
Investigation of the Factors Responsible for the Poor Oral Bioavailability of Acacetin in Rats: Physicochemical and Biopharmaceutical Aspects

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Animals
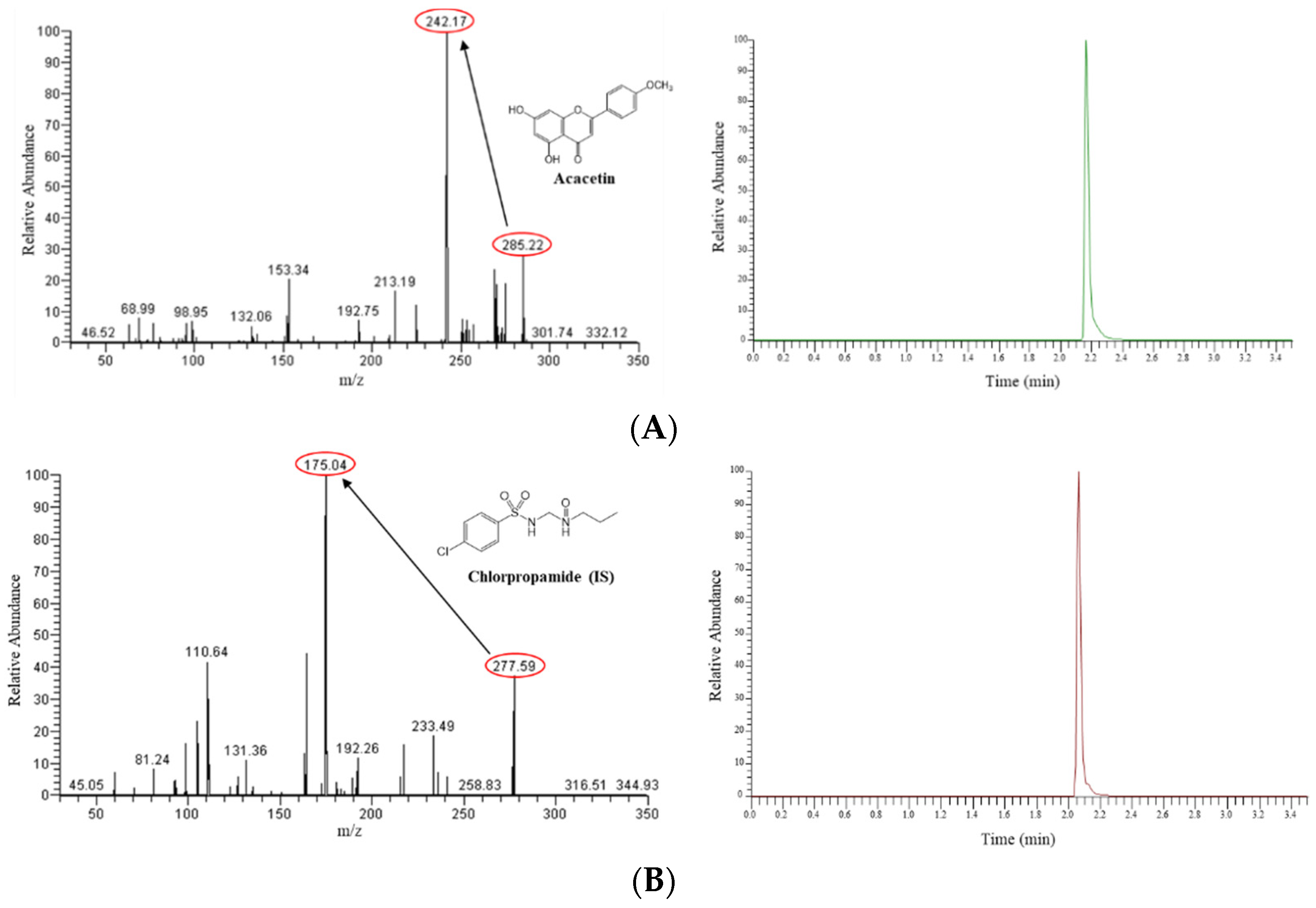
2.2. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Method for the Determination of Acacetin
2.3. Determination of Solubility, Distribution Coefficients, and Permeability
2.4. Determination of Plasma Protein Binding and Blood Distribution
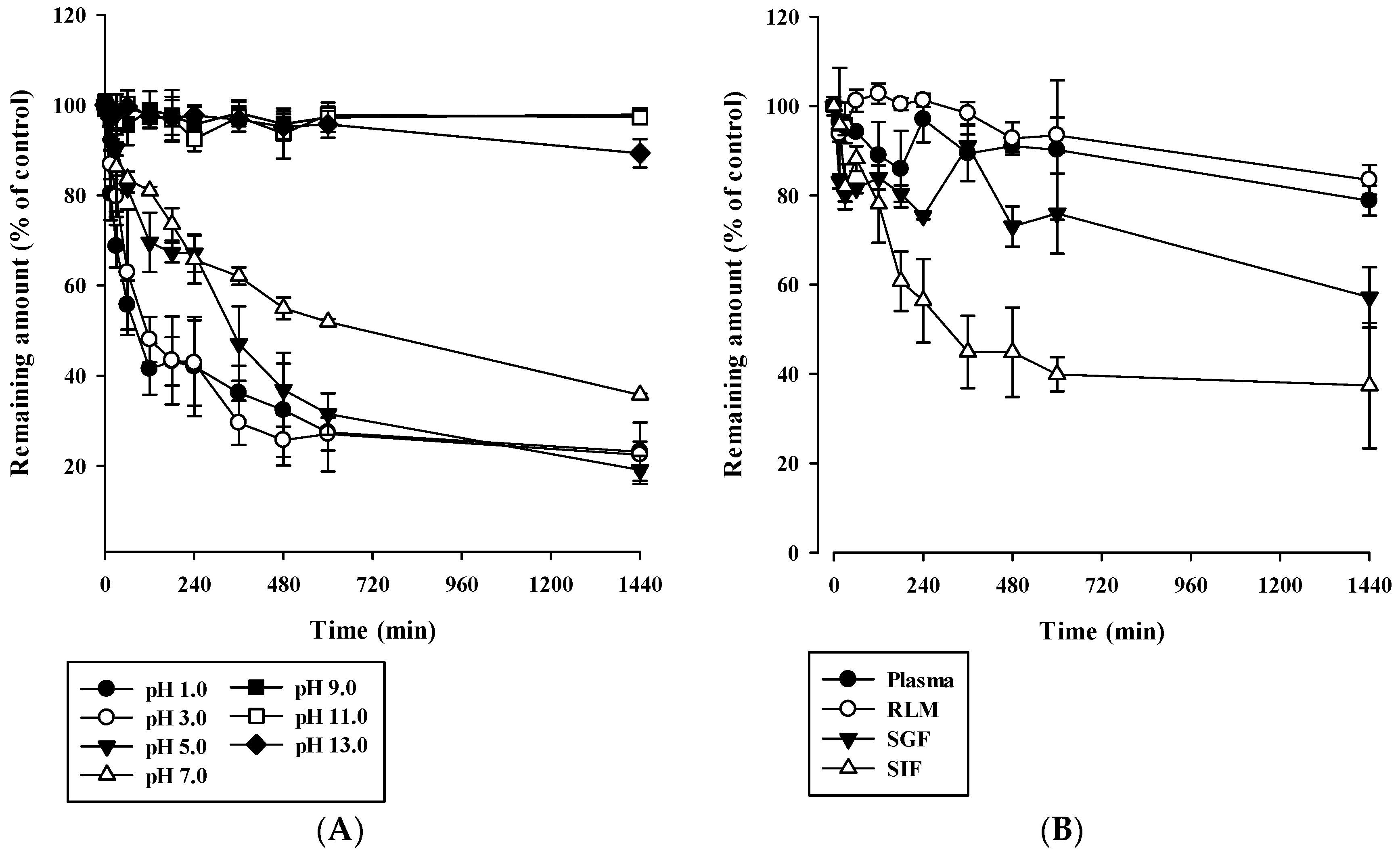
2.5. Stability Study
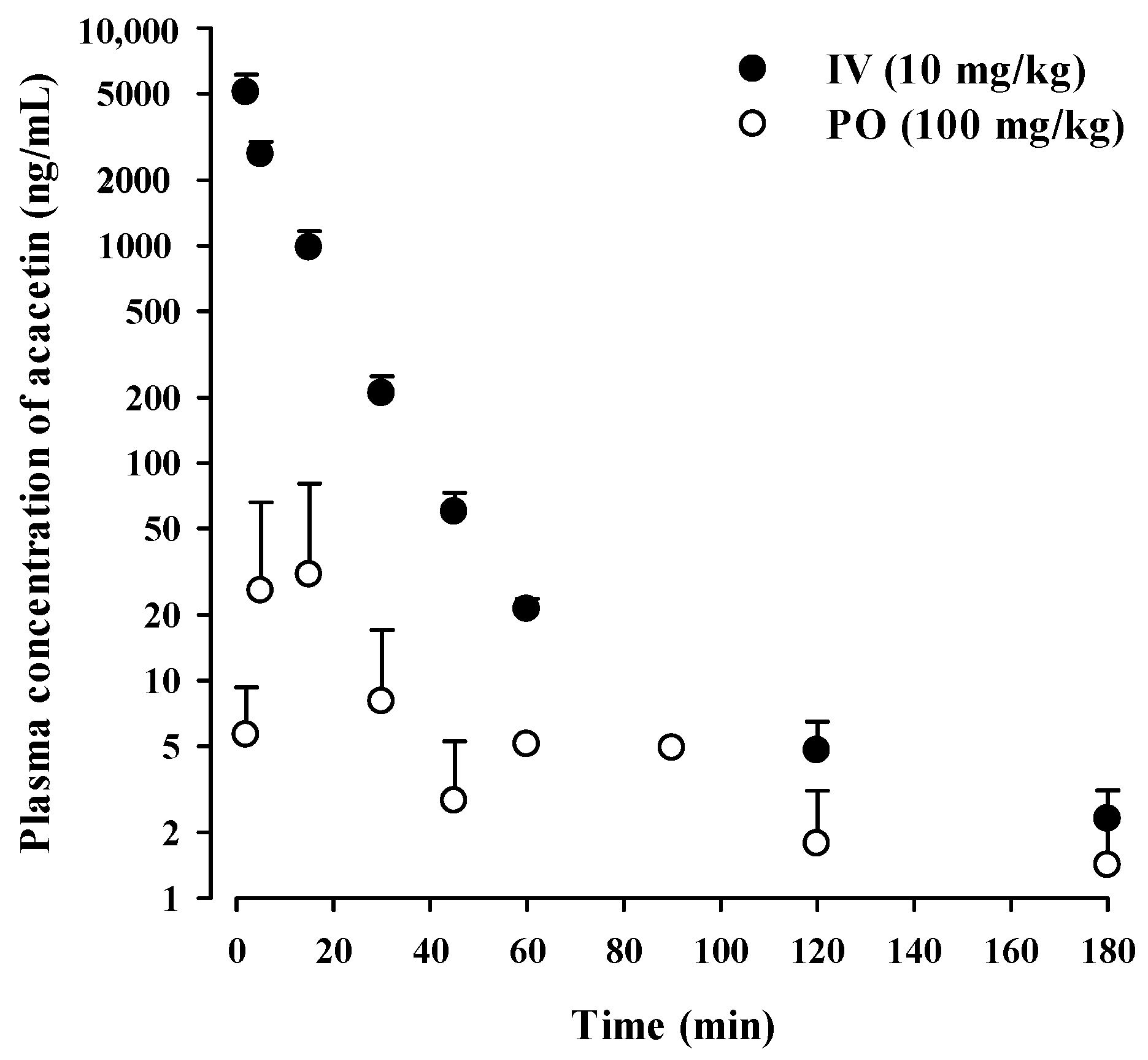
2.6. Intravenous and Oral Pharmacokinetic Studies in Rats
2.7. In Situ Closed-Loop Study
2.8. Metabolism Study in Rat Tissue S9 Fractions and RLM
2.9. Pharmacokinetic Analysis
2.10. Statistical Analysis
3. Results
3.1. LC-MS/MS Method for Acacetin
3.2. Physicochemical Properties of Acacetin
3.3. In Vitro Stability of Acacetin
3.4. In Vivo Pharmacokinetics of Acacetin in Rats
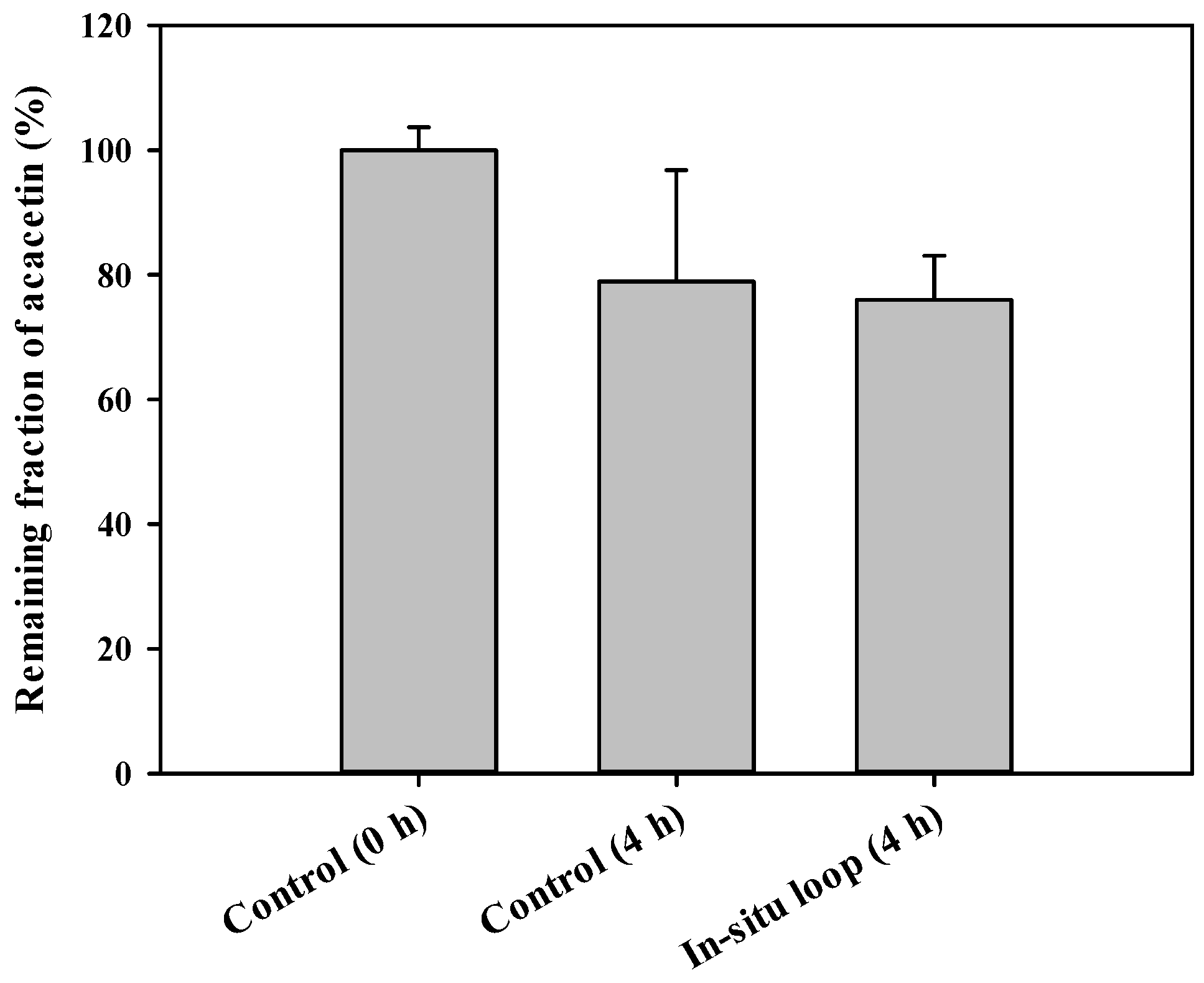
3.5. In Situ GI Absorption of Acacetin in Rats
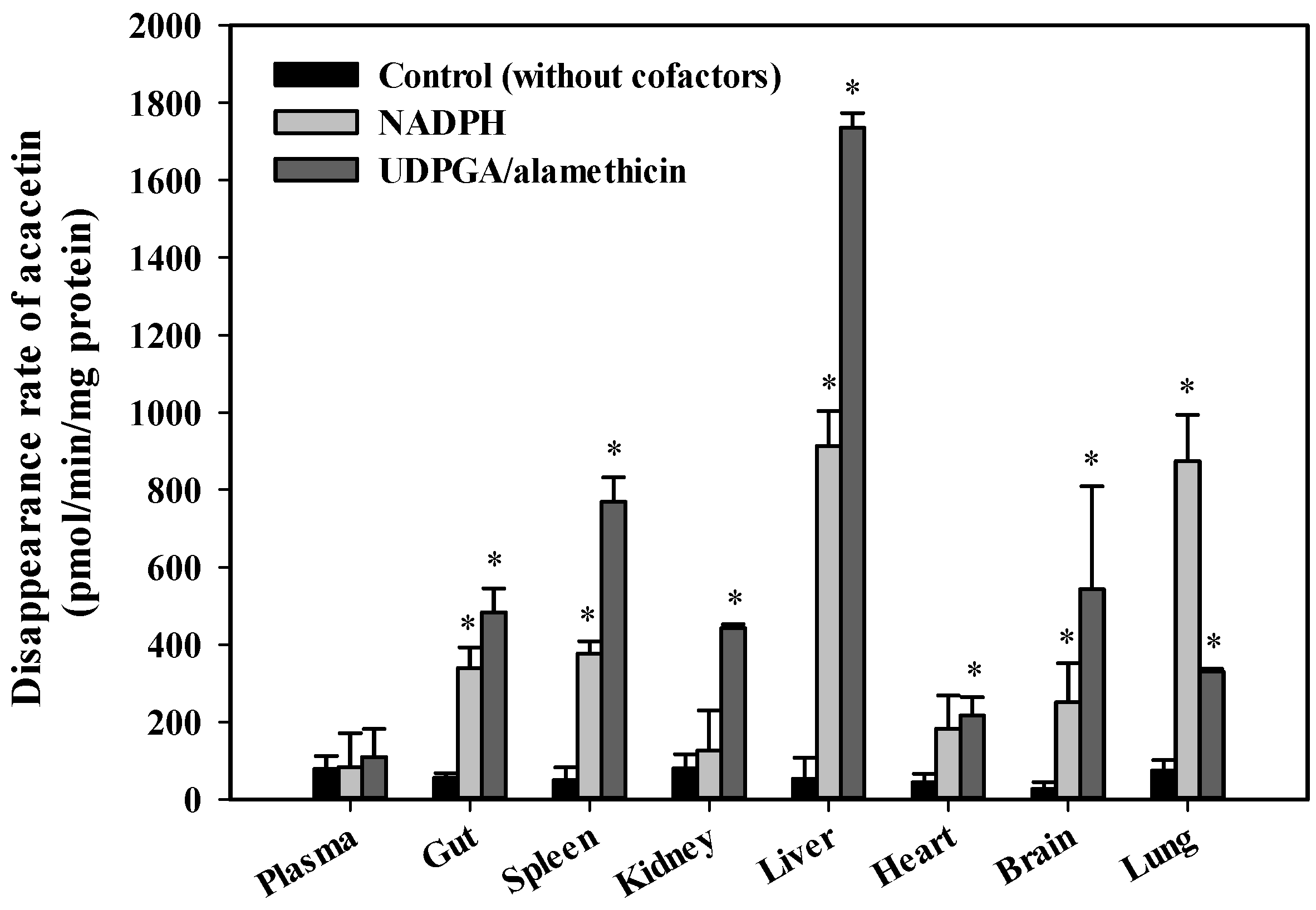
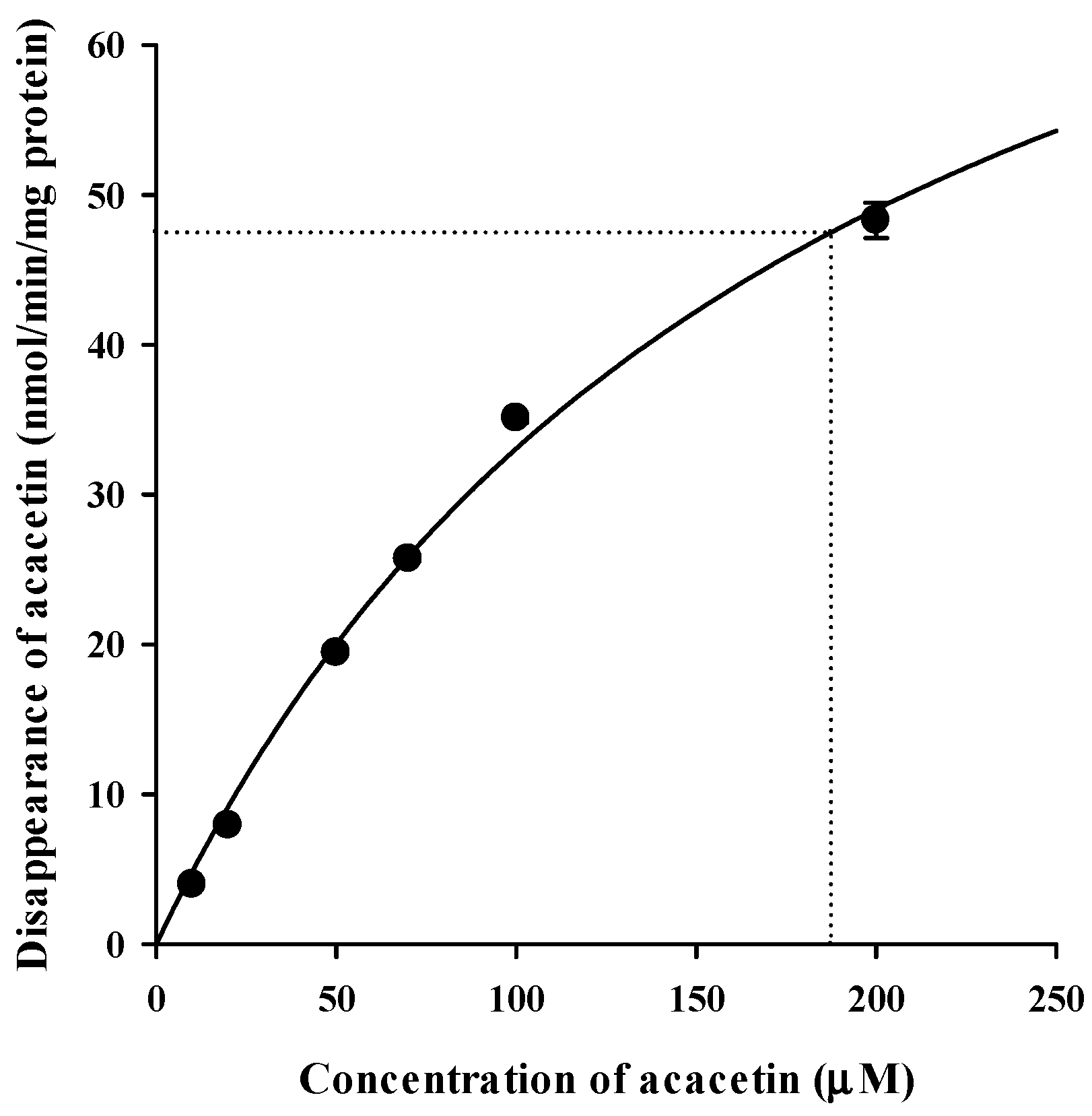
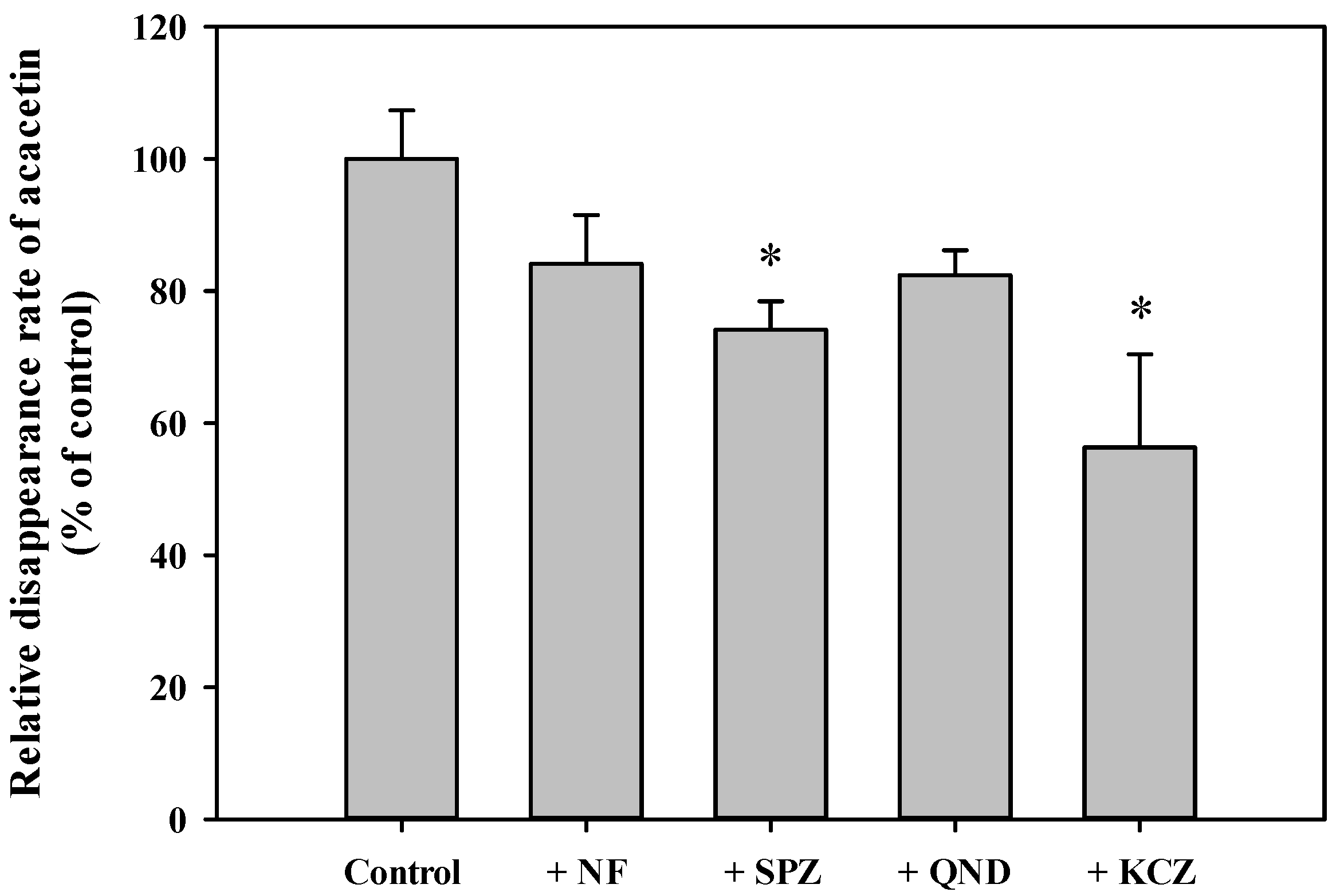
3.6. In Vitro Metabolism of Acacetin in Tissue S9 Fractions and RLM
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, S.B.; Lee, T.; Lee, H.S.; Song, C.K.; Cho, H.J.; Kim, D.D.; Maeng, H.J.; Yoon, I.S. Development and validation of a highly sensitive LC-MS/MS method for the determination of acacetin in human plasma and its application to a protein binding study. Arch. Pharm. Res. 2016, 39, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.H.; Li, X.; Chen, D.Y.; Zhang, N.; Wang, Y.; Shan, Y.; Hu, Y.; Xu, R.A.; Jin, J.; Ge, R.S. Determination of acacetin in rat plasma by UPLC-MS/MS and its application to a pharmacokinetic study. J. Chromatogr. B 2015, 986–987, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.H.; Yang, H.; Kim, J.E.; Lee, K.W. Safflower seed oil and its active compound acacetin inhibit UVB-induced skin photoaging. J. Microbiol. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.K.; Kim, J.E.; Lee, S.Y.; Lee, M.H.; Byun, S.; Kim, Y.A.; Lim, T.G.; Reddy, K.; Huang, Z.; Bode, A.M.; et al. The P110 subunit of PI3-K is a therapeutic target of acacetin in skin cancer. Carcinogenesis 2014, 35, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.R.; Park, C.G.; Jung, J.Y. Acacetin (5,7-dihydroxy-4′-methoxyflavone) exhibits in vitro and in vivo anticancer activity through the suppression of NF-kappaB/Akt signaling in prostate cancer cells. Int. J. Mol. Med. 2014, 33, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Chien, S.T.; Lin, S.S.; Wang, C.K.; Lee, Y.B.; Chen, K.S.; Fong, Y.; Shih, Y.W. Acacetin inhibits the invasion and migration of human non-small cell lung cancer A549 cells by suppressing the p38alpha MAPK signaling pathway. Mol. Cell Biochem. 2011, 350, 135–148. [Google Scholar] [CrossRef]
- Shim, H.Y.; Park, J.H.; Paik, H.D.; Nah, S.Y.; Kim, D.S.; Han, Y.S. Acacetin-induced apoptosis of human breast cancer MCF-7 cells involves caspase cascade, mitochondria-mediated death signaling and SAPK/JNK1/2-c-Jun activation. Mol. Cells 2007, 24, 95–104. [Google Scholar]
- Zhang, Q.; Zhu, L.; Gong, X.; Ruan, Y.; Yu, J.; Jiang, H.; Wang, Y.; Qi, X.; Lu, L.; Liu, Z. Sulfonation disposition of acacetin: In vitro and in vivo. J. Agric. Food Chem. 2017, 65, 4921–4931. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Sulaiman, S.A.; Azlan, S.A.; Gan, S.H. Two-year variations of phenolics, flavonoids and antioxidant contents in acacia honey. Molecules 2013, 18, 14694–14710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammad, A.; Odunola, O.A.; Ibrahim, M.A.; Sallau, A.B.; Erukainure, O.L.; Aimola, I.A.; Malami, I. Potential biological activity of acacia honey. Front. Biosci. 2016, 8, 351–357. [Google Scholar] [CrossRef]
- Meo, S.A.; Al-Asiri, S.A.; Mahesar, A.L.; Ansari, M.J. Role of honey in modern medicine. Saudi J. Biol. Sci. 2017, 24, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Thilakarathna, S.H.; Rupasinghe, H.P. Flavonoid bioavailability and attempts for bioavailability enhancement. Nutrients 2013, 5, 3367–3387. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Kim, J.E.; Kim, D.D.; Yoon, I.S. In vitro-in vivo extrapolation (IVIVE) for predicting human intestinal absorption and first-pass elimination of drugs: Principles and applications. Drug Dev. Ind. Pharm. 2014, 40, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Y.; Ning, E.; Peng, Y.; Zhang, J. Mechanisms of poor oral bioavailability of flavonoid morin in rats: From physicochemical to biopharmaceutical evaluations. Eur. J. Pharm. Sci. 2019, 128, 290–298. [Google Scholar] [CrossRef]
- Kim, J.E.; Cho, H.J.; Kim, J.S.; Shim, C.K.; Chung, S.J.; Oak, M.H.; Yoon, I.S.; Kim, D.D. The limited intestinal absorption via paracellular pathway is responsible for the low oral bioavailability of doxorubicin. Xenobiotica 2013, 43, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Yoon, I.S.; Cho, H.J.; Kim, D.H.; Choi, Y.H.; Kim, D.D. Emulsion-based colloidal nanosystems for oral delivery of doxorubicin: Improved intestinal paracellular absorption and alleviated cardiotoxicity. Int. J. Pharm. 2014, 464, 117–126. [Google Scholar] [CrossRef]
- Davidson, M.H.; Lukacsko, P.; Sun, J.X.; Phillips, G.; Walters, E.; Sterman, A.; Niecestro, R.; Friedhoff, L. A multiple-dose pharmacodynamic, safety, and pharmacokinetic comparison of extended- and immediate-release formulations of lovastatin. Clin. Ther. 2002, 24, 112–125. [Google Scholar] [CrossRef]
- Yanamandra, S.; Venkatesan, N.; Kadajji, V.G.; Wang, Z.; Issar, M.; Betageri, G.V. Proliposomes as a drug delivery system to decrease the hepatic first-pass metabolism: Case study using a model drug. Eur. J. Pharm. Sci. 2014, 64, 26–36. [Google Scholar] [CrossRef]
- Yin, J.; Ma, Y.; Liang, C.; Gao, J.; Wang, H.; Zhang, L. A systematic study of the metabolites of dietary acacetin in vivo and in vitro based on UHPLC-Q-TOF-MS/MS analysis. J. Agric. Food Chem. 2019, 67, 5530–5543. [Google Scholar] [CrossRef]
- Hodek, P.; Trefil, P.; Stiborova, M. Flavonoids-potent and versatile biologically active compounds interacting with cytochromes P450. Chem. Biol. Interact. 2002, 139, 1–21. [Google Scholar] [CrossRef]
- Dai, P.; Luo, F.; Wang, Y.; Jiang, H.; Wang, L.; Zhang, G.; Zhu, L.; Hu, M.; Wang, X.; Lu, L.; et al. Species- and gender-dependent differences in the glucuronidation of a flavonoid glucoside and its aglycone determined using expressed UGT enzymes and microsomes. Biopharm. Drug Dispos. 2015, 36, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Han, D.G.; Kim, K.S.; Seo, S.W.; Baek, Y.M.; Jung, Y.; Kim, D.D.; Yoon, I.S. A sensitive HPLC-FL method to simultaneously determine febuxostat and diclofenac in rat plasma: Assessment of metabolic drug interactions in vitro and in vivo. Anal. Methods 2020, 12, 2166–2175. [Google Scholar] [CrossRef]
- USA Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation. 2018. Available online: https://www.fda.gov/media/70858/download (accessed on 28 November 2019).
- Jang, J.H.; Jeong, S.H.; Cho, H.Y.; Lee, Y.B. Comparison of UPLC-MS/MS and HPLC-UV methods for the determination of zaltoprofen in human plasma. J. Pharm. Investig. 2019, 49, 613–624. [Google Scholar] [CrossRef]
- Seo, S.W.; Park, J.W.; Han, D.G.; Kim, J.M.; Kim, S.; Park, T.; Kang, K.H.; Yang, M.H.; Yoon, I.S. In vitro and in vivo assessment of metabolic drug interaction potential of dutasteride with ketoconazole. Pharmaceutics 2019, 11, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.B.; Kim, K.S.; Ryu, H.M.; Yoon, I.S.; Cho, H.J.; Chung, S.J.; Chong, S.; Kim, D.D. Comparison of saline vs. blood replenishment after blood sampling in a rat pharmacokinetic study. J. Pharm. Investig. 2019, 49, 543–551. [Google Scholar] [CrossRef]
- Jeong, S.H.; Jang, J.H.; Cho, H.Y.; Oh, I.J.; Lee, Y.B. A sensitive UPLC–ESI–MS/MS method for the quantification of cinnamic acid in vivo and in vitro: Application to pharmacokinetic and protein binding study in human plasma. J. Pharm. Investig. 2020, 50, 159–172. [Google Scholar] [CrossRef]
- Kiani, Y.S.; Jabeen, I. Lipophilic metabolic efficiency (LipMetE) and drug efficiency indices to explore the metabolic properties of the substrates of selected cytochrome P450 isoforms. ACS Omega 2020, 5, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- Lang, C.C.; Moreland, T.A.; Davey, P.G. Bioavailability of cefuroxime axetil: Comparison of standard and abbreviated methods. J. Antimicrob. Chemother. 1990, 25, 645–650. [Google Scholar] [CrossRef]
- Nayak, B.S.; Roy, H.; Dinda, S.C.; Ellaiah, P. Pharmacokinetic evaluation of flavonoid compound (acacetin) isolated from Gmelina arborea roxb. Res. J. Pharm. Technol. 2019, 12, 3659–3663. [Google Scholar] [CrossRef]
- Jiang, H.; Yu, J.; Zheng, H.; Chen, J.; Wu, J.; Qi, X.; Wang, Y.; Wang, X.; Hu, M.; Zhu, L.; et al. Breast cancer resistance protein and multidrug resistance protein 2 regulate the disposition of acacetin glucuronides. Pharm. Res. 2017, 34, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jia, P.; Zhang, X.; Zhang, Q.; Yang, H.; Shi, H.; Zhang, L. LC-MS/MS determination and pharmacokinetic study of seven flavonoids in rat plasma after oral administration of Cirsium japonicum DC. extract. J. Ethnopharmacol. 2014, 158, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Fagerholm, U. Prediction of human pharmacokinetics-gastrointestinal absorption. J. Pharm. Pharmacol. 2007, 59, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Hilgers, A.R.; Smith, D.P.; Biermacher, J.J.; Day, J.S.; Jensen, J.L.; Sims, S.M.; Adams, W.J.; Friis, J.M.; Palandra, J.; Hosley, J.D.; et al. Predicting oral absorption of drugs: A case study with a novel class of antimicrobial agents. Pharm. Res. 2003, 20, 1149–1155. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Y.; Du, F.; Yang, J.; Xu, F.; Niu, W.; Ren, Y.; Li, C. Intestinal absorption and presystemic elimination of various chemical constituents present in GBE50 extract, a standardized extract of Ginkgo biloba leaves. Curr. Drug Metab. 2012, 13, 494–509. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Park, J.W.; Yoon, I.S.; Kim, D.D. Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: Enhanced intestinal absorption and lymphatic uptake. Int. J. Nanomed. 2014, 9, 495–504. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nominal Concentration (ng/mL) | Precision (%) | Accuracy (%) | ||
|---|---|---|---|---|
| Intra-Day | Inter-Day | Intra-Day | Inter-Day | |
| LLOQ (0.5) | 15.2 | 14.6 | 97.5 | 104 |
| LQC (1) | 12.0 | 9.0 | 101 | 93.9 |
| MQC (100) | 4.8 | 4.1 | 93.8 | 92.0 |
| HQC (500) | 3.2 | 0.9 | 99.1 | 101 |
| Property | Parameter | Value |
|---|---|---|
| Lipophilicity | Log P | 3.51 ± 0.09 |
| Log D7.4 | 3.40 ± 0.20 | |
| Solubility (ng/mL) | Water | 40.1 ± 7.3 |
| pH 7.4 phosphate buffer | 12.1 ± 1.1 | |
| SGF | 59.3 ± 7.1 | |
| SIF | 119 ± 8 | |
| Permeability (×10−6 cm/s) | PAMPA | 8.06 ± 0.91 |
| Distribution | fuP | 0.0102 ± 0.0011 |
| fuMIC | 0.302 ± 0.021 | |
| fuSGF | 0.822 ± 0.026 | |
| fuSIF | 0.714 ± 0.009 | |
| RB | 1.04 ± 0.10 |
| Parameter | IV 10 mg/kg | PO 100 mg/kg |
|---|---|---|
| AUCinf (μg∙min/mL) | 51.3 ± 9.2 | ND b |
| AUClast (μg∙min/mL) | 0.771 ± 0.415 | |
| Vss (mL/kg) | 2220 ± 507 | |
| CL (mL/min/kg) | 199 ± 36 | |
| t1/2 (min) | 40.3 ± 7.2 | ND b |
| Tmax (min) | 5 (2–15) | |
| Cmax (ng/mL) | 52.1 ± 50.5 | |
| Ae24h (% of dose) | 2.50 ± 1.15 | 0.0601 ± 0.0634 |
| GI24h (% of dose) | 6.66 ± 5.34 | 29.3 ± 22.3 |
| F (%) a | 2.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, D.-G.; Cha, E.; Joo, J.; Hwang, J.S.; Kim, S.; Park, T.; Jeong, Y.-S.; Maeng, H.-J.; Kim, S.-B.; Yoon, I.-S. Investigation of the Factors Responsible for the Poor Oral Bioavailability of Acacetin in Rats: Physicochemical and Biopharmaceutical Aspects. Pharmaceutics 2021, 13, 175. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13020175
Han D-G, Cha E, Joo J, Hwang JS, Kim S, Park T, Jeong Y-S, Maeng H-J, Kim S-B, Yoon I-S. Investigation of the Factors Responsible for the Poor Oral Bioavailability of Acacetin in Rats: Physicochemical and Biopharmaceutical Aspects. Pharmaceutics. 2021; 13(2):175. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13020175
Chicago/Turabian StyleHan, Dong-Gyun, Eunju Cha, Jeongmin Joo, Ji Sun Hwang, Sanghyun Kim, Taeuk Park, Yoo-Seong Jeong, Han-Joo Maeng, Sang-Bum Kim, and In-Soo Yoon. 2021. "Investigation of the Factors Responsible for the Poor Oral Bioavailability of Acacetin in Rats: Physicochemical and Biopharmaceutical Aspects" Pharmaceutics 13, no. 2: 175. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13020175