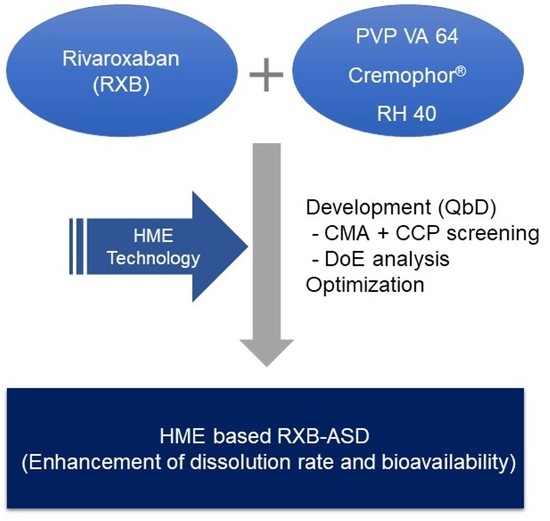
1. Introduction
Rivaroxaban (RXB), an anticoagulant agent and the active ingredient in Xarelto
® tablets, is used to treat deep vein thrombosis (DVT) and pulmonary embolism (PE). RXB does not require a cofactor for activity since RXB inhibits free factor Xa (FXa) and prothrombinase activity [
1] compared to anticoagulant drugs such as vitamin K and warfarin that are not widely used in the clinic due to their toxic side effects. However, RXB is classified as a Biopharmaceutics Classification System Class II (BCS Class II) drug and it has low solubility (20 μg/mL) in aqueous solutions. There have been several attempts in the preparation of polymeric amorphous solid dispersions (ASDs) containing RXB [
2,
3]. To improve the solubility, various techniques such as liposomes, nanosuspensions, solid dispersions, and cyclodextrin inclusions were tested [
4,
5,
6,
7]. Solvent evaporation, freeze-drying, supercritical fluid method, spray drying, and thermal melting are applied in the manufacture of solid dispersion systems [
8,
9,
10,
11,
12,
13,
14,
15]. Hot-melt extrusion (HME), used for ASDs of poorly water-soluble active pharmaceutical ingredients (APIs), is a complex commercialized technique. In comparison to traditional methods of preparation of ASDs, HME is the most promising solvent-free, continuous, industry feasible, and scalable process for preparation of ASDs [
16]. It is possible to improve the stability by preventing hydrolysis and oxidation and reducing residence times at high temperatures and screw rotation. During the preparation of such ASDs, the API is usually mixed/extruded with a molten of thermos-softened polymer.
Recently, many studies have been conducted on methods of preparing physically stable ASDs using polymer mixtures and the QbD approach, which allows for enhancing pharmaceutical development through design efforts from product development conceptualization to its commercialization [
17,
18,
19,
20,
21,
22]. The design of experiments (DoEs) for the drug development and product process is classified into screening design, factorial design, and response surface methodology (RSM). RSM is used to estimate possible effects, quadratic effects, the shape of the response surface, and interactions, and is positively applied to pharmaceutical development because there can be a variation of only one parameter at a time, keeping other parameters constant, although two or more variables can be studied simultaneously [
23,
24]. In addition, RSM has the advantage in terms of reduced process variability, higher percentage yields, lower treatment time, and cost-effectiveness. It estimates the relative significance of different variables.
However, a systemic attempt using the QbD to improve the solubility of RXB has not been reported in HME-based ASDs, although there are several approaches of preparation of polymeric ASDs containing RXB [
2,
3]. Therefore, HME-based ASD on RXB using optimal polymer combination was developed in terms of improvement of the dissolution behavior and oral bioavailability, based on the experimental design method of RSM.
2. Materials and Methods
2.1. Materials
RXB (purity > 98.5%; Hanseo Chemical, Pyeongtaek, Korea) and Xarelto® tablets (20 mg, Bayer, Luverkusen, Germany) were purchased.
Polyvinylpyrrolidone-vinyl acetate 64 (PVP VA 64, Kollidone® VA 64), Soluplus® (Polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft co-polymer), Cremophor® RH 40 (PEG-40 hydrogenated castor oil), polyvinylpyrrolidone K 90 (PVP 90), Solutol® HS 15 (Polyoxyl 15-hydroxystearate), and Kolliphor® 188 (Poloxamer 188) were obtained from BASF (Ludwigshafen, Germany). Polyvinylalcohol (PVA, MW 89,000–98,000) was purchased from Sigma (St. Louis, MO, USA), and polyethylene glycol (PEG) was purchased from Daejung (Seoul, Korea). Gelucire® 44/14 (Lauroyl polyoxy-32-glycerides) and Gelucire® 50/13 (Stearoyl macrogol-32 glycrides) were obtained from Gattefosse (Saint-Priest, France). Carboxymethyl cellulose (CMC) and hydroxypropyl cellulose (HPC) were purchased from Samchun (Pyeongtaek, Korea). Acetonitrile and methanol for HPLC were purchased from J.T. Baker (Seoul, Korea). All the other chemicals and reagents used were of analytical grade.
2.2. Before the Study
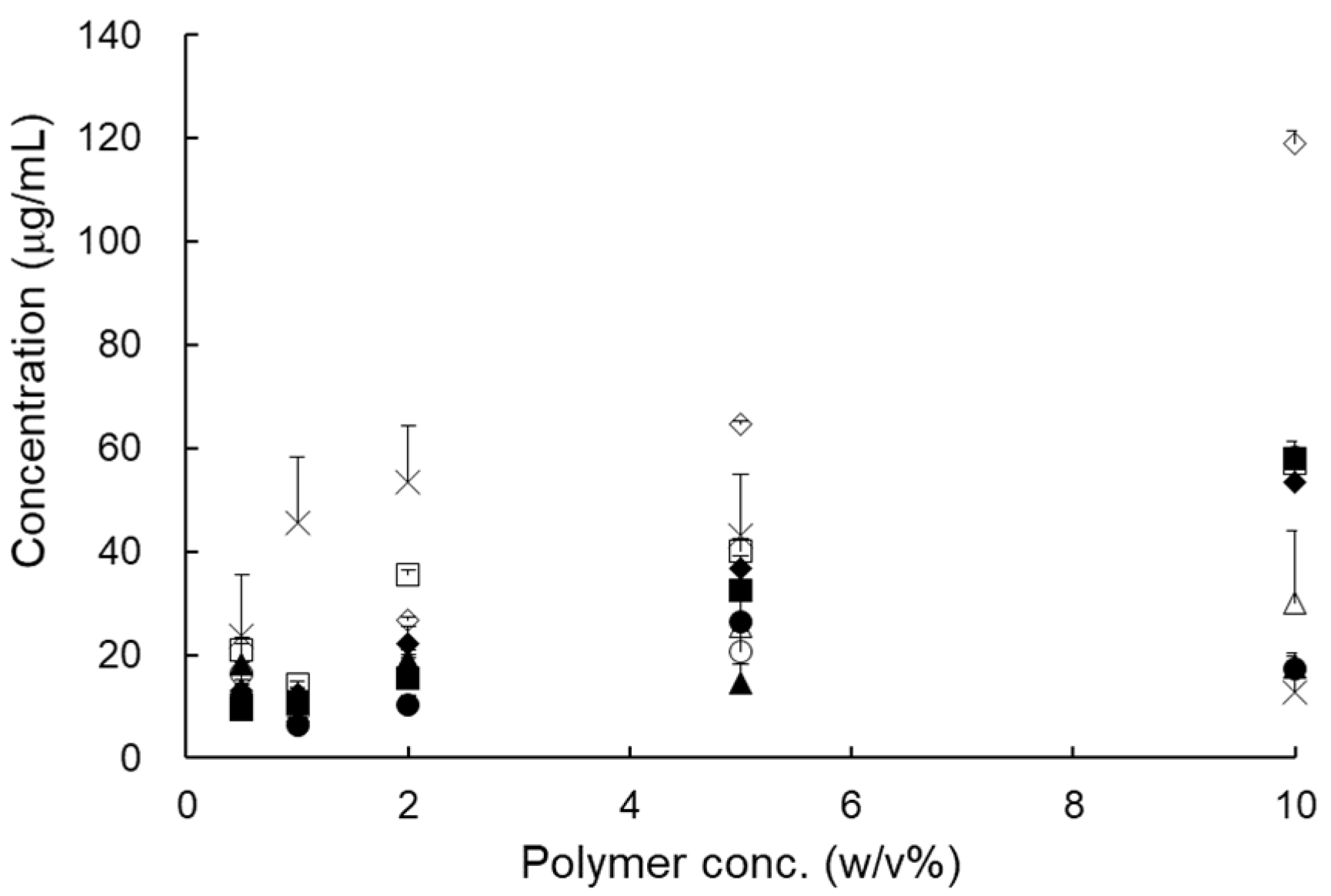
2.2.1. Selection of a Carrier
To study critical material attributes (CMA), first of all, a 1% aqueous solution containing various polymers and surfactants was prepared, and we added various amounts of RXB to determine how much of it dissolved in the solution. The mixtures were stirred continuously for 72 h at 25 ± 0.5 °C and centrifuged at 15,000 rpm for 5 min; then, the supernatants were filtered through membrane filters (0.45 μm, Whatman, PA, USA). The filtrates were diluted in solution (chloroform: methanol = 1:7 (v/v, %), and the concentration of RXB was quantified. Next, by changing the concentration of various polymers and surfactants in aqueous solution from 0.5% to 10% (w/w, i.e., 0.5%, 1%, 2%, 5%, and 10%), the maximum concentration of RXB that could be dissolved was determined.
The concentration of RXB was analyzed by an HPLC (Shimadzu, Japan) system; the system featured an LC10-AD isocratic pump, a SPD-10A VP variable spectrophotometric detector, and a Shimpak GIS ODS column (5 μm pore diameter; 4.6 × 150 mm, Shimadzu, Japan). An acetonitrile/water mixture (55/45, v/v, %) served as the mobile phase; the flow rate was 1.2 mL/min, and the detection wavelength was 250 nm. The RXB calibration curve was linear (r = 0.9999) over the concentration range of 0.625–200 µg/mL.
2.2.2. HME Process Condition
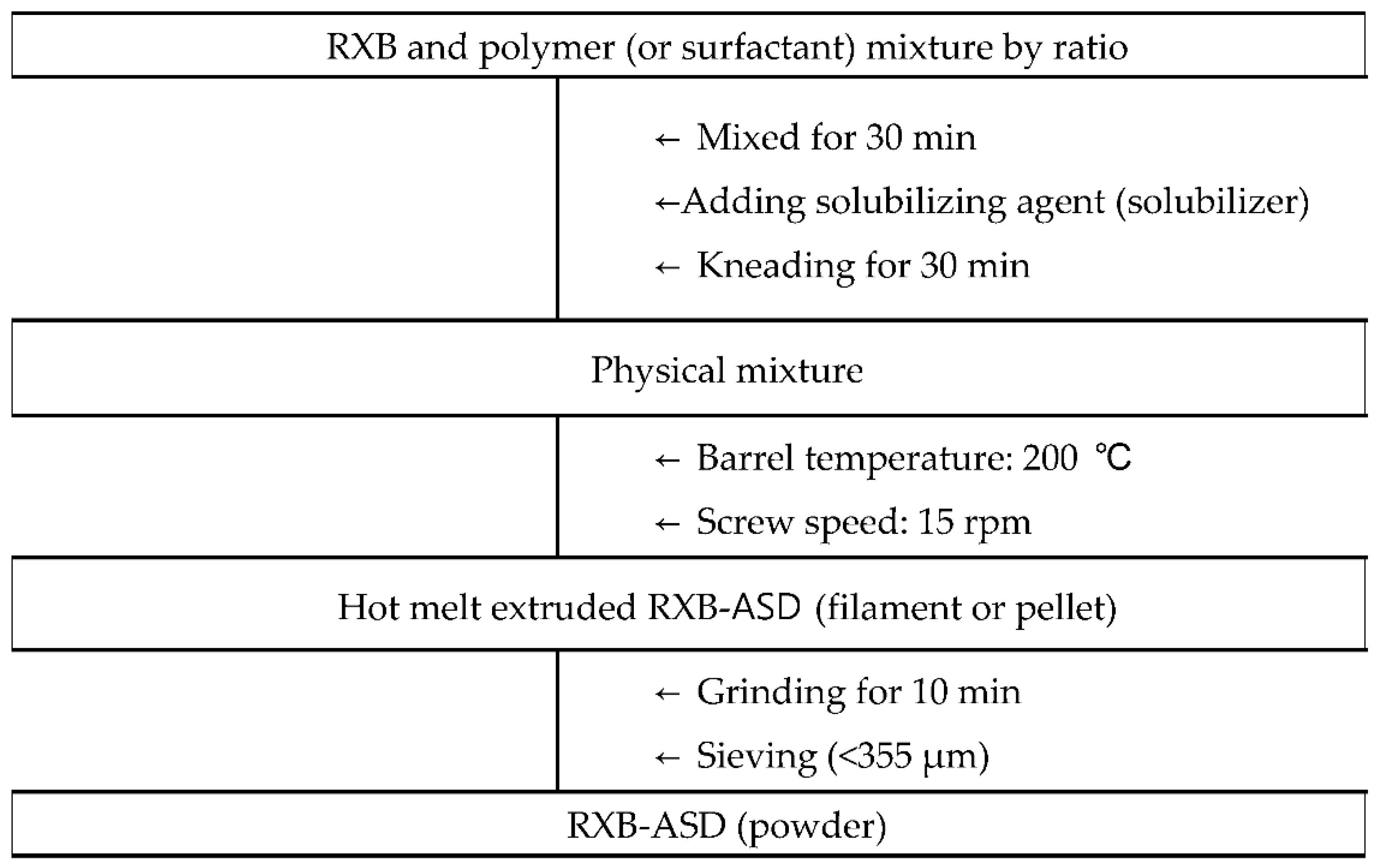
The solubility and dissolution of RXB-ASDs were selected as critical process parameters (CPP) in HME technology. RXB-ASDs were prepared by alternating the barrel temperatures between 140 °C, 180 °C, 200 °C, 220 °C, and 240 °C and the screw speeds between 10, 15, and 20 rpm for a Haake Mini CTW hot-melt extruder (Thermo Scientific Inc., Waltham, MA, USA) fixed at a weight polymer ratio of 1:4 (RXB: Soluplus®). The effect of screw speed on solubility was observed by adjusting the screw speed to 10, 15, or 20 rpm with the temperature fixed at 200 °C.
Next, after fixing the temperature and screw speed, the effects on the solubility and dissolution at 6 h in SGF (pH 6.8) of RXB-ASDs, prepared as shown in
Figure 1 with various polymer compositions (
Table 1), were evaluated.
2.2.3. Preformulation Study of RXB-ASDs Using a Full Factorial Design (FFD)
Based on the results of failure mode effect analysis (FMEA), with a preformulation experimental design with a hot-melt extruder at a 15 rpm screw speed, the effect of independent variables (PVP VA 64 ratio (X1, 1:1, 1:2, 1:4), total weight of Cremophor
® RH 40 (X2, 0, 10, 20
w/
w%) and barrel temperature (X3, 180, 200, 220 °C) on dependent variables (content (Y1) and the dissolution rate at 6 h in SIF (Y2)) was monitored (
Table 2).
2.3. Optimization by Central Composite Design (CCD)
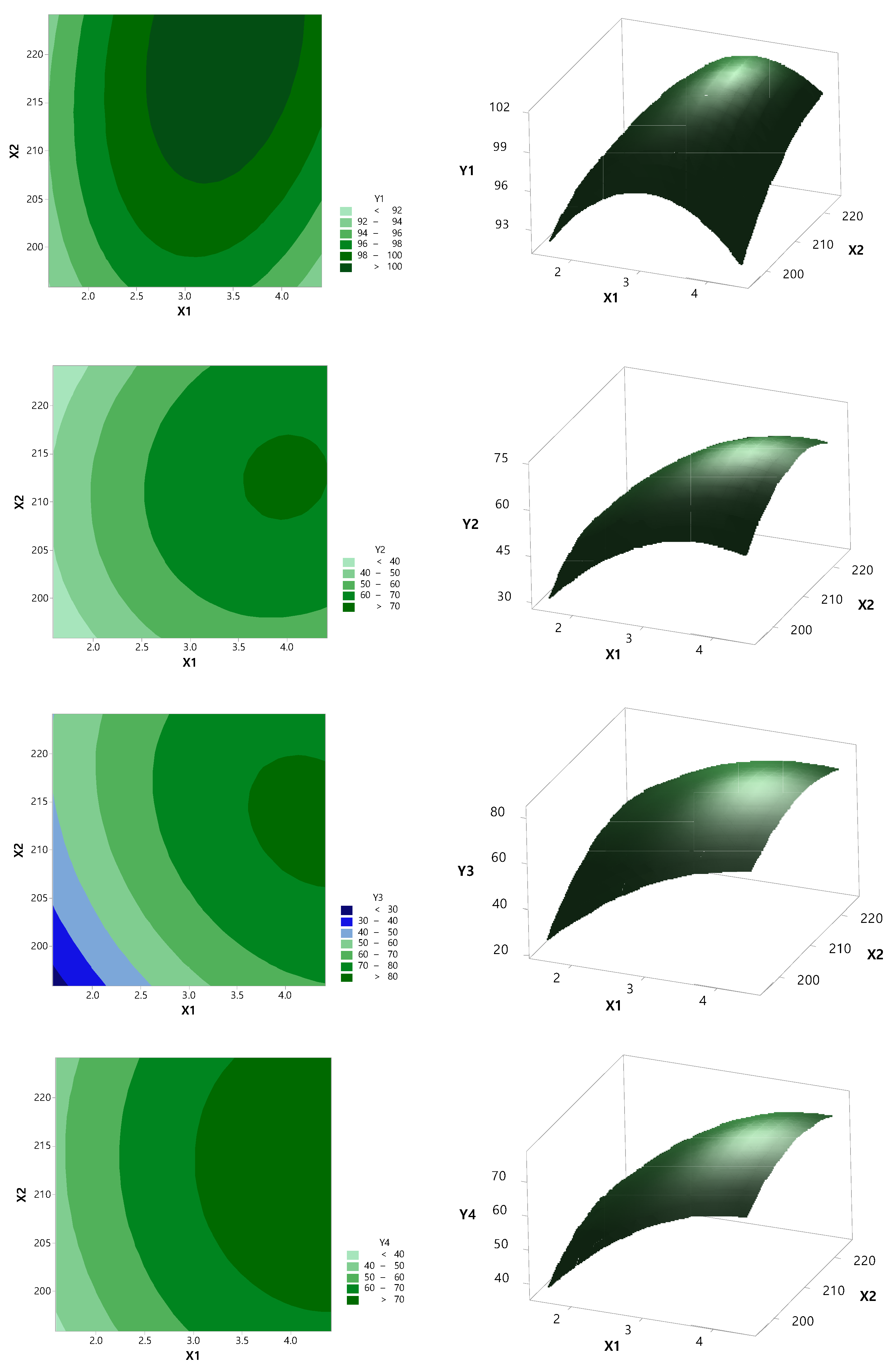
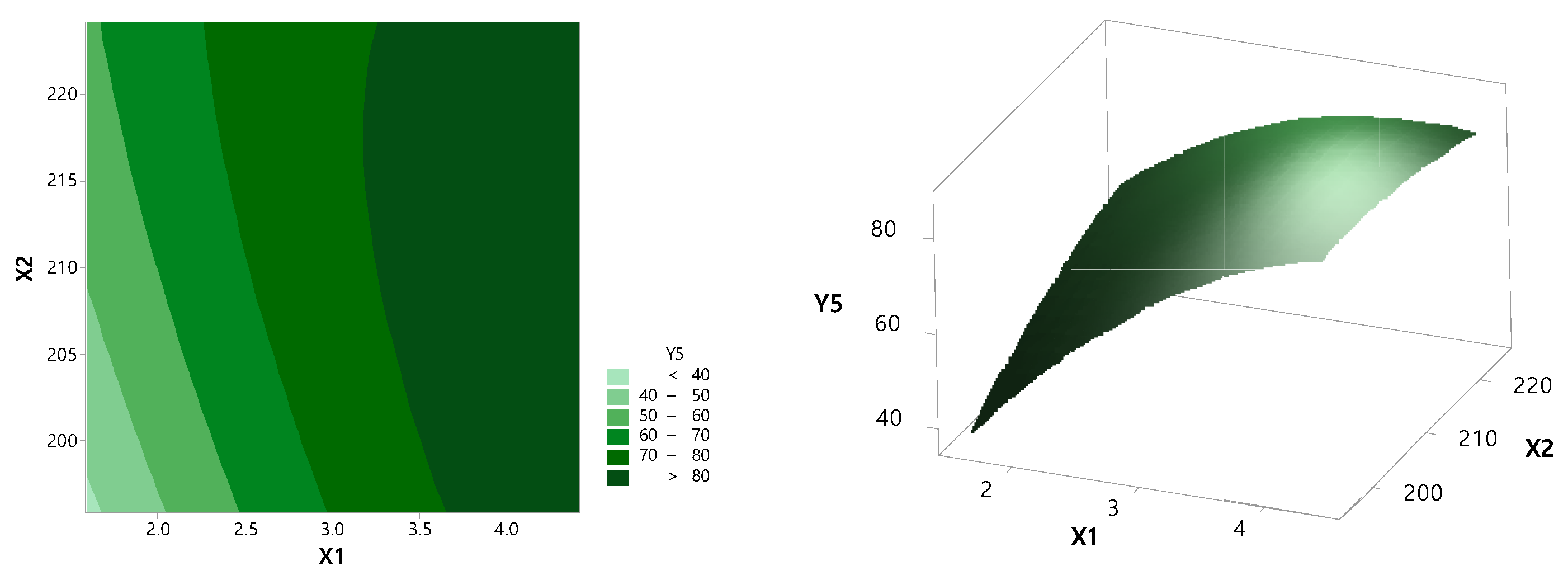
Based on preformulation results, the RXB-ASD experimental design and data analysis were conducted using the central composite design module in Minitab version 18 software (Minitab Inc., State College, PA, USA). The RSM central composite design method was used to optimize critical factors at 20% of Cremophor® RH 40 and 15 rpm screw speed. The effect of the independent variables such as the polymer ratio (X1, 1:2–1:4) and barrel temperature (X2, 200–220 °C) on the dependent variables such as the content (Y1) and dissolution rate at 2 h (Y2) and 6 h (Y3) in SGF (pH 1.2) and the dissolution rate at 2 h (Y4) and 6 h (Y5) in SIF (pH 6.8) was evaluated.
The linear equation of the model is as follows:
where Y is the response of the dependent variables associated with each factor-level combination; A0 is the intercept; A1, A2, A3, A4, and A5 are the regression coefficients; and X1 and X2 are the independent variables.
The data were fitted to a second-order polynomial equation, and regression coefficients were obtained. Analysis of variance (ANOVA) was conducted to evaluate the significance and adequacy of the developed regression model. The adequacy of the response surface models was clarified by the determination coefficient (R2) and the lack of fit.
One-way ANOVA and multiple regression analysis were performed to test the significance of the model and factor coefficients. The polynomial, plots, and two-contour plots also revealed the interactions between each independent variable.
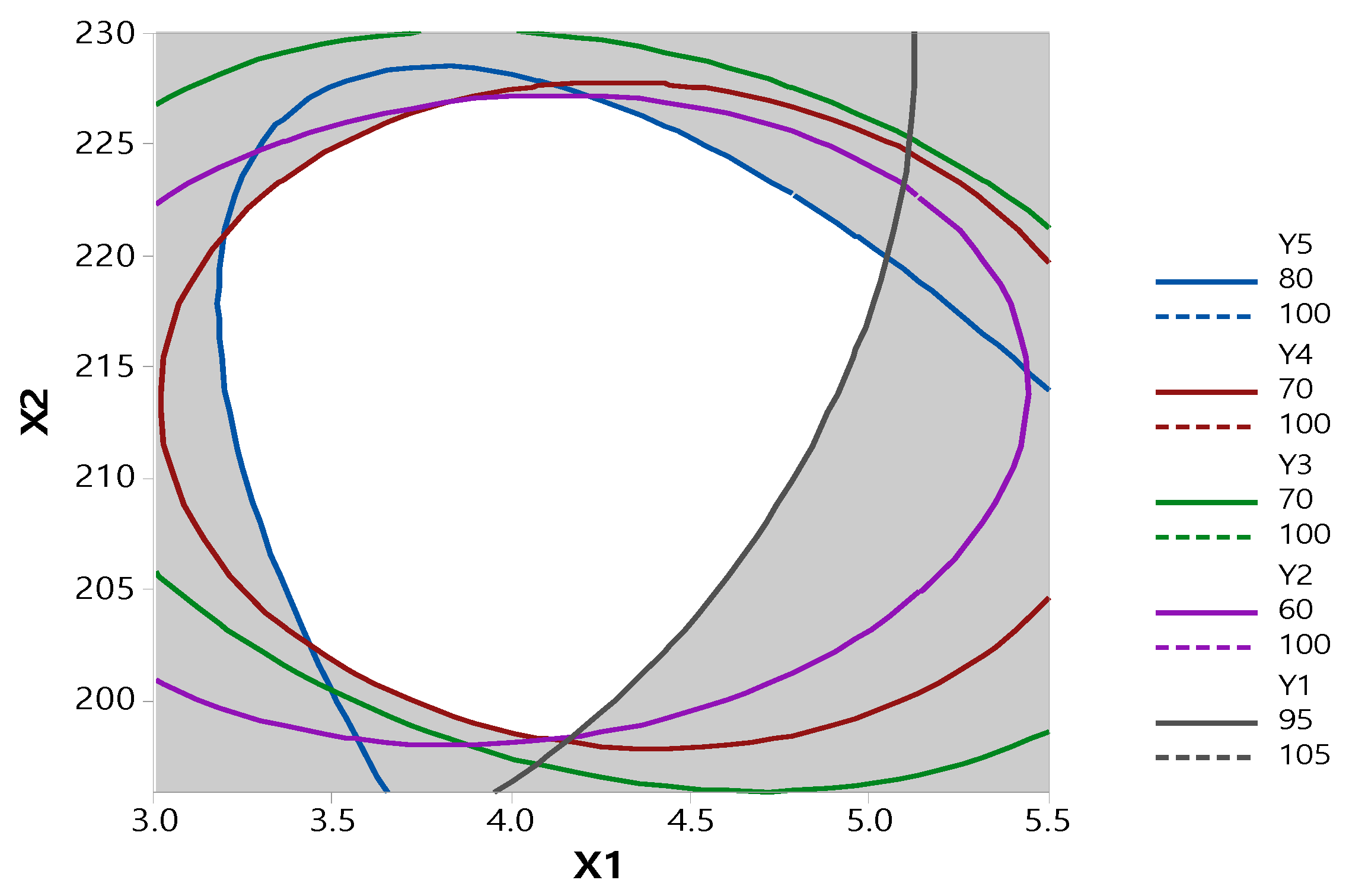
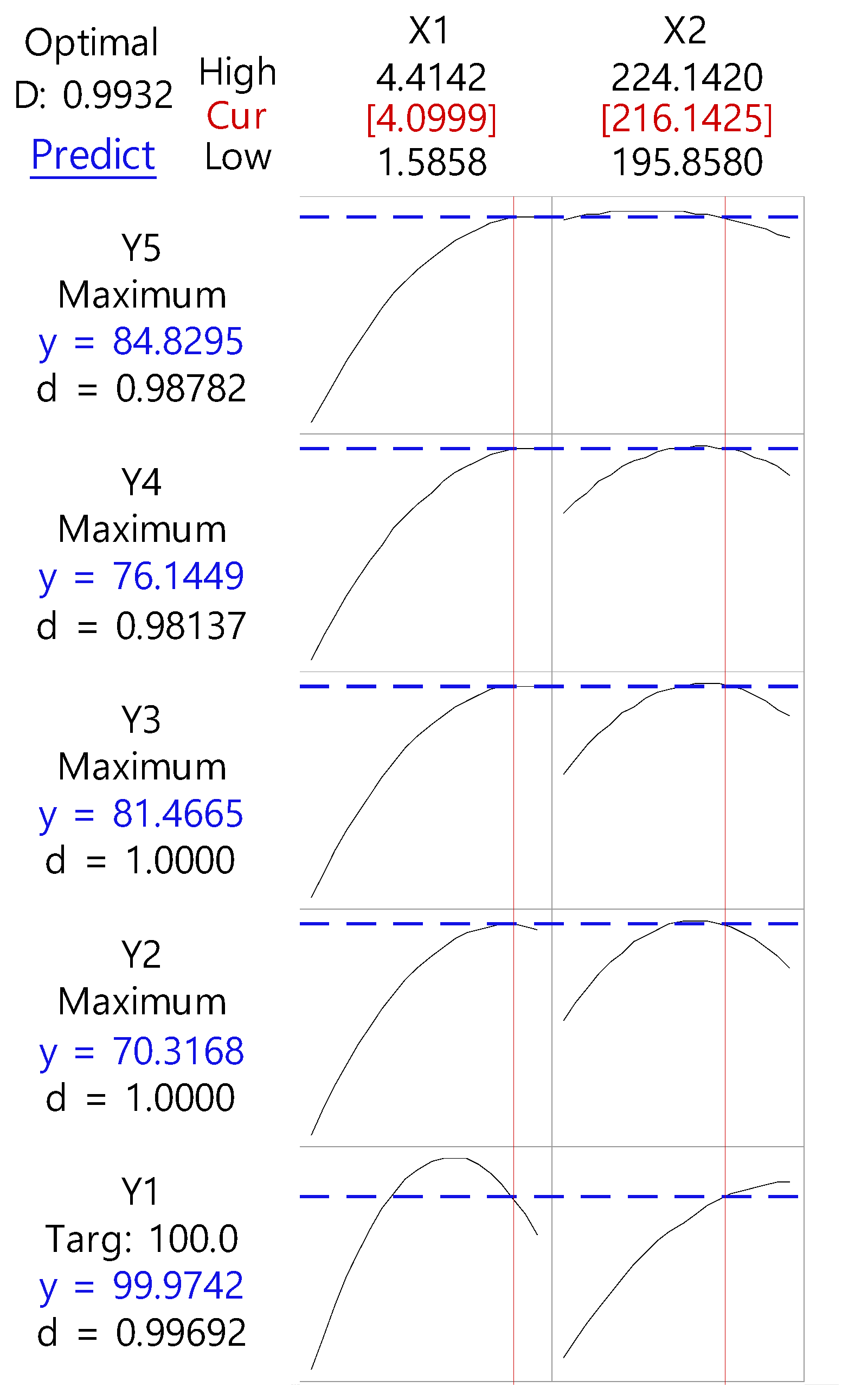
After generating the polynomial equations relating to the dependent and independent variables, optimization of the dependent variables (Y1, Y2, Y3, Y4, and Y5) was performed using a desirability function to obtain the levels of X1 and X2 that maximized the dependent variable.
2.4. Physicochemical Evaluation of RXB-ASD
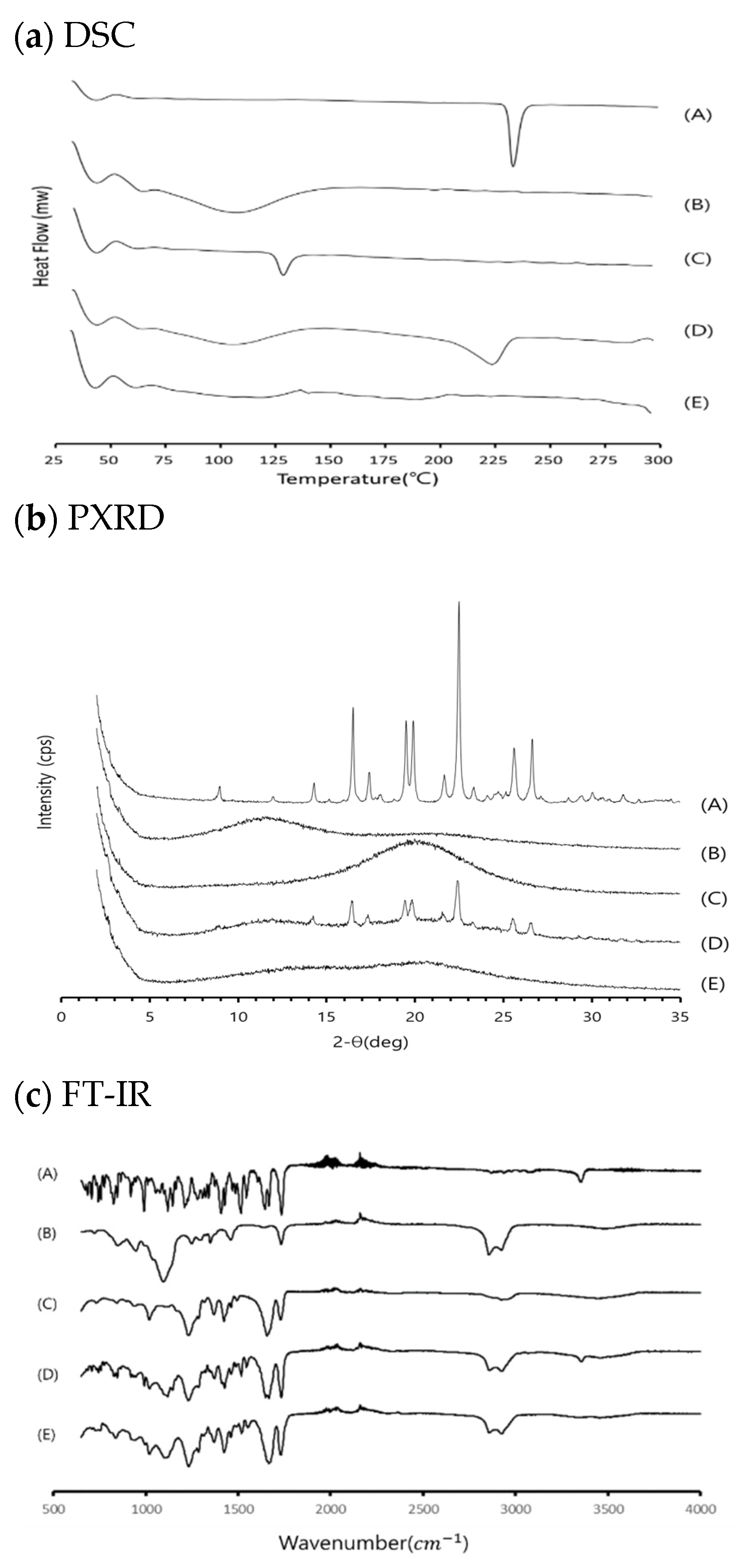
The calorimetric responses of samples were recorded on a differential scanning calorimetry (DSC) system (N-650, Scinco, Korea) equipped with a refrigerated cooling system, calibrated for temperature and heat flow using a high-purity indium standard. The sample cell was purged with dry nitrogen at a flow rate of 40 mL/min. The 3–5-mg samples were laid on crimped aluminum pans and measured at a heating rate of 20 °C/min up to 250 °C.
Powder X-ray diffraction (PXRD patterns of different samples were recorded at room temperature using a Smartlab X-ray diffractometer (Rigaku Mechatronics Co. Ltd., Tokyo, Japan) equipped with a 2θ compensating slit, using Cu Kα radiation (1.5406 Å) at 45 kV and 200 mA passing through a nickel filter with an IS slit (0.5°), a soller slit (5°), and receiving slit (20 mm). Samples were mounted on a zero-background sample holder and subjected to a continuous scan over a Bragg angle 2 θ range of 2–35° at a step size of 0.01° and scan rate of 5°/min. Obtained diffractograms were analyzed with the PDXL processing program.
To monitor the intermolecular interaction between drug and polymers, the crystalline RXB and ASD were monitored with FT-IR by a conventional KBr pellet method using an FTIR-4100 spectrophotometer (JASCO, Tokyo, Japan).
The surface morphology of powder samples was monitored under a scanning electron microscope (SEM) (S-3400, Hitachi, Ltd., Tokyo, Japan) with an excitation voltage of 25 kV. The powder samples were mounted onto a steel stage and sputter-coated with gold using ion sputtering (E-1010, Hitachi, Ltd.) prior to analysis.
2.5. Drug Content, In Vitro Dissolution Test, and Food Effect
For quantification of the drug content, RXB-ASDs containing 2 mg of RXB were dissolved in 2 mL of dimethyl sulfoxide. After stirring for 3 h, the solution was filtered with a membrane and diluted 100-fold (0.1 mL of filtrate in a total volume of 10 mL) in the solution (chloroform: methanol = 12.5:87.5, v/v%). The concentration of RXB in the diluted solution was quantified using HPLC.
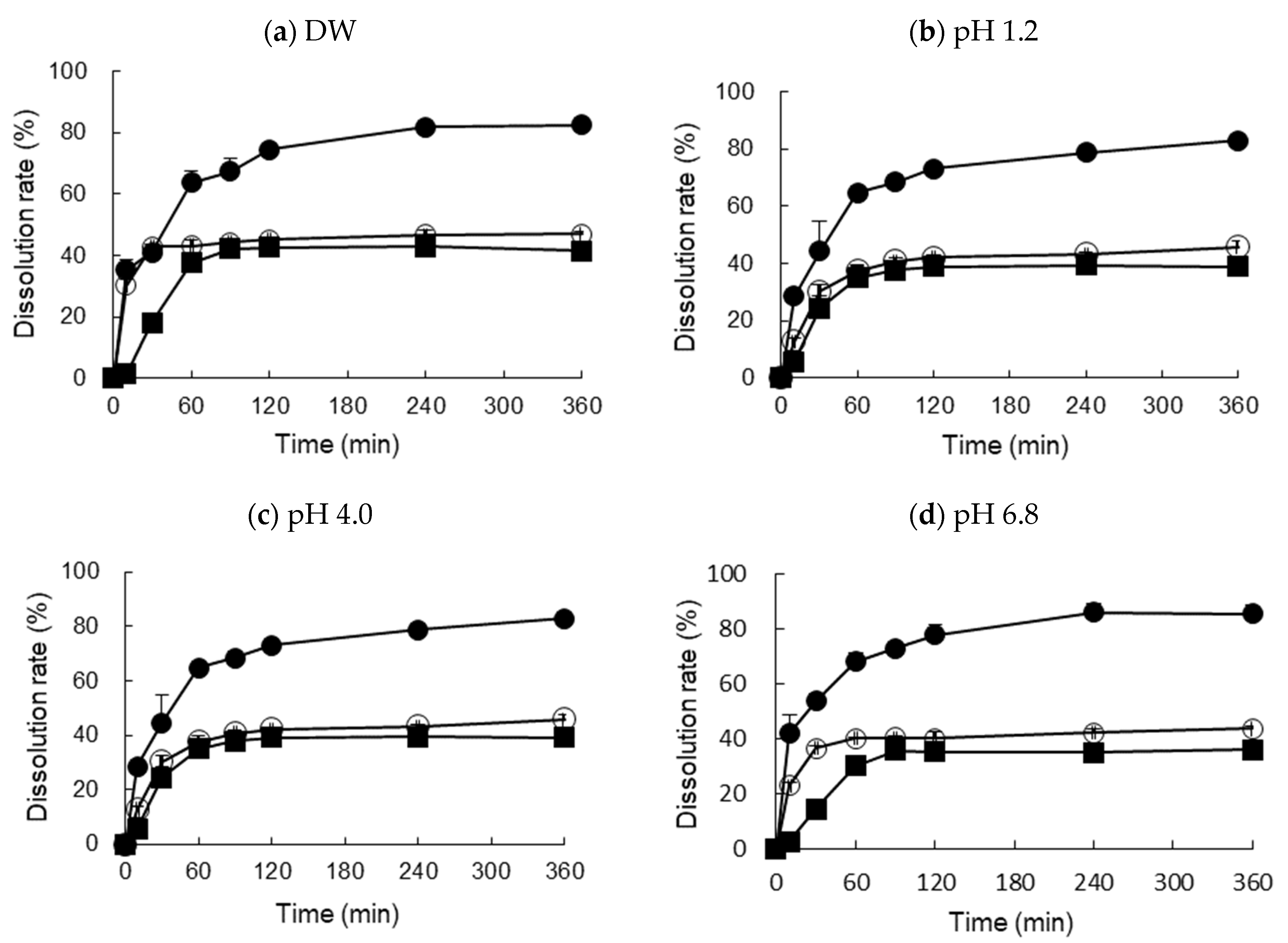
Dissolution was tested using a dissolution tester (DRS-14, Labbindia, India) with 900 mL distilled water (DW), SGF (pH 1.2), acetate buffer (pH 4.0), and SIF (pH 6.8), via agitation with a paddle at 75 rpm and 37 ± 0.5 °C.
The optimized RXB-ASD sample, placed within a hard gelatin capsule (size number 2) containing equivalent amounts of RXB (20 mg) in a sinker, was placed in a dissolution medium, and 5-mL aliquot samples were withdrawn at certain time intervals (0.10, 30, 60, 120, 240, and 360 min) and filtered using a membrane filter (0.45 μm, Whatman, PA, USA). The filtered samples were diluted with a chloroform and methanol mixture (1:7 v/v%), and the concentration of the drug was determined by HPLC. The release rates were compared with those of a conventional tablet (Xalretol®, 20 mg as RXB), with RXB powder as a negative control.
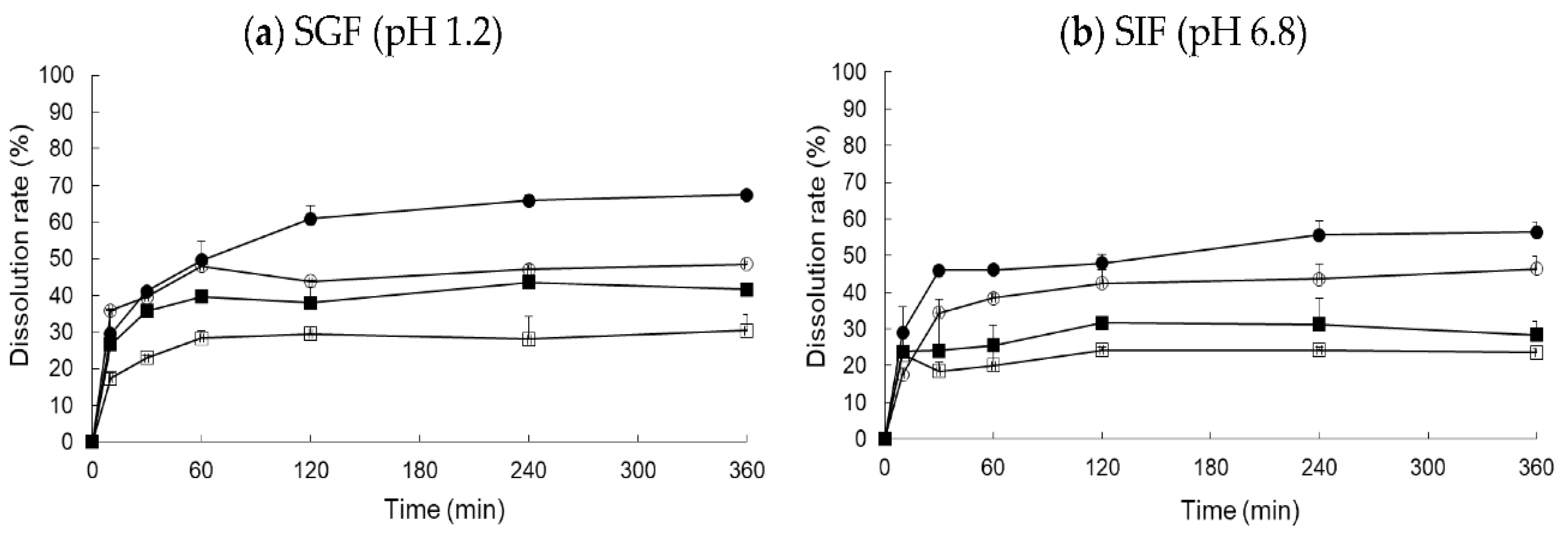
To investigate the effect of food on optimized RXB-ASD, the dissolution rate was monitored for fasted-state simulated gastric fluid (FaSSGF), fed-state simulated gastric fluid (FeSSGF), fasted-state simulated intestinal fluid (FaSSIF), and fed-state simulated intestinal fluid (FeSSIF).
The similarity of release profiles between the test preparation and reference preparation in different dissolution media was evaluated as the similarity factor,
f2. The relevant equation is as follows [
24,
25]:
where
n is the number of sampling time points, and
R and
T represent the cumulative dissolution of the drug at the specified time point in the respective reference formulation and the test formulation. The value of
f2 ranged from 0 to 100; when the value exceeded 50, the drug release profiles between the reference formulation and test formulation were considered to be similar.
2.6. Stability
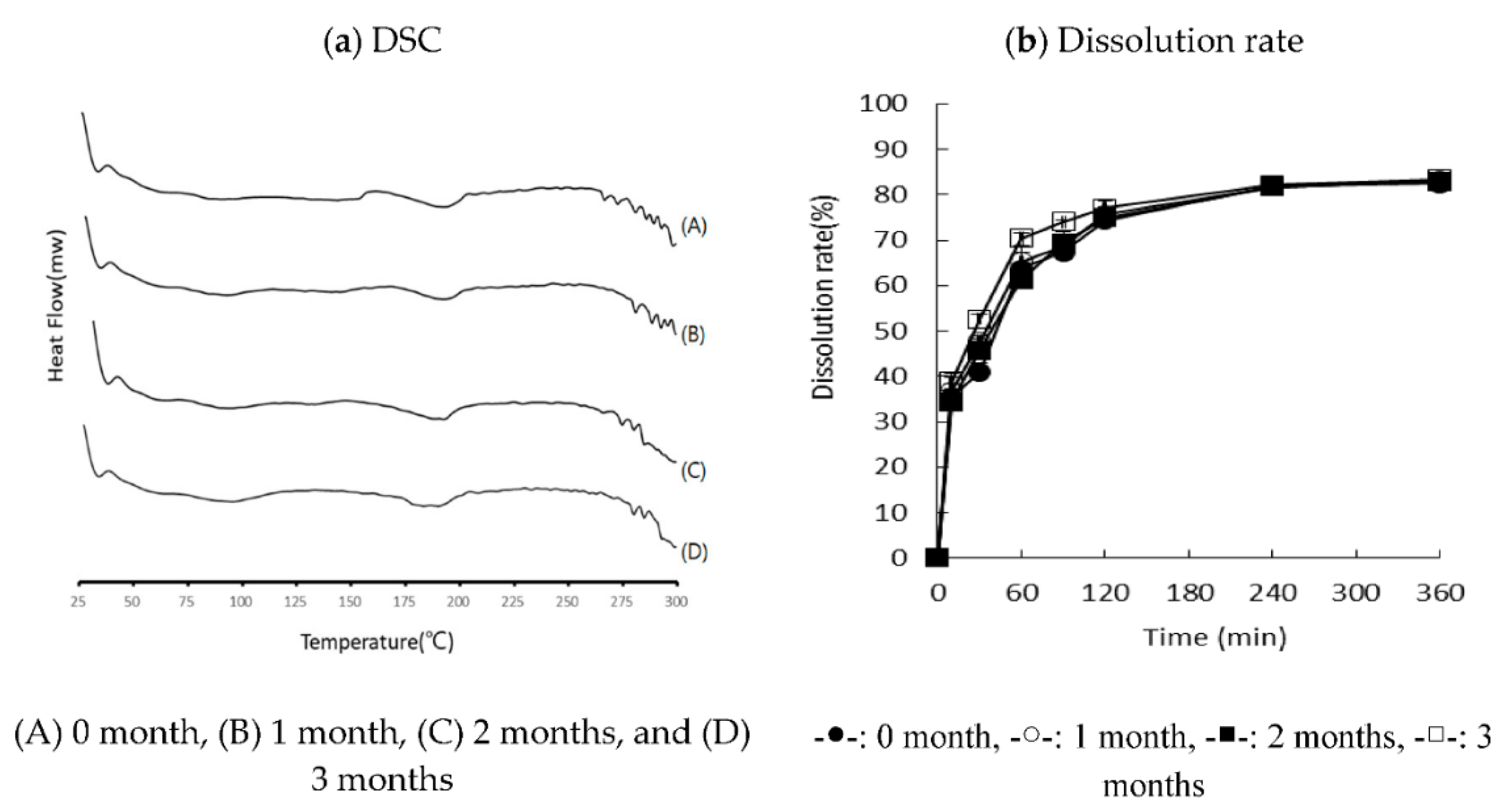
To check the stability, hard gelatin capsules (size number 2) filled with RXB-ASD were submitted to accelerated degenerative conditions (40 °C/75% RH). The appearance, drug content, and in vitro dissolution rates were evaluated for three months.
2.7. Application to Pharmacokinetic Study
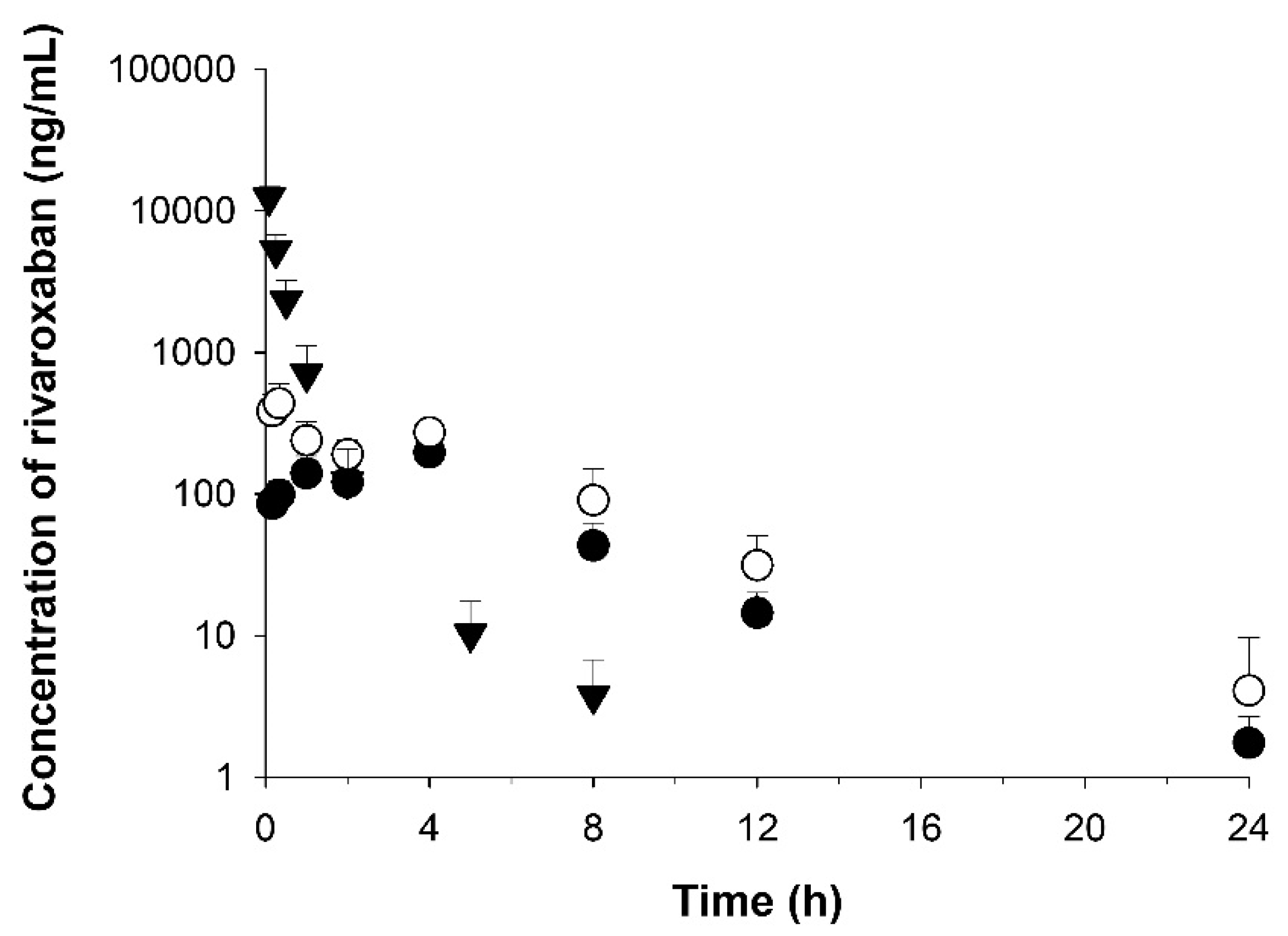
RXB and RXB-ASD were administered orally at dose of 10 mg/kg to elucidate the absorption in male Sprague–Dawley rats aged seven weeks and weighing 199–225 g (Orient Bio, Seongnam, Korea). In addition, to monitor the bioavailability, RXB-ASD was administered intravenously at dose of 2 mg/kg. For all experiments, the animals were kept in plastic cages with free access to a standard rat diet (PMI Nutrition International, Richmond, IN, USA) and water at a temperature of 20–26 °C, with a 12 h light–dark cycle and a relative humidity of 40–60% under the guidance of the Institutional Animal Care and Use Committee of Chungnam National University (202003A-CNU-055, 23 June and 8 August 2020, Daejeon, Korea).
Prior to dosing, animals were fasted for 14 h and provided with free access to water after a further 4 h. RXB was homogenized in normal saline and administered for oral administration at a volume of 5 mL/kg and solubilized in mixture composed of 10% DMSO, 40% PEG 400, and 50% normal saline for intravenous injection at a volume of 2 mL/kg. Blood samples (300 μL) were obtained from the jugular vein at 0.0167, 0.33, 1, 2, 4, 8, 12, and 24 h after oral dosing and at 0.083, 0.25, 0.5, 1, 2, 5, 8, and 24 h after intravenous dosing in four animals per group. The blood samples were immediately centrifuged at 17,600× g for 5 min, and the separated plasma samples were stored at −20 °C until analysis.
With respect to RXB in rat plasma, the bioanalytical method for RXB in rat plasma was adjusted and optimized based on a previously established method [
26]. LC used a 1200 series system from Agilent Technologies (Santa Clara, CA, USA) composed of a binary pump, degasser, autosampler, and column oven. A Zorbax phenyl column (50 × 2.1 mm, 5 µm particle size; Agilent, Santa Clara, CA, USA) was used with the mobile phase consisting of (A) 10 mM ammonium formate containing 0.1% formic acid of total volume in water (pH 4.5) and (B) methanol with gradient elution at a 0.3 mL/min flow rate. Samples (2 µL) were analyzed using the following isocratic mode for 3 min with a composition of 40% A and 60% B. The temperatures of the column oven and autosampler were maintained at 40 °C and 10 °C, respectively.
MS was performed on the API 4000 Qtrap LC-MS/MS system (AB Sciex, Framingham, MA, USA) operated in the negative ion mode. The ion source parameters were set as follows: curtain gas 20 psi, ion spray voltage 5500 V, ion source temperature 600 °C, nebulizing gas (GS1) 60 psi, drying gas (GS2) 50 psi. The MS parameters of declustering potential and collision energy for RXB were optimized at 86 V and 37 V, respectively, and those for the internal standard were optimized at 81 V and 25 V, respectively. The ion transitions in multiple-reaction monitoring (MRM) were monitored at m/z 436.2→145.0 for RXB and m/z 338.2→296.1 for linezolid, an internal standard. The data were acquired using Analyst (version 1.4.2) from AB Sciex.
The pharmacokinetic analysis was performed by a noncompartmental analysis using Phoenix WinNonlin
® 8.1 (Pharsight Corp., Cary, NC, USA). The peak plasma concentration (C
max) and the time to reach the peak concentration (T
max) were obtained directly from the profile of the time‒plasma concentration. The elimination rate constant (K
el) was determined by linear regression in the terminal phase. The half-life (T
1/2) in the terminal phase was calculated by dividing ln 2 by the K
el. In addition, we determined the systemic clearance (CL), the volume of distribution (V
d), and mean residence time (MRT). The area under the plasma concentration‒time curve from time zero to infinity (AUC
inf) was calculated via the linear trapezoidal rule and the standard area extrapolation method [
27].


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}