CHL-DPPC/DSPC-DPPE-GA liposomes coated with EAP | Colistin | 203 nm, −15.3 mV, EE = 51%, 20% release in 5 h in PBS or GIT-mimicking media |
| Oral delivery/intracellular infections | [22] |
| Red blood cell (RBC)-mimetic hybrid liposome composed of lipid S100-DSPE-PEG2000 | Polymyxin B | ~150 nm, −28 mV |
| Antivirulence therapy | [49] |
| Coencapsulated liposomes | |||||
| CHL-HSPC-DMPG/DSPG Liposomes | Ciprofloxacin Colistin | ~100 nm, anionic, EE = 67% (colistin), EE = 90% (ciprofloxacin), 50–80% release in 30 min then sustained release |
| Pulmonary infections | [50,53] |
| CHL-HSPC-DSPG-PEG liposomal powder formulation | Ciprofloxacin Colistin | 141–378 nm, −21.0 to −9.2 mV, EE = 47–59% (colistin), EE = 32–71% (ciprofloxacin) |
| Pulmonary infections | [51,52] |
| CHL-PC-OA liposomes decorated with AMP2 or AMP3 | Vancomycin AMP2/AMP3 | 137–387 nm, −9.8 to +1.8 mV EE = 27–64%, 49–67% release in 8 h at pH 6, 18–23% release in 8 h at pH 7.4 |
| Intracellular infections | [23] |
| CHL-SPC liposomes incorporating DP7-CHL | Azithromycin DP-7 | 100–106 nm, +3.7 to +5.3 mV, EE = 97–98% (AZT), DL = 5% (DP7-CHL), ~50% release in 96 h (sustained release) |
| Topical infections | [54] |
| Niosomes | |||||
| Niosomes composed of Span60 and cholesterol | Polymyxin B | 257 nm, −22.5 mV, EE = 72%, stability in SGF (86.22% in SGF pH 1.2 and 78.5% in SIF pH 6.8) |
| Oral delivery/intestinal infections | [56] |
| Solid lipid nanoparticles (sLNPs) | |||||
| GeleolTM-lecithin-Kolliphor® RH40-Transcutol® sLNPs | Lacticin 3147 | 81–85 nm, EE = 16% (Ltnα), EE = 84% (Ltnβ) |
| Oral delivery/intestinal infections | [60] |
| Crodacol® CS90/Crodacol® C90-Lipoid® S75 SPC sLNPs complexed with sodium alginate | Polymyxin B | 203–574 nm, −40.7 to −24.1 mV, EE = 93–94% |
| Topical infections | [61] |
| Glyceryl monostearate-PC-PVA sLNPs | LL-37 Serpin A1 | 210–232 nm, −20 to −16 mV, EE = 82–89%, 14% release in 24 h followed by slow sustained release over 15 days |
| Topical infections | [62] |
| Nanostructured lipid carriers (NLCs) | |||||
| Precirol® ATO 5-Miglyol 812-Polysorbate 80-Poloxamer 188 NLCs | Colistin | 300–427 nm, negatively charged, EE = 80–95%, sustained release with >50% release in 24 h |
| Pulmonary infections/biofilm removal | [64,65] |
| Precirol® ATO 5-Miglyol 182 N/F-Tween® 80-Poloxamer 188 NLCs | Colistin | 354 nm, −20.4 mV, EE = 95%, 80% release in 5 h and 92% in 24 h |
| Systemic/pulmonary infections | [66] |
| Precirol® ATO 5-Miglyol 812N-Tween® 80-Poloxamer 188 NLCs | LL-37 | 274 nm, −31.6 mV, EE = 96%, DL = 17% |
| Topical infections/chronic wounds | [67] |
| Coencapsulated NLCs | |||||
| CP-CCP-Lipoid® S100 SPC-PL-SLS NLCs coated with polymyxin B | Dexamethasone acetate Polymyxin B | 231–256 nm, −2.1 to +3.5 mV, EE = 94% (dexamethasone acetate), EE = 99% (polymyxin B coating) |
| Ocular infections accompanied by inflammation | [68] |
| Softisan154-MCT-Kolliphor® P188 NLCs coated polymyxin B and surface modified with chitosan or dextran | Buparvaquone Polymyxin B | 184 nm, −20.1 mV (BPQ-NLC-PB-[chitosan]); 209 nm, +31.1 mV (BPQ-NLC-PB-[dextran]); 172 nm, −30.9 mV (BPQ-NLC), EE = 99.3–99.7% (BPQ) |
| Intracellular infections | [69] |
| Lipid nanocapsules (LNCs) | |||||
| Labrafac WL1349-Lipoid S75-Kolliphor HS15-NaCl LNCs (adsorption strategy) | AA230 DPK-060 LL-37 | 60–77 nm, −3.7 to −0.8 mV, EE = 26–35% |
| MDR infections | [71] |
| Reverse micelles in Oleic Plurol-Kolliphor HS-15-Labrafac WL 1349-NaCl-DSS LNCs | AP138 | 63 nm, −25.6 mV, EE = 98%, 50% release in 2 h, 100% release in 24 h |
| Topical infections | [72] |
| ML-Solutol® HS15-Labrafac® WL1349-NaCl LNCs | AP114 AP138 | 36–37 nm, AE = 34–62%, DL =1–3% |
| Topical infections | [73] |
| ML-Solutol® HS15-Labrafac® CC-NaCl LNCs | DPK-060 LL-37 | 32–135 nm, +5 to +20 mV, AE = 28–42% (DPK-060), AE = 72–77% (LL-37), sustained release |
| Topical infections | [74] |
| Cubosomes | |||||
| Capmul-90 EP/NF cubosomes in Poloxamer 407 gel | DPK-060 | 200–300 nm (cubosomes), 50–70% release cubosomes in 24 h |
| Topical infections | [77] |
| GMO-Lutrol F127 cubosomes and GMO-OA-Lutrol F127 hexasomes | AP114 DPK-060 LL-37 | 87–111 nm, −11.1 mV and EE = 27% (AP114), +0.9 mV and EE = 50% (DPK060), +4.5 mV and EE = 81% (LL-37) |
| MDR infections | [78] |
| GMO-Poloxamer 407 cubosomes | LL-37 | 130 nm, no release in 24 h |
| Topical infections | [79] |
| Micelles | |||||
| SDCS micelles | Polymyxin B | 126–189 nm, −7.4 to −4.9 mV, EE = 48–57%, >80% release in plasma in 24 h, sustained release |
| MDR Gram-negative infections | [82,83] |
| DSPE-PEG2000 micelles | Aurein-derived AMPs | 12–14 nm |
| Topical infections | [84] |
| DP7-CHL micelles | DP7 | 36 nm, +43.8 mV |
| Systemic infections | [81] |
3.2. Polymeric Nanoparticles
3.2.1. Synthetic Nanoparticles
PLGA Nanoparticles
PLA Nanoparticles
Poly(l-lactic acid-co-d,l-mandelic acid) Nanoparticles
3.2.2. Natural Nanoparticles
Chitosan Nanoparticles
Pectin Nanoparticles
3.2.3. Future Perspective of Polymeric Nanoparticles
| Type of Nanoparticles and Particle Composition | AMP | Physicochemical Properties (Size, Surface Charge, Encapsulation Efficiency, Release) | In Vitro and In Vivo Results | Application | Refs. |
|---|---|---|---|---|---|
| Synthetic AMP-loaded nanoparticles | |||||
| PLGA | Esculentin | 261–282 nm, −0.7 to −0.8 mV, EE = 100%, LC: 2%, 60% released after 3 h, then sustained for 3 days |
| Systemic/lung infection | [91] |
| PLGA | LL-37 | 304 nm, −21 mV, EE = 70%, LC = 1%, ~40% burst release, then 14 day sustained release |
| Topical/wound infection | [90] |
| PLGA | G17 and G19 | 284–291 nm, +7.3 to +12.9 mV, EE = 90%, LC: 0.6–0.9%, 45% released after 1 h, controlled release up to 48 h |
| Topical/wound infection | [93] |
| PLGA | HHC10 | 320 nm, +13.3 mV, EE = 54%, 42% release up to 10 h followed by plateau phase |
| Systemic infection | [94] |
| PLGA with N-acetylcysteine coating | IDR-1018 | 5.1–6.2 μm, EE = 59–62%, sustained release for up to 48 h followed by a controlled release of peptide for up to 120 h |
| Systemic/lung infection | [21] |
| PLGA | K4 | 416 nm, +1 mV, 89% peptide conjugation |
| Topical/chronic wound infection | [92] |
| PLGA | Plectasin | 215 nm, −18 mV, EE: 71–90%, 77% release after 1 h, rest was released over 24 h |
| Systemic infection | [110] |
| PLGA/PLA | GIBIM-P5S9K | 258–352 nm, +22.7 to +29.4 mV, EE = 55–75%, 50% peptide release after 8 h and a successive slower release phase |
| Topical infection | [96] |
| Poly(LA-co-MA) | BF-30 | 2.75 µm, EE = 92%, LC = 8%, no initial burst release, only controlled release of peptide was observed after 25 days |
| Topical infection | [100] |
| Natural AMP-loaded nanoparticles | |||||
| PLGA-chitosan composite | KSL | 61–67 µm, EE = 70–93%, LC = 1.7–3.7%, 25–35% released after 10 days, and 80–90% released after 80 days |
| Topical infection in oral cavity | [103] |
| Carboxymethyl chitosan | OH-CATH30 | 258 nm, 30.2 mV, EE = 82%, LC = 33%, near-linear release with 70% released at 24 h |
| Topical/skin infection | [102] |
| Pectin | Nisin | 200–500 nm, −20–−45 mV, EE = 100% |
| Food preservation | [106] |
3.3. Polymeric Nanogels
3.3.1. Natural Cationic Polymer-Based Nanogels
3.3.2. Natural Anionic Polymer-Based Nanogels
3.3.3. Synthetic Polymer-Based Nanogels
| Type of Nanoparticles and Particle Composition | AMP | Physicochemical Properties (Size, Surface Charge, Encapsulation Efficiency, Release) | In Vitro/In Vivo Results | Application | Refs. |
|---|---|---|---|---|---|
| Natural cationic polymer-based | |||||
| Chitosan: tripolyphosphate | Temporin B | 185 nm, +8.8 mV, up to 75% EE, burst effect + gradual release (17% over 15 days) |
| Topical infections | [123] |
| Chitosan | Pep-H | 244 nm, +12 mV, 72% EE, 30% burst release, up to 50% released over 72 h |
| Intracellular infections | [125] |
| Chitosan: tripolyphosphate | Cryptdin-2 | 105 nm, −22 mV, 60% EE and 65% in vitro release in 4.5 h |
| Intestinal infections | [124] |
| Chitosan and poly-γ-glutamic acid | LL-37 | 793–2128 nm, −36 to +50 mV, 23–76% EE, 90% released in 10 h | N/A | Infections | [121] |
| 2,3-Dimethyl maleic anhydride grafted chito-oligosaccharide | Polymyxin B | 154 nm, −8.7 mV |
| Systemic infections | [141] |
| Natural anionic polymer-based | |||||
| Alginate | Polymyxin B | 100–125 nm, −7 to −35 mV, ~90% EE |
| Infections | [142] |
| Alginate | Pep19–2.5 | 342–841 nm, released in pancreatic fluid in 1 h |
| Gastrointestinal infections | [127] |
| Octenyl succinic anhydride-modified HA | Novicidin | 80–144 nm, −24 to −57 mV, 15–71% EE Complete release over 12 days |
| Systemic infections | [130] |
| Octenyl succinic anhydride-modified HA | DJK-5 | 174–194 nm, −11.6 to −9.5 mV, 33–60% EE, complete release in 48 h |
| Systemic or topical infections | [19] |
| Octenyl succinic anhydride-modified HA | LBP-3 | 155–250 nm, −10 to −28 mV, 37–90% EE |
| Systemic or pulmonary infections | [129] |
| Poly-L-lysine cross-linked HA | Vancomycin GFP | 120 nm, −15.4 to −35 mV, DL of 4%, complete release in 48 h |
| Intracellular of pulmonary infections | [131] |
| Oleyamine-modified HA | Vancomycin | 201–360 nm, −17.6 to −20.4 mV, 26–43% EE, drug release over 72 h |
| Infections | [143] |
| 11-Amino-1-undecanethiol hydrochloride-modified HA | LLKKK18 | 533 nm, +2.4 mV, approx. 70% EE |
| Intracellular infections | [132] |
| PEG-poly(glutamic acid) | MSI-78 | 80–120 nm, −16 to −38 mV, 75–87% EE, approx. 80% released in 4 days |
| Systemic infections | [144] |
| Synthetic polymer-based | |||||
| Poly (styrene sulfonate) | Polymyxin B | 166–186 nm, −40 mV, approx. 80% released |
| Infections | [145] |
| PAA-g-PNIPAAm polyelectrolyte complex | E5 | N/A |
| Infections | [139] |
| PEG20K-hbG3-OH dendritic nanogels | DPK-060 LL-37 | 205–331 nm, −5 to +5 mV, 40–60 μM/0.1 wt % |
| Systemic or topical infections | [146] |
| Poly(ethyl acrylate-co-methacrylic acid) microgels | DPK-060 LL-37 | 50–260 nm, −10 to −30 mV, 35–75% peptide released in 2 h |
| Systemic infections | [140] |
| Poly (EA/MAA/BDDA) microgels | EFK17 | N/A size, −30 mV, 60–100% release in 1 h |
| Infections | [147] |
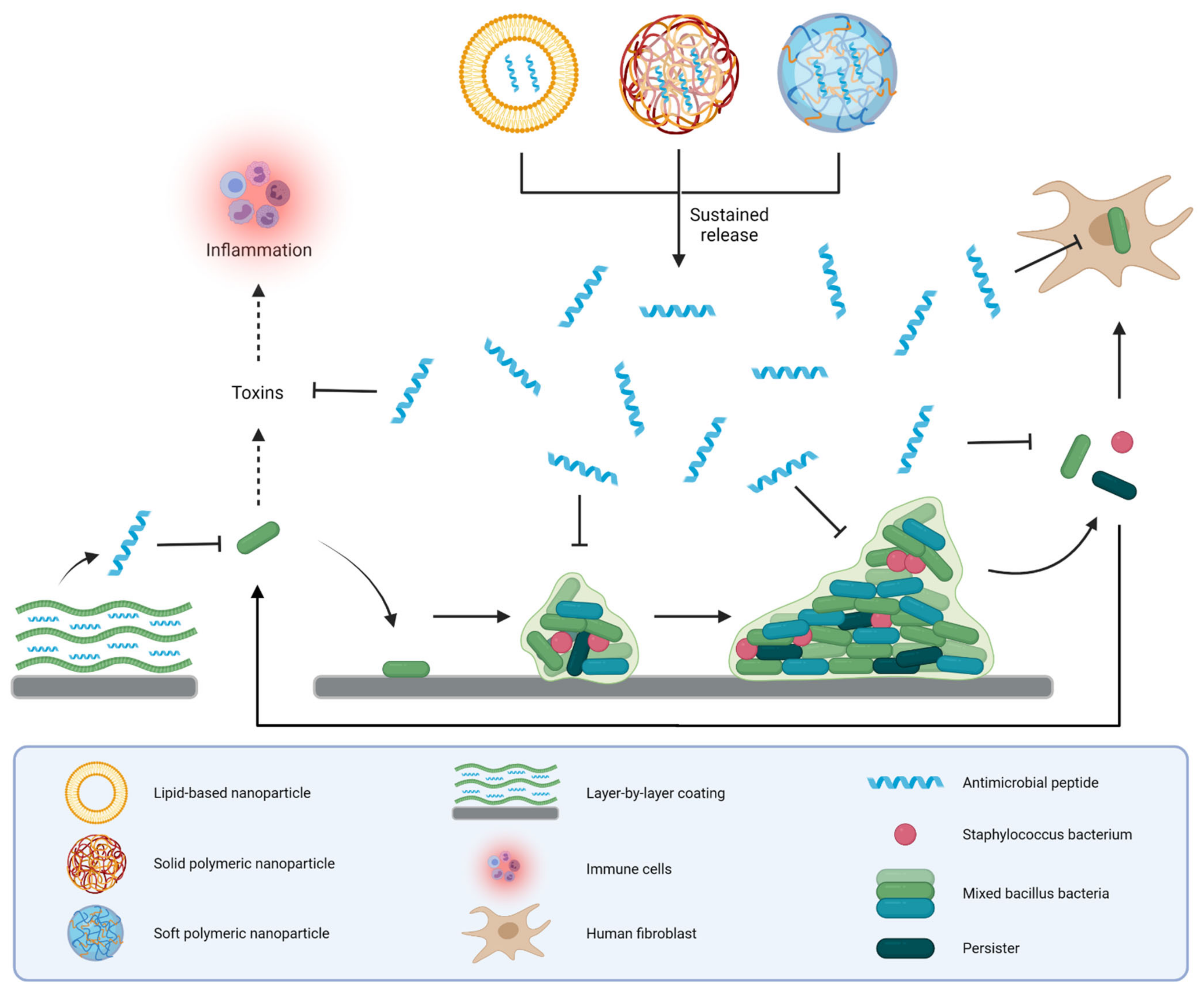
3.4. Polymeric AMP Coatings
3.4.1. Thin Layer Polymeric Coatings
3.4.2. Polymeric Brushes
3.4.3. Layer-by-Layer Coatings
| AMP | Coating and Release | Surface | Antibacterial Activities In Vitro and In Vivo | Refs. |
|---|---|---|---|---|
| Thin layer polymeric coatings | ||||
| Dhvar 5 and hLf1-11 | Chitosan by spin coating | Titanium, Gold |
| [151] |
| CRW | Polydopamine coating | Silicone |
| [154] |
| HHC-36 | Hydrogel-polydopamine coating; 37% burst release in 24 h, sustained release for 20 days | Silicone catheter |
| [155] |
| SESB2V | Polydopamine coating | Titanium |
| [156] |
| Polymeric brushes | ||||
| Tet213 | Poly(DMA-co-APMA) copolymer brush | Titanium |
| [157] |
| E6 and Tet20 | Poly(DMA-co-APMA) copolymer brush | Titanium, polystyrene nanoparticles, quartz |
| [158] |
| Peptide | Block copolymer Pluronic F-127 | Silicone |
| [159] |
| Peptide E6 | PDMA-co-APMA brush | Polyurethane |
| [149] |
| CysLasio-III | Allyl glycidyl ether and PEG coupling | Catheter |
| [160] |
| Chain201D | Tetra(ethylene) glycol-terminated self-assembled monolayers | Gold |
| [148] |
| hLf1-11 | Silanization vs brush of DMA-APMA copolymer prepared by SI-ATRP | Titanium |
| [161] |
| D-GL13K | Engineered protein polymers (brush) | Titanium |
| [162] |
| Layer-by-layer coatings | ||||
| Ponericin G1 | Sequential deposition of poly beta amino ester/polyanion/ponericin G1/polyanion; release up to 10 days | Silicone catheters |
| [163] |
| β-peptide (ACHC-β3hVal-β3hLys)3 | ~700 nm thick multilayer PGA PLL coating; release over 4 months | Catheter |
| [164] |
| HHC-36 | 3-layered system; AMP in each layer; burst release, then steady release up to 5 days | Titanium |
| [165] |
| Tet213 | Multilayered peptide-functionalized collagen; AMP released over at least 28 days | Titanium |
| [26] |
| OP-145 | PLEX; two layers; In 48 h, 55% release, then 1% daily release for 30 days | Titanium-aluminum (7%)-niobium (6%) |
| [25] |
| SAAP-145 | PLEX; 5 layers; initial burst >50%, constant release of 0.6% daily up to day 30 | Titanium |
| [166] |
| SAAP-276 | PLEX; 5 layers; initial burst >50%, constant release of 0.6% daily up to day 30 | Titanium |
| [166] |
4. Discussion
4.1. Key Challenges in Bringing AMP-Based Nanoformulations to the Clinic
4.1.1. Lack of Standardized Tests
4.1.2. Lack of Shelf-Stable Formulations and Evaluations Thereof
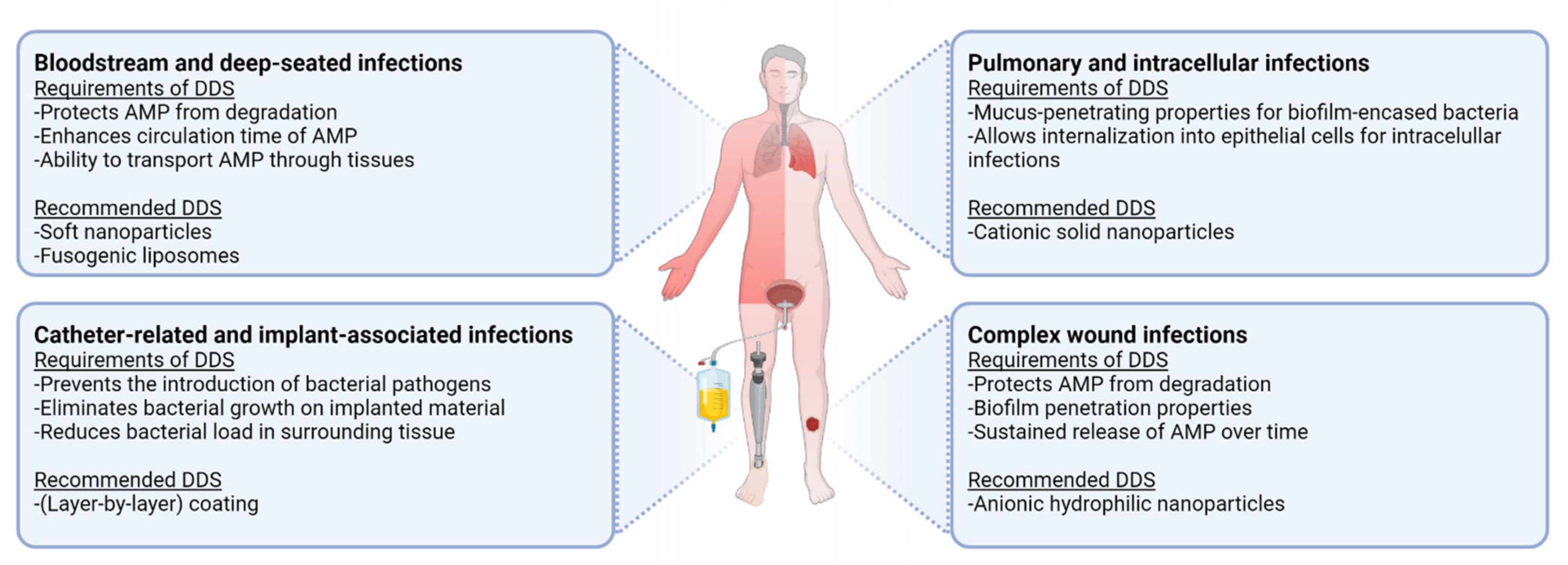
4.2. Clinical Applications of AMP Delivery Systems
4.2.1. Bloodstream and Deep-Seated Infections
4.2.2. Catheter-Related and Implant-Associated Infections
4.2.3. Pulmonary and Intracellular Infections
4.2.4. Complex Wound Infections
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tenover, F.C. Mechanisms of Antimicrobial Resistance in Bacteria. Am. J. Med. 2006, 119, S3–S10. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef]
- Sulaiman, J.E.; Lam, H. Evolution of Bacterial Tolerance Under Antibiotic Treatment and Its Implications on the Development of Resistance. Front. Microbiol. 2021, 12, 617412. [Google Scholar] [CrossRef] [PubMed]
- Kamaruzzaman, N.F.; Kendall, S.; Good, L. Targeting the hard to reach: Challenges and novel strategies in the treatment of intracellular bacterial infections. Br. J. Pharmacol. 2016, 174, 2225–2236. [Google Scholar] [CrossRef]
- Li, W.; Separovic, F.; O’Brien-Simpson, N.M.; Wade, J.D. Chemically modified and conjugated antimicrobial peptides against superbugs. Chem. Soc. Rev. 2021, 50, 4932–4973. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Scheper, H.; Wubbolts, J.M.; Verhagen, J.A.M.; de Visser, A.W.; van der Wal, R.J.P.; Visser, L.G.; de Boer, M.G.J.; Nibbering, P.H. SAAP-148 Eradicates MRSA Persisters Within Mature Biofilm Models Simulating Prosthetic Joint Infection. Front. Microbiol. 2021, 12, 625952. [Google Scholar] [CrossRef] [PubMed]
- De Breij, A.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; de Boer, L.; Koning, R.I.; Ravensbergen, E.; Franken, M.; van der Heijde, T.; Boekema, B.K.; et al. The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci. Transl. Med. 2018, 10, 423. [Google Scholar] [CrossRef] [Green Version]
- Di Luca, M.; Maccari, G.; Maisetta, G.; Batoni, G. BaAMPs: The database of biofilm-active antimicrobial peptides. Biofouling 2015, 31, 193–199. [Google Scholar] [CrossRef]
- Yasir, M.; Willcox, M.D.P.; Dutta, D. Action of Antimicrobial Peptides against Bacterial Biofilms. Materials 2018, 11, 2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, Y.; Papo, N.; Shai, Y. Endotoxin (Lipopolysaccharide) Neutralization by Innate Immunity Host-Defense Peptides. J. Biol. Chem. 2006, 281, 1636–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Mansour, S.C.; Pena, O.M.; Hancock, R.E. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Barreto-Santamaría, A.; Patarroyo, M.E.; Curtidor, H. Designing and optimizing new antimicrobial peptides: All targets are not the same. Crit. Rev. Clin. Lab. Sci. 2019, 56, 351–373. [Google Scholar] [CrossRef]
- Ulvatne, H. Antimicrobial peptides: Potential use in skin infections. Am. J. Clin. Dermatol. 2003, 4, 591–595. [Google Scholar] [CrossRef]
- Kang, S.-J.; Park, S.J.; Mishig-Ochir, T.; Lee, B.-J. Antimicrobial peptides: Therapeutic potentials. Expert Rev. Anti-Infect. Ther. 2014, 12, 1477–1486. [Google Scholar] [CrossRef]
- Månsson, R.; Frenning, G.; Malmsten, M. Factors Affecting Enzymatic Degradation of Microgel-Bound Peptides. Biomacromolecules 2013, 14, 2317–2325. [Google Scholar] [CrossRef]
- Klodzinska, S.; Pletzer, D.; Rahanjam, N.; Rades, T.; Hancock, R.; Nielsen, H.M. Hyaluronic acid-based nanogels improve in vivo compatibility of the anti-biofilm peptide DJK-5. Nanomed. Nanotechnol. Biol. Med. 2019, 20, 102022. [Google Scholar] [CrossRef]
- Ron-Doitch, S.; Sawodny, B.; Kühbacher, A.; David, M.M.N.; Samanta, A.; Phopase, J.; Burger-Kentischer, A.; Griffith, M.; Golomb, G.; Rupp, S. Reduced cytotoxicity and enhanced bioactivity of cationic antimicrobial peptides liposomes in cell cultures and 3D epidermis model against HSV. J. Control. Release 2016, 229, 163–171. [Google Scholar] [CrossRef]
- Sharma, A.; Vaghasiya, K.; Gupta, P.; Singh, A.K.; Gupta, U.D.; Verma, R.K. Dynamic mucus penetrating microspheres for efficient pulmonary delivery and enhanced efficacy of host defence peptide (HDP) in experimental tuberculosis. J. Control. Release 2020, 324, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Menina, S.; Eisenbeis, J.; Kamal MA, M.; Koch, M.; Bischoff, M.; Gordon, S.; Loretz, B.; Lehr, C.-M. Bioinspired Liposomes for Oral Delivery of Colistin to Combat Intracellular Infections by Salmonella enterica. Adv. Healthc. Mater. 2019, 8, e1900564. [Google Scholar] [CrossRef] [Green Version]
- Faya, M.; Hazzah, H.A.; Omolo, C.A.; Agrawal, N.; Maji, R.; Walvekar, P.; Mocktar, C.; Nkambule, B.; Rambharose, S.; Albericio, F.; et al. Novel formulation of antimicrobial peptides enhances antimicrobial activity against methicillin-resistant Staphylococcus aureus (MRSA). Amino Acids 2020, 52, 1439–1457. [Google Scholar] [CrossRef]
- Riool, M.; De Breij, A.; Drijfhout, J.W.; Nibbering, P.H.; Zaat, S.A.J. Antimicrobial Peptides in Biomedical Device Manufacturing. Front. Chem. 2017, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- de Breij, A.; Riool, M.; Kwakman, P.; de Boer, L.; Cordfunke, R.; Drijfhout, J.; Cohen, O.; Emanuel, N.; Zaat, S.; Nibbering, P.; et al. Prevention of Staphylococcus aureus biomaterial-associated infections using a polymer-lipid coating containing the antimicrobial peptide OP-145. J. Control. Release 2016, 222, 1–8. [Google Scholar] [CrossRef]
- Shi, J.; Liu, Y.; Wang, Y.; Zhang, J.; Zhao, S.; Yang, G. Biological and immunotoxicity evaluation of antimicrobial peptide-loaded coatings using a layer-by-layer process on titanium. Sci. Rep. 2015, 5, 16336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K. Controlled drug delivery systems: Past forward and future back. J. Control. Release 2014, 190, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Peulen, T.-O.; Wilkinson, K.J. Diffusion of Nanoparticles in a Biofilm. Environ. Sci. Technol. 2011, 45, 3367–3373. [Google Scholar] [CrossRef]
- Battaglia, L.; Gallarate, M. Lipid nanoparticles: State of the art, new preparation methods and challenges in drug delivery. Expert Opin. Drug Deliv. 2012, 9, 497–508. [Google Scholar] [CrossRef]
- Has, C.; Sunthar, P. A comprehensive review on recent preparation techniques of liposomes. J. Liposome Res. 2020, 30, 336–365. [Google Scholar] [CrossRef]
- Kousalová, J.; Etrych, T. Polymeric Nanogels as Drug Delivery Systems. Physiol. Res. 2018, 67, S305–S317. [Google Scholar] [CrossRef]
- Li, C.; Obireddy, S.R.; Lai, W.-F. Preparation and use of nanogels as carriers of drugs. Drug Deliv. 2021, 28, 1594–1602. [Google Scholar] [CrossRef]
- Vauthier, C.; Bouchemal, K. Methods for the Preparation and Manufacture of Polymeric Nanoparticles. Pharm. Res. 2008, 26, 1025–1058. [Google Scholar] [CrossRef] [PubMed]
- Vert, M.; Doi, Y.; Hellwich, K.-H.; Hess, M.; Hodge, P.; Kubisa, P.; Rinaudo, M.; Schué, F. Terminology for biorelated polymers and applications (IUPAC Recommendations 2012). Pure Appl. Chem. 2012, 84, 377–410. [Google Scholar] [CrossRef]
- Park, K. Nanotechnology: What it can do for drug delivery. J. Control. Release 2007, 120, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, S.; Wu, S.Y. The use of lipid-based nanocarriers for targeted pain therapies. Front. Pharmacol. 2013, 4, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Sun, J.; Liang, D. Aggregation, Fusion, and Leakage of Liposomes Induced by Peptides. Langmuir 2014, 30, 7334–7342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rozek, A.; Hancock, R. Interaction of Cationic Antimicrobial Peptides with Model Membranes. J. Biol. Chem. 2001, 276, 35714–35722. [Google Scholar] [CrossRef] [Green Version]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Khan, O.; Chaudary, N. The Use of Amikacin Liposome Inhalation Suspension (Arikayce) in the Treatment of Refractory Nontuberculous Mycobacterial Lung Disease in Adults. Drug Des. Dev. Ther. 2020, 14, 2287–2294. [Google Scholar] [CrossRef]
- Li, Y.; Tang, C.; Zhang, E.; Yang, L. Electrostatically entrapped colistin liposomes for the treatment of Pseudomonas aeruginosa infection. Pharm. Dev. Technol. 2017, 22, 436–444. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Matsui, Y.; Yamamoto, H.; Kawashima, Y. Mucoadhesive properties of carbopol or chitosan-coated liposomes and their effectiveness in the oral administration of calcitonin to rats. J. Control. Release 2003, 86, 235–242. [Google Scholar] [CrossRef]
- Fang, Y.; Lou, M.-M.; Li, B.; Xie, G.-L.; Wang, F.; Zhang, L.-X.; Luo, Y.-C. Characterization of Burkholderia cepacia complex from cystic fibrosis patients in China and their chitosan susceptibility. World J. Microbiol. Biotechnol. 2010, 26, 443–450. [Google Scholar] [CrossRef]
- De Paz, L.E.C.; Resin, A.; Howard, K.A.; Sutherland, D.S.; Wejse, P.L. Antimicrobial effect of chitosan nanoparticles on Streptococcus mutans biofilms. Appl. Environ. Microbiol. 2011, 77, 3892–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, S.; Seino, H.; Akiyama, Y.; Nonaka, I. Chitosan: A biocompatible material for oral and intravenous administrations. In Progress in Biomedical Polymers; Gebelein, C.G., Dunn, R.L., Eds.; Springer: Boston, MA, USA, 1990. [Google Scholar]
- Laverde-Rojas, V.; Liscano, Y.; Rivera-Sánchez, S.P.; Ocampo-Ibáñez, I.D.; Betancourt, Y.; Alhajj, M.J.; Yarce, C.J.; Salamanca, C.H.; Oñate-Garzón, J. Antimicrobial Contribution of Chitosan Surface-Modified Nanoliposomes Combined with Colistin against Sensitive and Colistin-Resistant Clinical Pseudomonas aeruginosa. Pharmaceutics 2020, 13, 41. [Google Scholar] [CrossRef]
- Aboumanei, M.H.; Mahmoud, A.F.; Motaleb, M.A. Formulation of chitosan coated nanoliposomes for the oral delivery of colistin sulfate: In vitro characterization, 99mTc-radiolabeling and in vivo biodistribution studies. Drug Dev. Ind. Pharm. 2021, 47, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhu, Y.; Luan, P.; Xu, J.; Ru, G.; Fu, J.-G.; Sang, N.; Xiong, Y.; He, Y.; Lin, G.-Q.; et al. Bacteria-Anchoring Hybrid Liposome Capable of Absorbing Multiple Toxins for Antivirulence Therapy of Escherichia coli Infection. ACS Nano 2021, 15, 4173–4185. [Google Scholar] [CrossRef]
- Wang, S.; Yu, S.; Lin, Y.; Zou, P.; Chai, G.; Yu, H.H.; Wickremasinghe, H.; Shetty, N.; Ling, J.; Li, J.; et al. Co-Delivery of Ciprofloxacin and Colistin in Liposomal Formulations with Enhanced In Vitro Antimicrobial Activities against Multidrug Resistant Pseudomonas aeruginosa. Pharm. Res. 2018, 35, 187. [Google Scholar] [CrossRef]
- Yu, S.; Wang, S.; Zou, P.; Chai, G.; Lin, Y.-W.; Velkov, T.; Li, J.; Pan, W.; Zhou, Q.T. Inhalable liposomal powder formulations for co-delivery of synergistic ciprofloxacin and colistin against multi-drug resistant gram-negative lung infections. Int. J. Pharm. 2019, 575, 118915. [Google Scholar] [CrossRef]
- Yu, S.; Yuan, H.; Chai, G.; Peng, K.; Zou, P.; Li, X.; Li, J.; Zhou, F.; Chan, H.-K.; Zhou, Q.T. Optimization of inhalable liposomal powder formulations and evaluation of their in vitro drug delivery behavior in Calu-3 human lung epithelial cells. Int. J. Pharm. 2020, 586, 119570. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.; Park, H.; Yu, S.; Zhou, F.; Li, J.; Xu, Q.; Zhou, Q. Evaluation of co-delivery of colistin and ciprofloxacin in liposomes using an in vitro human lung epithelial cell model. Int. J. Pharm. 2019, 569, 118616. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Z.; Wang, X.; Chen, Y.; Wu, F.; Men, K.; Xu, T.; Luo, Y.; Yang, L. Novel antimicrobial peptide-modified azithromycin-loaded liposomes against methicillin-resistant Staphylococcus aureus. Int. J. Nanomed. 2016, 11, 6781–6794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoee, S.; Yaghoobian, M. Niosomes: A novel approach in modern drug delivery systems. In Nanostructures for Drug Delivery; Elsevier: Amsterdam, The Netherlands, 2017; pp. 207–237. [Google Scholar]
- Chauhan, M.K.; Bhatt, N. Bioavailability Enhancement of Polymyxin B With Novel Drug Delivery: Development and Optimization Using Quality-by-Design Approach. J. Pharm. Sci. 2018, 108, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021. [Google Scholar] [CrossRef]
- Fang, C.L.; Al-Suwayeh, S.A.; Fang, J.Y. Nanostructured lipid carriers (NLCs) for drug delivery and targeting. Recent Pat. Nanotechnol. 2013, 7, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, S.; Shanmuganathan, S.; Anbarasan, B.; Sandhya, K. Nanostructured lipid carriers (NLCs): A Novel based nano carrier for drug delivery and drug targeting. Adv. J. Pharm. Life Sci. Res. 2016, 4, 17–24. [Google Scholar]
- Ryan, A.; Patel, P.; O’Connor, P.M.; Ross, R.P.; Hill, C.; Hudson, S.P. Pharmaceutical design of a delivery system for the bacteriocin lacticin 3147. Drug Deliv. Transl. Res. 2021, 11, 1735–1751. [Google Scholar] [CrossRef]
- Severino, P.; Silveira, E.F.; Loureiro, K.; Chaud, M.V.; Antonini, D.; Lancellotti, M.; Sarmento, V.H.; da Silva, C.F.; Santana, M.H.A.; Souto, E.B. Antimicrobial activity of polymyxin-loaded solid lipid nanoparticles (PLX-SLN): Characterization of physicochemical properties and in vitro efficacy. Eur. J. Pharm. Sci. 2017, 106, 177–184. [Google Scholar] [CrossRef]
- Fumakia, M.; Ho, E.A. Nanoparticles Encapsulated with LL37 and Serpin A1 Promotes Wound Healing and Synergistically Enhances Antibacterial Activity. Mol. Pharm. 2016, 13, 2318–2331. [Google Scholar] [CrossRef]
- Salvi, V.R.; Pawar, P. Nanostructured lipid carriers (NLC) system: A novel drug targeting carrier. J. Drug Deliv. Sci. Technol. 2019, 51, 255–267. [Google Scholar] [CrossRef]
- Serramitjana, E.S.; Fusté, E.; Martínez-Garriga, B.; Merlos, A.; Pastor, M.; Pedraz, J.; Esquisabel, A.; Bachiller, D.; Vinuesa, T.; Viñas, M. Killing effect of nanoencapsulated colistin sulfate on Pseudomonas aeruginosa from cystic fibrosis patients. J. Cyst. Fibros. 2015, 15, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Sans-Serramitjana, E.; Jorba, M.; Pedraz, J.L.; Vinuesa, T.; Viñas, M. Determination of the spatiotemporal dependence of Pseudomonas aeruginosa biofilm viability after treatment with NLC-colistin. Int. J. Nanomed. 2017, 12, 4409–4413. [Google Scholar] [CrossRef] [Green Version]
- Pastor, M.; Basas, J.; Vairo, C.; Gainza, G.; Moreno-Sastre, M.; Gomis, X.; Fleischer, A.; Palomino, E.; Bachiller, D.; Gutiérrez, F.B.; et al. Safety and effectiveness of sodium colistimethate-loaded nanostructured lipid carriers (SCM-NLC) against P. aeruginosa: In vitro and in vivo studies following pulmonary and intramuscular administration. Nanomed. Nanotechnol. Biol. Med. 2019, 18, 101–111. [Google Scholar] [CrossRef]
- Garcia-Orue, I.; Gainza, G.; Girbau, C.; Alonso, R.; Aguirre, J.J.; Pedraz, J.L.; Igartua, M.; Hernandez, R.M. LL37 loaded nanostructured lipid carriers (NLC): A new strategy for the topical treatment of chronic wounds. Eur. J. Pharm. Biopharm. 2016, 108, 310–316. [Google Scholar] [CrossRef]
- Rocha, E.D.; Ferreira, M.R.S.; Neto, E.D.S.; Barbosa, E.J.; Löbenberg, R.; Lourenço, F.R.; Bou-Chacra, N. Enhanced In Vitro Antimicrobial Activity of Polymyxin B–Coated Nanostructured Lipid Carrier Containing Dexamethasone Acetate. J. Pharm. Innov. 2020, 16, 125–135. [Google Scholar] [CrossRef]
- Monteiro, L.M.; Löbenberg, R.; Fotaki, N.; Araujo, G.; Cotrim, P.C.; Bou-Chacra, N. Co-delivery of buparvaquone and polymyxin B in a nanostructured lipid carrier for leishmaniasis treatment. J. Glob. Antimicrob. Resist. 2019, 18, 279–283. [Google Scholar] [CrossRef]
- Huynh, N.; Passirani, C.; Saulnier, P.; Benoit, J. Lipid nanocapsules: A new platform for nanomedicine. Int. J. Pharm. 2009, 379, 201–209. [Google Scholar] [CrossRef]
- Matougui, N.; Groo, A.-C.; Umerska, A.; Cassisa, V.; Saulnier, P. A comparison of different strategies for antimicrobial peptides incorporation onto/into lipid nanocapsules. Nanomedicine 2019, 14, 1647–1662. [Google Scholar] [CrossRef] [PubMed]
- Groo, A.-C.; Matougui, N.; Umerska, A.; Saulnier, P. Reverse micelle-lipid nanocapsules: A novel strategy for drug delivery of the plectasin derivate AP138 antimicrobial peptide. Int. J. Nanomed. 2018, 13, 7565–7574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umerska, A.; Cassisa, V.; Bastiat, G.; Matougui, N.; Nehme, H.; Manero, F.; Eveillard, M.; Saulnier, P. Synergistic interactions between antimicrobial peptides derived from plectasin and lipid nanocapsules containing monolaurin as a cosurfactant against Staphylococcus aureus. Int. J. Nanomed. 2017, 12, 5687–5699. [Google Scholar] [CrossRef] [Green Version]
- Rozenbaum, R.T.; Su, L.; Umerska, A.; Eveillard, M.; Håkansson, J.; Mahlapuu, M.; Huang, F.; Liu, J.; Zhang, Z.; Shi, L.; et al. Antimicrobial synergy of monolaurin lipid nanocapsules with adsorbed antimicrobial peptides against Staphylococcus aureus biofilms in vitro is absent in vivo. J. Control. Release 2018, 293, 73–83. [Google Scholar] [CrossRef]
- Garg, G.; Saraf, S.; Saraf, S. Cubosomes: An overview. Biol. Pharm. Bull. 2007, 30, 350–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barriga, H.M.; Holme, M.N.; Stevens, M.M. Cubosomes: The Next Generation of Smart Lipid Nanoparticles? Angew. Chem. Int. Ed. 2019, 58, 2958–2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Håkansson, J.; Ringstad, L.; Umerska, A.; Johansson, J.; Andersson, T.; Boge, L.; Rozenbaum, R.T.; Sharma, P.; Tollbäck, P.; Björn, C.; et al. Characterization of the in vitro, ex vivo, and in vivo Efficacy of the Antimicrobial Peptide DPK-060 Used for Topical Treatment. Front. Cell. Infect. Microbiol. 2019, 9, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boge, L.; Bysell, H.; Ringstad, L.; Wennman, D.; Umerska, A.; Cassisa, V.; Eriksson, J.; Joly-Guillou, M.-L.; Edwards, K.; Andersson, M. Lipid-Based Liquid Crystals as Carriers for Antimicrobial Peptides: Phase Behavior and Antimicrobial Effect. Langmuir 2016, 32, 4217–4228. [Google Scholar] [CrossRef] [Green Version]
- Boge, L.; Hallstensson, K.; Ringstad, L.; Johansson, J.; Andersson, T.; Davoudi, M.; Larsson, P.T.; Mahlapuu, M.; Håkansson, J.; Andersson, M. Cubosomes for topical delivery of the antimicrobial peptide LL-37. Eur. J. Pharm. Biopharm. 2019, 134, 60–67. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Van Der Mei, H.C.; Ren, Y.; Busscher, H.J.; Shi, L. Lipid-Based Antimicrobial Delivery-Systems for the Treatment of Bacterial Infections. Front. Chem. 2020, 7, 872. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, F.; Wu, L.; Tian, Y.; Zhou, B.; Zhang, X.; Huang, R.; Yu, C.; He, G.; Yang, L. Novel Self-Assembled Micelles Based on Cholesterol-Modified Antimicrobial Peptide (DP7) for Safe and Effective Systemic Administration in Animal Models of Bacterial Infection. Antimicrob. Agents Chemother. 2018, 62, e00368-18. [Google Scholar] [CrossRef] [Green Version]
- Madhumanchi, S.; Suedee, R.; Nakpheng, T.; Tinpun, K.; Temboot, P.; Srichana, T. Binding interactions of bacterial lipopolysaccharides to polymyxin B in an amphiphilic carrier ‘sodium deoxycholate sulfate’. Colloids Surf. B Biointerfaces 2019, 182, 110374. [Google Scholar] [CrossRef]
- Temboot, P.; Kaewpaiboon, S.; Tinpun, K.; Nakpeng, T.; Khalil, R.; Ul-Haq, Z.; Thamlikitkul, V.; Tiengrim, S.; Srichana, T. Potential of sodium deoxycholate sulfate as a carrier for polymyxin B: Physicochemical properties, bioactivity and in vitro safety. J. Drug Deliv. Sci. Technol. 2020, 58, 101779. [Google Scholar] [CrossRef]
- Kumar, P.; Pletzer, D.; Haney, E.F.; Rahanjam, N.; Cheng, J.T.J.; Yue, M.; Aljehani, W.; Hancock, R.; Kizhakkedathu, J.N.; Straus, S.K. Aurein-Derived Antimicrobial Peptides Formulated with Pegylated Phospholipid Micelles to Target Methicillin-Resistant Staphylococcus aureus Skin Infections. ACS Infect. Dis. 2019, 5, 443–453. [Google Scholar] [CrossRef]
- Li, Y.; Huang, L.; Tang, C.; Zhang, E.; Ding, L.; Yang, L. Preparation and characterisation of the colistin-entrapped liposome driven by electrostatic interaction for intravenous administration. J. Microencapsul. 2016, 33, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.J.; Mai, S.M.; Fairclough, J.P.A.; Hamley, I.W. Structures of amphiphilic block copolymers in their liquid and solid states. In Amphiphilic Block Copolymers; Alexandridis, P., Lindman, B., Eds.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2000; pp. 151–167. [Google Scholar]
- Durham, O.Z.; Poetz, K.L.; Shipp, D.A. Polyanhydride Nanoparticles: Thiol-Ene ‘Click’ Polymerizations Provide Functionalized and Cross-Linkable Nanoparticles with Tuneable Degradation Times. Aust. J. Chem. 2017, 70, 735–742. [Google Scholar] [CrossRef]
- Lichtenthaler, F.W. Carbohydrates: Occurrence, Structures and Chemistry. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Liu, Z.; Jiao, Y.; Wang, Y.; Zhou, C.; Zhang, Z. Polysaccharides-based nanoparticles as drug delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 1650–1662. [Google Scholar] [CrossRef]
- Chereddy, K.K.; Her, C.-H.; Comune, M.; Moia, C.; Lopes, A.; Porporato, P.E.; Vanacker, J.; Lam, M.C.; Steinstraesser, L.; Sonveaux, P.; et al. PLGA nanoparticles loaded with host defense peptide LL37 promote wound healing. J. Control. Release 2014, 194, 138–147. [Google Scholar] [CrossRef]
- Casciaro, B.; D’Angelo, I.; Zhang, X.; Loffredo, M.R.; Conte, G.; Cappiello, F.; Quaglia, F.; Di, Y.-P.P.; Ungaro, F.; Mangoni, M.L. Poly(lactide-co-glycolide) Nanoparticles for Prolonged Therapeutic Efficacy of Esculentin-1a-Derived Antimicrobial Peptides against Pseudomonas aeruginosa Lung Infection: In Vitro and in Vivo Studies. Biomacromolecules 2019, 20, 1876–1888. [Google Scholar] [CrossRef]
- Vijayan, A.; James, P.P.; Nanditha, C.; Kumar, G.V. Multiple cargo deliveries of growth factors and antimicrobial peptide using biodegradable nanopolymer as a potential wound healing system. Int. J. Nanomed. 2019, 14, 2253–2263. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Sequeda, N.; Ruiz, J.; Ortiz, C.; Urquiza, M.; Torres, R. Potent and Specific Antibacterial Activity against Escherichia coli O157:H7 and Methicillin Resistant Staphylococcus aureus (MRSA) of G17 and G19 Peptides Encapsulated into Poly-Lactic-Co-Glycolic Acid (PLGA) Nanoparticles. Antibiotics 2020, 9, 384. [Google Scholar] [CrossRef]
- Sharma, A.; Vaghasiya, K.; Ray, E.; Verma, R.K. Nano-encapsulated HHC10 host defense peptide (HDP) reduces the growth of Escherichia coli via multimodal mechanisms. Artif. Cells Nanomed. Biotechnol. 2018, 46, S156–S165. [Google Scholar] [CrossRef] [Green Version]
- Zaaba, N.F.; Jaafar, M. A review on degradation mechanisms of polylactic acid: Hydrolytic, photodegradative, microbial, and enzymatic degradation. Polym. Eng. Sci. 2020, 60, 2061–2075. [Google Scholar] [CrossRef]
- Cruz, J.; Flórez, J.; Torres, R.; Urquiza, M.; Gutiérrez, J.A.; Guzmán, F.; Ortiz, C.C. Antimicrobial activity of a new synthetic peptide loaded in polylactic acid or poly(lactic-co-glycolic) acid nanoparticles against Pseudomonas aeruginosa, Escherichia coli O157:H7 and methicillin resistant Staphylococcus aureus (MRSA). Nanotechnology 2017, 28, 135102. [Google Scholar] [CrossRef]
- Panyam, J.; Williams, D.; Dash, A.; Leslie-Pelecky, D.; Labhasetwar, V. Solid-state Solubility Influences Encapsulation and Release of Hydrophobic Drugs from PLGA/PLA Nanoparticles. J. Pharm. Sci. 2004, 93, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Tao, Y.; Pan, Y.; Qu, W.; Cheng, G.; Huang, L.; Chen, D.; Wang, X.; Liu, Z.; Yuan, Z. Biodegradable nanoparticles for intracellular delivery of antimicrobial agents. J. Control. Release 2014, 187, 101–117. [Google Scholar] [CrossRef]
- Tsuji, H.; Eto, T.; Sakamoto, Y. Synthesis and Hydrolytic Degradation of Substituted Poly(DL-Lactic Acid)s. Materials 2011, 4, 1384–1398. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, S.; Li, H.; Bao, Y.; Chen, H.; Yuan, M. Preparation, Characterization and Biological Activity of Cathelicidin-BF-30-Loaded Poly(LA-co-MA) Microspheres. J. Nanosci. Nanotechnol. 2019, 19, 2435–2442. [Google Scholar] [CrossRef]
- Teixeira, M.C.; Carbone, C.; Sousa, M.C.; Espina, M.; Garcia, M.L.; Sanchez-Lopez, E.; Souto, E.B. Nanomedicines for the Delivery of Antimicrobial Peptides (AMPs). Nanomaterials 2020, 10, 560. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Zhan, B.; Zhang, W.; Qin, D.; Xia, G.; Zhang, H.; Peng, M.; Li, S.-A.; Zhang, Y.; Gao, Y.; et al. Carboxymethyl chitosan nanoparticles loaded with bioactive peptide OH-CATH30 benefit nonscar wound healing. Int. J. Nanomed. 2018, 13, 5771–5786. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Na, R.; Wang, X.; Liu, H.; Zhao, L.; Sun, X.; Ma, G.; Cui, F. Fabrication of Antimicrobial Peptide-Loaded PLGA/Chitosan Composite Microspheres for Long-Acting Bacterial Resistance. Molecules 2017, 22, 1637. [Google Scholar] [CrossRef] [Green Version]
- Sriamornsak, P. Application of pectin in oral drug delivery. Expert Opin. Drug Deliv. 2011, 8, 1009–1023. [Google Scholar] [CrossRef]
- Jonassen, H.; Treves, A.; Kjøniksen, A.-L.; Smistad, G.; Hiorth, M. Preparation of Ionically Cross-Linked Pectin Nanoparticles in the Presence of Chlorides of Divalent and Monovalent Cations. Biomacromolecules 2013, 14, 3523–3531. [Google Scholar] [CrossRef]
- Krivorotova, T.; Cirkovas, A.; Maciulyte, S.; Staneviciene, R.; Budriene, S.; Serviene, E.; Sereikaite, J. Nisin-loaded pectin nanoparticles for food preservation. Food Hydrocoll. 2016, 54, 49–56. [Google Scholar] [CrossRef]
- Thote, A.J.; Chappell, J.T., Jr.; Kumar, R.; Gupta, R.B. Reduction in the initial-burst release by surface crosslinking of PLGA microparticles containing hydrophilic or hydrophobic drugs. Drug Dev. Ind. Pharm. 2005, 31, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Bertram, J.P.; Jay, S.; Hynes, S.R.; Robinson, R.; Criscione, J.M.; Lavik, E.B. Functionalized poly(lactic-co-glycolic acid) enhances drug delivery and provides chemical moieties for surface engineering while preserving biocompatibility. Acta Biomater. 2009, 5, 2860–2871. [Google Scholar] [CrossRef] [Green Version]
- Sebti, T.; Amighi, K. Preparation and in vitro evaluation of lipidic carriers and fillers for inhalation. Eur. J. Pharm. Biopharm. 2006, 63, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Water, J.; Smart, S.; Franzyk, H.; Foged, C.; Nielsen, H.M. Nanoparticle-mediated delivery of the antimicrobial peptide plectasin against Staphylococcus aureus in infected epithelial cells. Eur. J. Pharm. Biopharm. 2015, 92, 65–73. [Google Scholar] [CrossRef] [PubMed]
- González-Alvarez, M.; González-Alvarez, I.; Bermejo, M. Hydrogels: An interesting strategy for smart drug delivery. Ther. Deliv. 2013, 4, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.K.; Siegwart, D.J.; Matyjaszewski, K. Synthesis and Biodegradation of Nanogels as Delivery Carriers for Carbohydrate Drugs. Biomacromolecules 2007, 8, 3326–3331. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Vinogradov, S.V. Nanogels as Pharmaceutical Carriers: Finite Networks of Infinite Capabilities. Angew. Chem. Int. Ed. 2009, 48, 5418–5429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, C.; Pereira, P.; Gama, M. Self-Assembled Hydrogel Nanoparticles for Drug Delivery Applications. Materials 2010, 3, 1420–1460. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Park, K. Environment-sensitive hydrogels for drug delivery. Adv. Drug Deliv. Rev. 2001, 53, 321–339. [Google Scholar] [CrossRef]
- Canal, T.; Peppas, N.A. Correlation between mesh size and equilibrium degree of swelling of polymeric networks. J. Biomed. Mater. Res. 1989, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Liao, W.; Wang, W.; Zhou, J.; Tan, W.; Xiang, W.; Zhang, J.; Guo, L.; Chen, T.; Ma, D.; et al. Genipin-crosslinked carboxymethyl chitosan nanogel for lung-targeted delivery of isoniazid and rifampin. Carbohydr. Polym. 2018, 197, 403–413. [Google Scholar] [CrossRef]
- Schatz, C.; Lucas, J.-M.; Viton, C.; Domard, A.; Pichot, A.C.; Delair, T. Formation and Properties of Positively Charged Colloids Based on Polyelectrolyte Complexes of Biopolymers. Langmuir 2004, 20, 7766–7778. [Google Scholar] [CrossRef]
- Elgadir, M.; Uddin, S.; Ferdosh, S.; Adam, A.; Chowdhury, A.J.K.; Sarker, Z.I. Impact of chitosan composites and chitosan nanoparticle composites on various drug delivery systems: A review. J. Food Drug Anal. 2015, 23, 619–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Liu, Y.; Liu, W.; Lu, C.; Wang, L. Chitosan microparticles ionically cross-linked with poly (gamma-glutamic acid) as antimicrobial peptides and nitric oxide delivery systems. Biochem. Eng. J. 2015, 95, 78–85. [Google Scholar] [CrossRef]
- Calvo, P.; Remuñán-López, C.; Vila-Jato, J.L.; Alonso, M.J. Chitosan and Chitosan/Ethylene Oxide-Propylene Oxide Block Copolymer Nanoparticles as Novel Carriers for Proteins and Vaccines. Pharm. Res. 1997, 14, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Piras, A.M.; Emaisetta, G.; Esandreschi, S.; Egazzarri, M.; Ebartoli, C.; Grassi, L.; Eesin, S.; Chiellini, F.; Ebatoni, G. Chitosan nanoparticles loaded with the antimicrobial peptide temporin B exert a long-term antibacterial activity in vitro against clinical isolates of Staphylococcus epidermidis. Front. Microbiol. 2015, 6, 372. [Google Scholar] [CrossRef] [Green Version]
- Rishi, P.; Bhogal, A.; Arora, S.; Pandey, S.K.; Verma, I.; Kaur, I.P. Improved oral therapeutic potential of nanoencapsulated cryptdin formulation against Salmonella infection. Eur. J. Pharm. Sci. 2015, 72, 27–33. [Google Scholar] [CrossRef]
- Sharma, R.; Raghav, R.; Priyanka, K.; Rishi, P.; Sharma, S.; Srivastava, S.; Verma, I. Exploiting chitosan and gold nanoparticles for antimycobacterial activity of in silico identified antimicrobial motif of human neutrophil peptide-1. Sci. Rep. 2019, 9, 7866. [Google Scholar] [CrossRef] [Green Version]
- Lehr, C.-M.; Bouwstra, J.A.; Schacht, E.H.; Junginger, H.E. In vitro evaluation of mucoadhesive properties of chitosan and some other natural polymers. Int. J. Pharm. 1992, 78, 43–48. [Google Scholar] [CrossRef]
- Kuhlmann, N.; Nehls, C.; Heinbockel, L.; Correa, W.; Moll, R.; Gutsmann, T.; Hübner, C.; Englisch, U.; Brandenburg, K. Encapsulation and release of as pidasept peptides in polysaccharide formulation for oral application. Eur. J. Pharm. Sci. 2021, 158, 105687. [Google Scholar] [CrossRef]
- Kheirollahpour, M.; Mehrabi, M.; Dounighi, N.M.; Mohammadi, M.; Masoudi, A. Nanoparticles and Vaccine Development. Pharm. Nanotechnol. 2020, 8, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Kłodzińska, S.N.; Molchanova, N.; Franzyk, H.; Hansen, P.R.; Damborg, P.; Nielsen, H.M. Biopolymer nanogels improve antibacterial activity and safety profile of a novel lysine-based α-peptide/β-peptoid peptidomimetic. Eur. J. Pharm. Biopharm. 2018, 128, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Water, J.J.; Kim, Y.; Maltesen, M.J.; Franzyk, H.; Foged, C.; Nielsen, H.M. Hyaluronic acid-based nanogels produced by microfluidics-facilitated self-assembly improves the safety profile of the cationic host defence peptide novicidin. Pharm. Res. 2015, 32, 2727–2735. [Google Scholar]
- Simonson, A.W.; Lawanprasert, A.; Goralski, T.D.; Keiler, K.; Medina, S.H. Bioresponsive peptide-polysaccharide nanogels—A versatile delivery system to augment the utility of bioactive cargo. Nanomed. Nanotechnol. Biol. Med. 2019, 17, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.P.; Gonçalves, C.; Costa, C.; Sousa, J.; Gomes, A.R.; Castro, G.; Pedrosa, J.; Appelberg, R.; Gama, F.M. Delivery of LLKKK18 loaded into self-assembling hyaluronic acid nanogel for tuberculosis treatment. J. Control. Release 2016, 235, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Montanari, E.; D’Arrigo, G.; Di Meo, C.; Virga, A.; Coviello, T.; Passariello, C.; Matricardi, P. Chasing bacteria within the cells using levofloxacin-loaded hyaluronic acid nanohydrogels. Eur. J. Pharm. Biopharm. 2014, 87, 518–523. [Google Scholar] [CrossRef]
- Kłodzińska, S.N.; Wan, F.; Jumaa, H.; Sternberg, C.; Rades, T.; Nielsen, H.M. Utilizing nanoparticles for improving anti-biofilm effects of azithromycin: A head-to-head comparison of modified hyaluronic acid nanogels and coated poly (lactic-co-glycolic acid) nanoparticles. J. Colloid Interface Sci. 2019, 555, 595–606. [Google Scholar] [CrossRef]
- Felgentreff, K.; Beisswenger, C.; Griese, M.; Gulder, T.; Bringmann, G.; Bals, R. The antimicrobial peptide cathelicidin interacts with airway mucus. Peptides 2006, 27, 3100–3106. [Google Scholar] [CrossRef]
- Montanari, E.; Di Meo, C.; Sennato, S.; Francioso, A.; Marinelli, A.L.; Ranzo, F.; Schippa, S.; Coviello, T.; Bordi, F.; Matricardi, P. Hyaluronan-cholesterol nanohydrogels: Characterisation and effectiveness in carrying alginate lyase. New Biotechnol. 2017, 37, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I. Alginates. In Biomaterials-Novel Materials from Biological Sources; Byrom, D., Ed.; Palgrave Macmillan: London, UK, 1991; pp. 307–331. [Google Scholar]
- Borro, B.C.; Bohr, A.; Bucciarelli, S.; Boetker, J.P.; Foged, C.; Rantanen, J.; Malmsten, M. Microfluidics-based self-assembly of peptide-loaded microgels: Effect of three dimensional (3D) printed micromixer design. J. Colloid Interface Sci. 2019, 538, 559–568. [Google Scholar] [CrossRef]
- Masuda, T.; Shimada, N.; Maruyama, A. A Thermoresponsive Cationic Comb-Type Copolymer Enhances Membrane Disruption Activity of an Amphiphilic Peptide. Biomacromolecules 2018, 19, 1333–1339. [Google Scholar] [CrossRef]
- Nordström, R.; Nyström, L.; Andrén, O.C.; Malkoch, M.; Umerska, A.; Davoudi, M.; Schmidtchen, A.; Malmsten, M. Membrane interactions of microgels as carriers of antimicrobial peptides. J. Colloid Interface Sci. 2018, 513, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Chai, M.; Gao, Y.; Liu, J.; Deng, Y.; Hu, D.; Jin, Q.; Ji, J. Polymyxin B-Polysaccharide Polyion Nanocomplex with Improved Biocompatibility and Unaffected Antibacterial Activity for Acute Lung Infection Management. Adv. Healthc. Mater. 2020, 9, 1901542. [Google Scholar] [CrossRef]
- Borro, B.C.; Toussaint, M.S.; Bucciarelli, S.; Malmsten, M. Effects of charge contrast and composition on microgel formation and interactions with bacteria-mimicking liposomes. Biochim. Biophys. Acta Gen. Subj. 2019, 1865, 129485. [Google Scholar] [CrossRef]
- Walvekar, P.; Gannimani, R.; Salih, M.; Makhathini, S.; Mocktar, C.; Govender, T. Self-assembled oleyamine grafted hyaluronic acid polymerosomes for delivery of vancomycin against methicillin resistant Staphylococcus aureus (MRSA). Colloids Surf. B Biointerfaces 2019, 182, 110388. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Feng, S.; Qie, J.; Wei, X.; Yan, H.; Liu, K. Polyion complexes of a cationic antimicrobial peptide as a potential systemically administered antibiotic. Int. J. Pharm. 2019, 554, 284–291. [Google Scholar] [CrossRef]
- Insua, I.; Majok, S.; Peacock, A.; Krachler, A.M.; Fernandez-Trillo, F. Preparation and antimicrobial evaluation of polyion complex (PIC) nanoparticles loaded with polymyxin B. Eur. Polym. J. 2017, 87, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordström, R.; Andrén, O.C.; Singh, S.; Malkoch, M.; Davoudi, M.; Schmidtchen, A.; Malmsten, M. Degradable dendritic nanogels as carriers for antimicrobial peptides. J. Colloid Interface Sci. 2019, 554, 592–602. [Google Scholar] [CrossRef]
- Singh, S.; Datta, A.; Borro, B.C.; Davoudi, M.; Schmidtchen, A.; Bhunia, A.; Malmsten, M. Conformational Aspects of High Content Packing of Antimicrobial Peptides in Polymer Microgels. ACS Appl. Mater. Interfaces 2017, 9, 40094–40106. [Google Scholar] [CrossRef]
- Monteiro, C.; Costa, F.; Pirttilä, A.M.; Tejesvi, M.V.; Martins, M.C.L. Prevention of urinary catheter-associated infections by coating antimicrobial peptides from crowberry endophytes. Sci. Rep. 2019, 9, 10753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Lo, J.C.; Yan, M.; Yang, X.; Brooks, D.; Hancock, R.; Lange, D.; Kizhakkedathu, J.N. Anti-adhesive antimicrobial peptide coating prevents catheter associated infection in a mouse urinary infection model. Biomaterials 2017, 116, 69–81. [Google Scholar] [CrossRef]
- Costa, F.; Maia, S.; Gomes, J.; Gomes, P.; Martins, M.C.L. Characterization of hLF1-11 immobilization onto chitosan ultrathin films, and its effects on antimicrobial activity. Acta Biomater. 2014, 10, 3513–3521. [Google Scholar] [CrossRef] [Green Version]
- Costa, F.M.; Maia, S.R.; Gomes, P.A.; Martins, M.C.L. Dhvar5 antimicrobial peptide (AMP) chemoselective covalent immobilization results on higher antiadherence effect than simple physical adsorption. Biomaterials 2015, 52, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Willcox, M.; Hume, E.; Aliwarga, Y.; Kumar, N.; Cole, N. A novel cationic-peptide coating for the prevention of microbial colonization on contact lenses. J. Appl. Microbiol. 2008, 105, 1817–1825. [Google Scholar] [CrossRef]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-Inspired Surface Chemistry for Multifunctional Coatings. Science 2007, 318, 426–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.; Chua, R.R.Y.; Ho, B.; Tambyah, P.A.; Hadinoto, K.; Leong, S.S.J. Development of a catheter functionalized by a polydopamine peptide coating with antimicrobial and antibiofilm properties. Acta Biomater. 2015, 15, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Yue, K.; Kazemzadeh-Narbat, M.; Liu, Y.; Khalilpour, A.; Li, B.; Zhang, Y.S.; Annabi, N.; Khademhosseini, A. Mussel-Inspired Multifunctional Hydrogel Coating for Prevention of Infections and Enhanced Osteogenesis. ACS Appl. Mater. Interfaces 2017, 9, 11428–11439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.W.; Goh, T.W.; Saraswathi, P.; Nyein, C.L.; Setiawan, M.; Riau, A.; Lakshminarayanan, R.; Liu, S.; Tan, D.; Beuerman, R.W.; et al. Effectiveness of Antimicrobial Peptide Immobilization for Preventing Perioperative Cornea Implant-Associated Bacterial Infection. Antimicrob. Agents Chemother. 2014, 58, 5229–5238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Yu, K.; Kindrachuk, J.; Brooks, D.E.; Hancock, R.E.W.; Kizhakkedathu, J.N. Antibacterial Surfaces Based on Polymer Brushes: Investigation on the Influence of Brush Properties on Antimicrobial Peptide Immobilization and Antimicrobial Activity. Biomacromolecules 2011, 12, 3715–3727. [Google Scholar] [CrossRef]
- Yu, K.; Lo, J.C.Y.; Mei, Y.; Haney, E.F.; Siren, E.; Kalathottukaren, M.T.; Hancock, R.E.; Lange, D.; Kizhakkedathu, J.N. Toward Infection-Resistant Surfaces: Achieving High Antimicrobial Peptide Potency by Modulating the Functionality of Polymer Brush and Peptide. ACS Appl. Mater. Interfaces 2015, 7, 28591–28605. [Google Scholar] [CrossRef] [PubMed]
- Muszanska, A.K.; Rochford, E.T.J.; Gruszka, A.; Bastian, A.A.; Busscher, H.J.; Norde, W.; Van Der Mei, H.C.; Herrmann, A. Antiadhesive Polymer Brush Coating Functionalized with Antimicrobial and RGD Peptides to Reduce Biofilm Formation and Enhance Tissue Integration. Biomacromolecules 2014, 15, 2019–2026. [Google Scholar] [CrossRef]
- Mishra, B.; Basu, A.; Chua, R.R.Y.; Saravanan, R.; Tambyah, P.A.; Ho, B.; Chang, M.W.; Leong, S.S.J. Site specific immobilization of a potent antimicrobial peptide onto silicone catheters: Evaluation against urinary tract infection pathogens. J. Mater. Chem. B 2014, 2, 1706–1716. [Google Scholar] [CrossRef] [Green Version]
- Godoy-Gallardo, M.; Mas-Moruno, C.; Yu, K.; Manero, J.M.; Gil, F.J.; Kizhakkedathu, J.N.; Rodriguez, D. Antibacterial Properties of hLf1–11 Peptide onto Titanium Surfaces: A Comparison Study Between Silanization and Surface Initiated Polymerization. Biomacromolecules 2015, 16, 483–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, S.; Ibañez-Fonseca, A.; Aparicio, C.; Rodríguez-Cabello, J.C. Antibiofilm coatings based on protein-engineered polymers and antimicrobial peptides for preventing implant-associated infections. Biomater. Sci. 2020, 8, 2866–2877. [Google Scholar] [CrossRef]
- Shukla, A.; Fleming, K.E.; Chuang, H.F.; Chau, T.M.; Loose, C.R.; Stephanopoulos, G.N.; Hammond, P.T. Controlling the release of peptide antimicrobial agents from surfaces. Biomaterials 2010, 31, 2348–2357. [Google Scholar] [CrossRef] [PubMed]
- Raman, N.; Lee, M.R.; Palecek, S.P.; Lynn, D.M. Polymer multilayers loaded with antifungal beta-peptides kill planktonic Candida albicans and reduce formation of fungal biofilms on the surfaces of flexible catheter tubes. J. Control. Release 2014, 191, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazemzadeh-Narbat, M.; Lai, B.F.; Ding, C.; Kizhakkedathu, J.N.; Hancock, R.; Wang, R. Multilayered coating on titanium for controlled release of antimicrobial peptides for the prevention of implant-associated infections. Biomaterials 2013, 34, 5969–5977. [Google Scholar] [CrossRef] [PubMed]
- Riool, M.; De Breij, A.; De Boer, L.; Kwakman, P.H.S.; Cordfunke, R.A.; Cohen, O.; Malanovic, N.; Emanuel, N.; Lohner, K.; Drijfhout, J.W.; et al. Controlled Release of LL-37-Derived Synthetic Antimicrobial and Anti-Biofilm Peptides SAAP-145 and SAAP-276 Prevents Experimental Biomaterial-Associated Staphylococcus aureus Infection. Adv. Funct. Mater. 2017, 27. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Burian, A.; Wagner, C.; Stanek, J.; Manafi, M.; Böhmdorfer, M.; Jäger, W.; Zeitlinger, M. Plasma protein binding may reduce antimicrobial activity by preventing intra-bacterial uptake of antibiotics, for example clindamycin. J. Antimicrob. Chemother. 2011, 66, 134–137. [Google Scholar] [CrossRef] [Green Version]
- Moncla, B.J.; Pryke, K.; Rohan, L.C.; Graebing, P.W. Degradation of naturally occurring and engineered antimicrobial peptides by proteases. Adv. Biosci. Biotechnol. 2011, 2, 404–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappen, F.G.; Qian, W.; Aleksejūnienė, J.; Leonardo, R.D.T.; Leonardo, M.R.; Haapasalo, M. Inhibition of Sodium Hypochlorite Antimicrobial Activity in the Presence of Bovine Serum Albumin. J. Endod. 2010, 36, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Kopper, P.M.P.; Quintana, R.M.; Jardine, A.P.; Montagner, F.; Parolo, C.C.F.; Morgental, R.D. Effect of human, dentin, albumin and lipopolysaccharide on the antibacteerial activity of endodontic activity of endodontic irrigants. J. Conserv. Dent. 2017, 20, 341–345. [Google Scholar] [CrossRef]
- Starr, C.G.; Wimley, W.C. Antimicrobial peptides are degraded by the cytosolic proteases of human erythrocytes. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2319–2326. [Google Scholar] [CrossRef] [PubMed]
- McCrudden, M.T.C.; McLean, D.T.F.; Zhou, M.; Shaw, J.; Linden, G.; Irwin, C.R.; Lundy, F.T. The Host Defence Peptide LL-37 is Susceptible to Proteolytic Degradation by Wound Fluid Isolated from Foot Ulcers of Diabetic Patients. Int. J. Pept. Res. Ther. 2014, 20, 457–464. [Google Scholar] [CrossRef]
- Dijksteel, G.S.; Ulrich, M.M.; Nibbering, P.H.; Cordfunke, R.A.; Drijfhout, J.W.; Middelkoop, E.; Boekema, B.K. The functional stability, bioactivity and safety profile of synthetic antimicrobial peptide SAAP-148. J. Microbiol. Antimicrob. 2020, 12, 70–80. [Google Scholar]
- de Breij, A.; Haisma, E.M.; Rietveld, M.; El Ghalbzouri, A.; van den Broek, P.J.; Dijkshoorn, L.; Nibbering, P.H. Three-dimensional human skin equivalent as a tool to study Acinetobacter baumannii colonization. Antimicrob. Agents Chemother. 2012, 56, 2459–2464. [Google Scholar] [CrossRef] [Green Version]
- Gloede, J.; Scheerans, C.; Derendorf, H.; Kloft, C. In vitro pharmacodynamic models to determine the effect of antibacterial drugs. J. Antimicrob. Chemother. 2009, 65, 186–201. [Google Scholar] [CrossRef] [Green Version]
- Dijksteel, G.S.; Ulrich, M.M.W.; Vlig, M.; Nibbering, P.H.; Cordfunke, R.A.; Drijfhout, J.W.; Middelkoop, E.; Boekema, B.K.H.L. Potential factors contributing to the poor antimicrobial efficacy of SAAP-148 in a rat wound infection model. Ann. Clin. Microbiol. Antimicrob. 2019, 18, 38. [Google Scholar] [CrossRef] [PubMed]
- Boekema, B.; Pool, L.; Ulrich, M. The effect of a honey based gel and silver sulphadiazine on bacterial infections of in vitro burn wounds. Burns 2013, 39, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Masson-Meyers, D.S.; Andrade, T.A.M.; Caetano, G.F.; Guimaraes, F.R.; Leite, M.N.; Leite, S.N.; Frade, M.A.C. Experimental models and methods for cutaneous wound healing assessment. Int. J. Exp. Pathol. 2020, 101, 21–37. [Google Scholar] [CrossRef]
- Semaniakou, A.; Croll, R.P.; Chappe, V. Animal Models in the Pathophysiology of Cystic Fibrosis. Front. Pharmacol. 2019, 9, 1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagens, W.I.; Oomen, A.G.; de Jong, W.H.; Cassee, F.R.; Sips, A.J. What do we (need to) know about the kinetic properties of nanoparticles in the body? Regul. Toxicol. Pharmacol. 2007, 49, 217–229. [Google Scholar] [CrossRef]
- Kroll, A.; Pillukat, M.H.; Hahn, D.; Schnekenburger, J. Current in vitro methods in nanoparticle risk assessment: Limitations and challenges. Eur. J. Pharm. Biopharm. 2009, 72, 370–377. [Google Scholar] [CrossRef]
- Khan, A.; Xu, M.; Wang, T.; You, C.; Wang, X.; Ren, H.; Zhou, H.; Khan, A.; Han, C.; Li, P. Catechol cross-linked antimicrobial peptide hydrogels prevent multidrug-resistant Acinetobacter baumannii infection in burn wounds. Biosci. Rep. 2019, 39, BSR20190504. [Google Scholar] [CrossRef] [Green Version]
- Nithya, S.; Nimal, T.; Baranwal, G.; Suresh, M.K.; Anju, C.P.; Kumar, V.A.; Mohan, C.G.; Jayakumar, R.; Biswas, R. Preparation, characterization and efficacy of lysostaphin-chitosan gel against Staphylococcus aureus. Int. J. Biol. Macromol. 2018, 110, 157–166. [Google Scholar] [CrossRef]
- Obuobi, S.A.O.; Voo, Z.X.; Low, M.W.; Czarny, B.; Selvarajan, V.; Ibrahim, N.L.; Yang, Y.Y.; Ee, P.L.R. Phenylboronic Acid Functionalized Polycarbonate Hydrogels for Controlled Release of Polymyxin B in Pseudomonas aeruginosa Infected Burn Wounds. Adv. Healthc. Mater. 2018, 7, e1701388. [Google Scholar] [CrossRef]
- Wasiak, J.; Cleland, H.; Campbell, F.; Spinks, A. Dressings for superficial and partial thickness burns. Cochrane Database Syst. Rev. 2013, CD002106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anselmo, A.C.; Zhang, M.; Kumar, S.; Vogus, D.R.; Menegatti, S.; Helgeson, M.E.; Mitragotri, S. Elasticity of Nanoparticles Influences Their Blood Circulation, Phagocytosis, Endocytosis, and Targeting. ACS Nano 2015, 9, 3169–3177. [Google Scholar] [CrossRef]
- Bakker-Woudenberg, I.A.J.M.; Mouton, J.W.; Woodle, M.C.; Storm, G.; Lokerse, A.F.; Kate, M.T.T. Liposomes with Prolonged Blood Circulation and Selective Localization in Klebsiella pneumoniae-Infected Lung Tissue. J. Infect. Dis. 1993, 168, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Gajendiran, M.; Divakar, S.; Raaman, N.; Balasubramanian, S. In vitro drug release behavior, mechanism and antimicrobial activity of rifampicin loaded low molecular weight PLGA-PEG-PLGA triblock copolymeric nanospheres. Curr. Drug Deliv. 2013, 10, 722–731. [Google Scholar] [CrossRef]
- Venturoli, D.; Rippe, B. Ficoll and dextran vs. globular proteins as probes for testing glomerular permselectivity: Effects of molecular size, shape, charge, and deformability. Am. J. Physiol. Physiol. 2005, 288, F605–F613. [Google Scholar] [CrossRef]
- Yuan, F. Transvascular drug delivery in solid tumors. Semin. Radiat. Oncol. 1998, 8, 164–175. [Google Scholar] [CrossRef]
- Edetsberger, M.; Gaubitzer, E.; Valic, E.; Waigmann, E.; Köhler, G. Detection of nanometer-sized particles in living cells using modern fluorescence fluctuation methods. Biochem. Biophys. Res. Commun. 2005, 332, 109–116. [Google Scholar] [CrossRef]
- De Jong, W.H.; Borm, P.J.A. Drug delivery and nanoparticles:applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panyam, J.; Zhou, W.Z.; Prabha, S.; Sahoo, S.K.; Labhasetwar, V. Rapid endo-lysosomal escape of poly (DL-lactide-co-glycolide) nanoparticles: Implications for drug and gene delivery. FASEB J. 2002, 16, 1217–1226. [Google Scholar] [CrossRef]
- Konan, Y.N.; Chevallier, J.; Gurny, R.; Allémann, E. Encapsulation of p-THPP into Nanoparticles: Cellular Uptake, Subcellular Localization and Effect of Serum on Photodynamic Activity. Photochem. Photobiol. 2003, 77, 638. [Google Scholar] [CrossRef]
- Wang, W.-X.; Gao, J.-Q.; Liang, W.-Q. Chitosan-coated liposomes for intracellular oligonucleotides delivery: Characteristics and cell uptake behavior. Drug Deliv. 2011, 18, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-S.; Kim, C.-S.; Lee, K.-M. The intracellular uptake ability of chitosan-coated poly (D,L-lactide-co-glycolide) nanoparticles. Arch. Pharm. Res. 2008, 31, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Forier, K.; Raemdonck, K.; De Smedt, S.; Demeester, J.; Coenye, T.; Braeckmans, K. Lipid and polymer nanoparticles for drug delivery to bacterial biofilms. J. Control. Release 2014, 190, 607–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shariat, S.; Badiee, A.; Jaafari, M.R.; Mortazavi, S.A. Optimization of a Method to Prepare Liposomes Containing HER2/Neu-Derived Peptide as a Vaccine Delivery System for Breast Cancer. IJPR 2014, 13, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Walboomers, X.F.; Jansen, J.A.; Yang, F. Influence of formulation parameters on encapsulation of doxycycline in PLGA microspheres prepared by double emulsion technique for the treatment of periodontitis. J. Drug Deliv. Sci. Technol. 2019, 52, 263–271. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Gent, M.E.; Ali, M.; Nibbering, P.H.; Kłodzińska, S.N. Current Advances in Lipid and Polymeric Antimicrobial Peptide Delivery Systems and Coatings for the Prevention and Treatment of Bacterial Infections. Pharmaceutics 2021, 13, 1840. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111840
van Gent ME, Ali M, Nibbering PH, Kłodzińska SN. Current Advances in Lipid and Polymeric Antimicrobial Peptide Delivery Systems and Coatings for the Prevention and Treatment of Bacterial Infections. Pharmaceutics. 2021; 13(11):1840. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111840
Chicago/Turabian Stylevan Gent, Miriam E., Muhanad Ali, Peter H. Nibbering, and Sylvia N. Kłodzińska. 2021. "Current Advances in Lipid and Polymeric Antimicrobial Peptide Delivery Systems and Coatings for the Prevention and Treatment of Bacterial Infections" Pharmaceutics 13, no. 11: 1840. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111840