
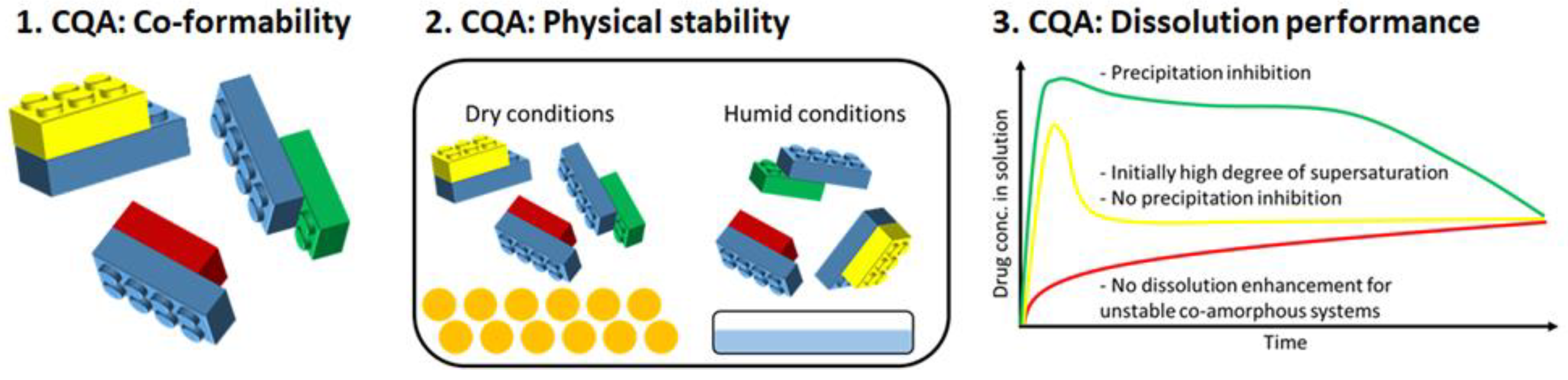
Co-Amorphous Drug Formulations in Numbers: Recent Advances in Co-Amorphous Drug Formulations with Focus on Co-Formability, Molar Ratio, Preparation Methods, Physical Stability, In Vitro and In Vivo Performance, and New Formulation Strategies


Abstract
:
1. Introduction
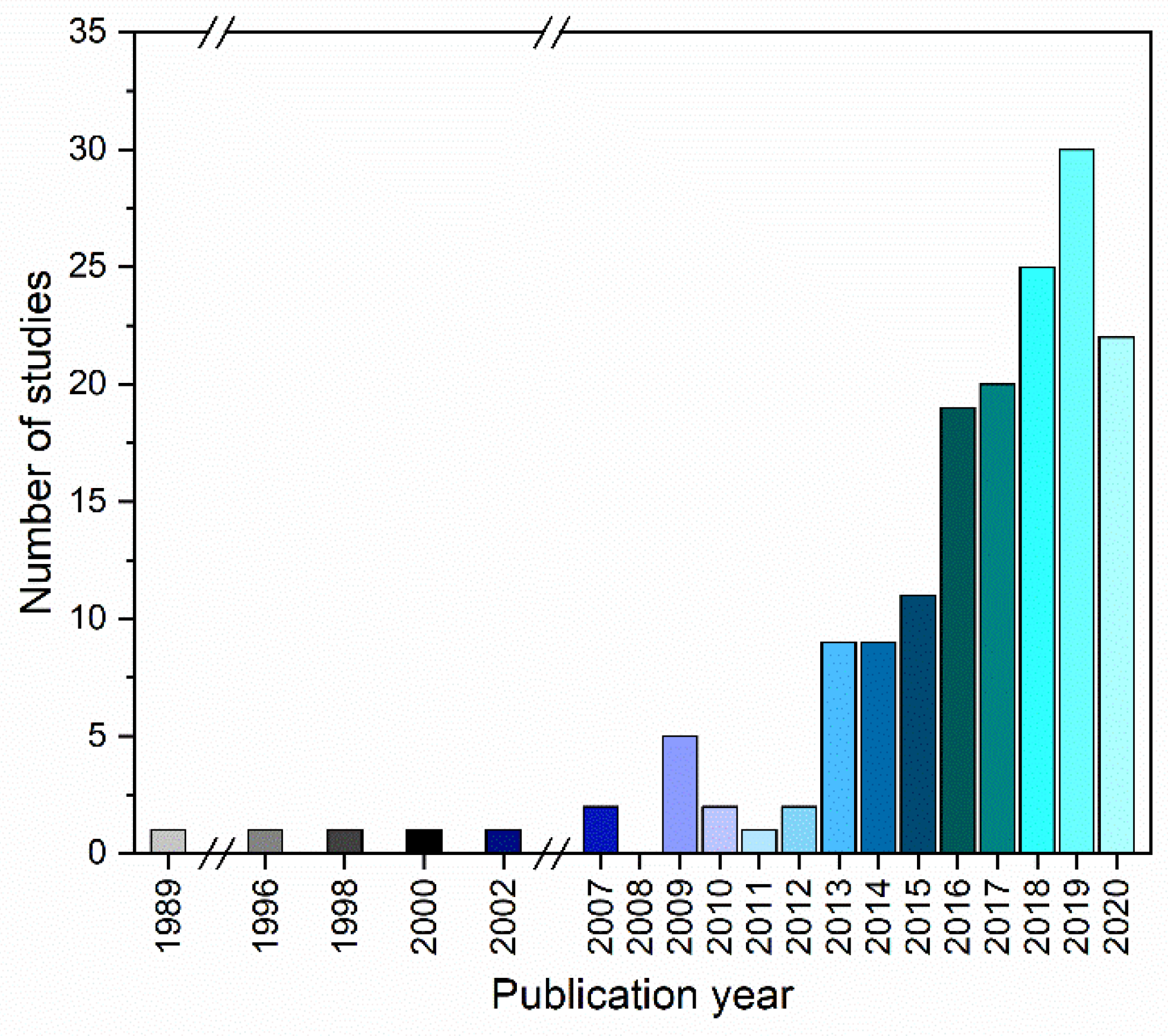
2. Research Interest in the Field of CAMS
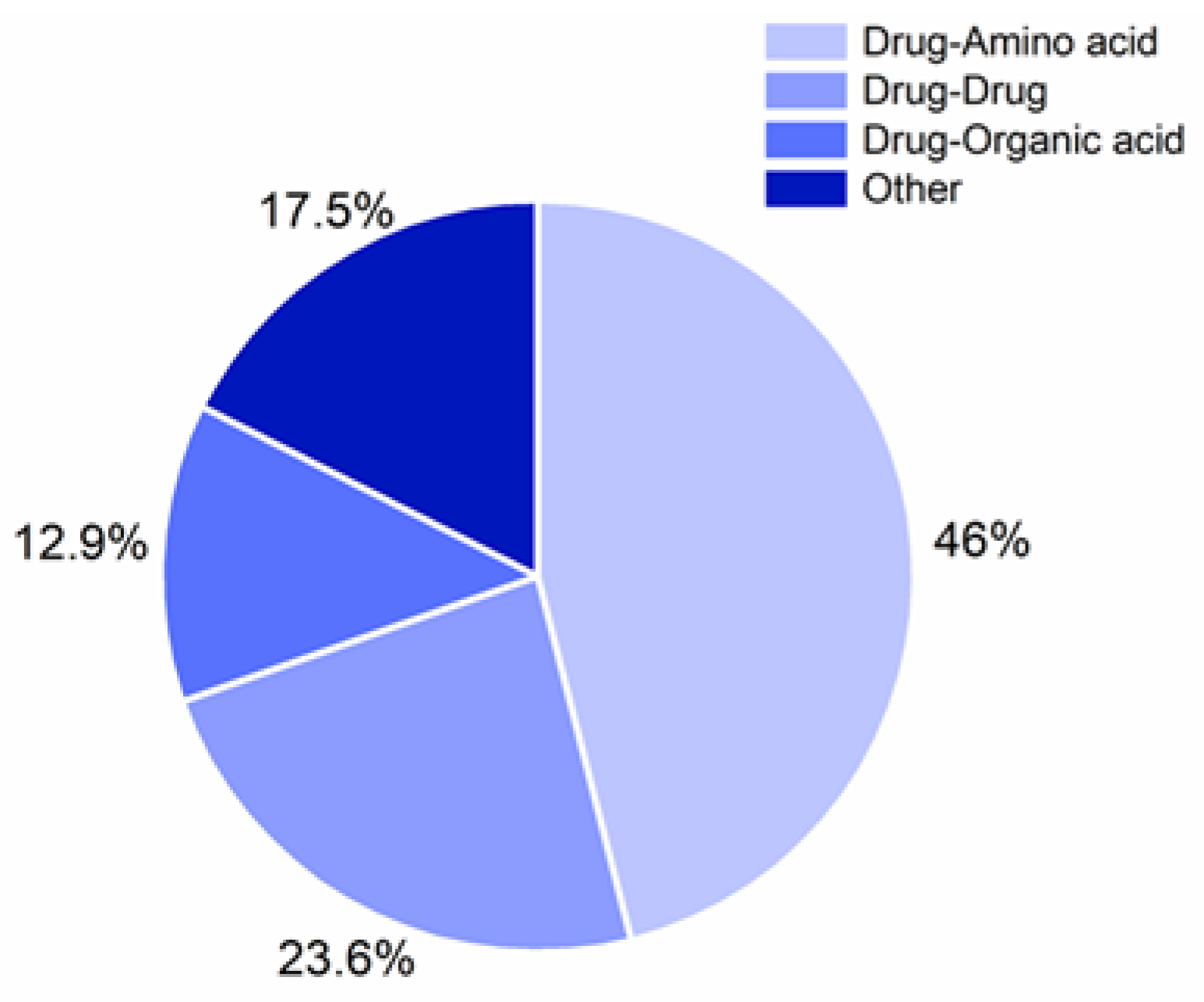
3. Classes of Investigated CAMS
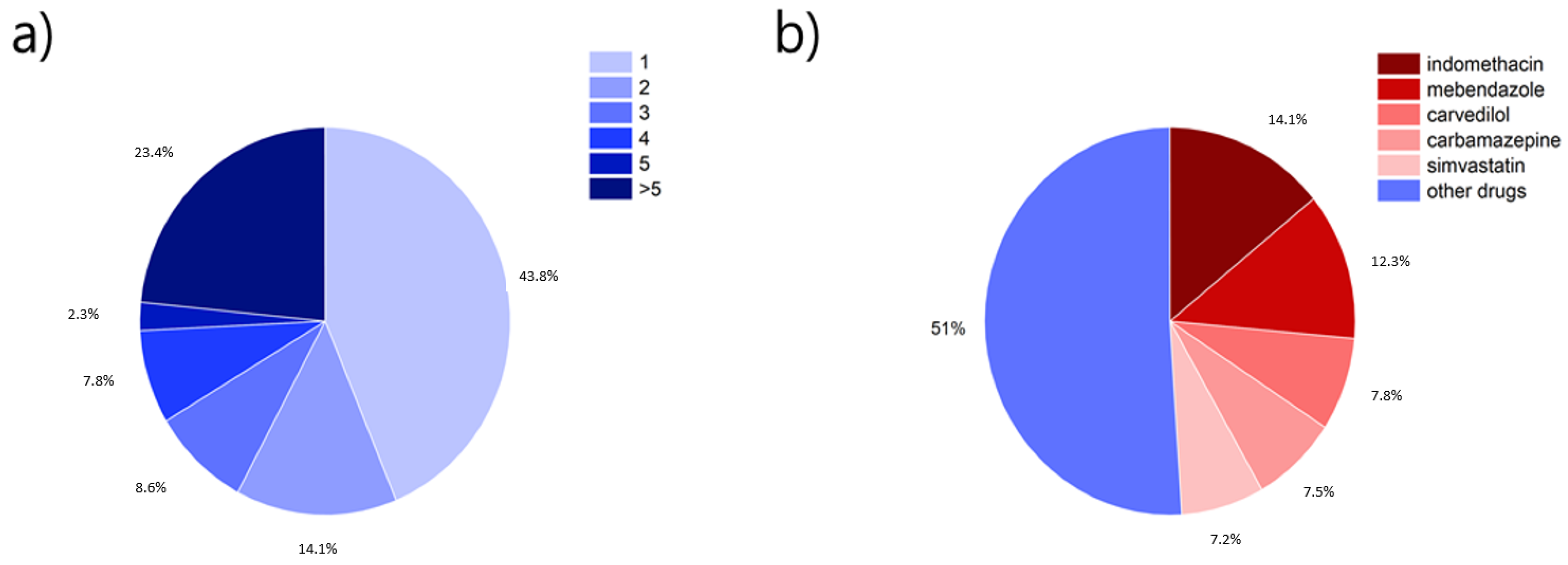
4. Drugs Investigated for the Formation of CAMS
5. Co-Former Selection of Investigated CAMS
5.1. Methods for Co-Former Selection
5.1.1. Prediction of the Miscibility of Two Components
5.1.2. Physicochemical Properties of Co-Formers
5.2. Stabilization Mechanism of CAMS
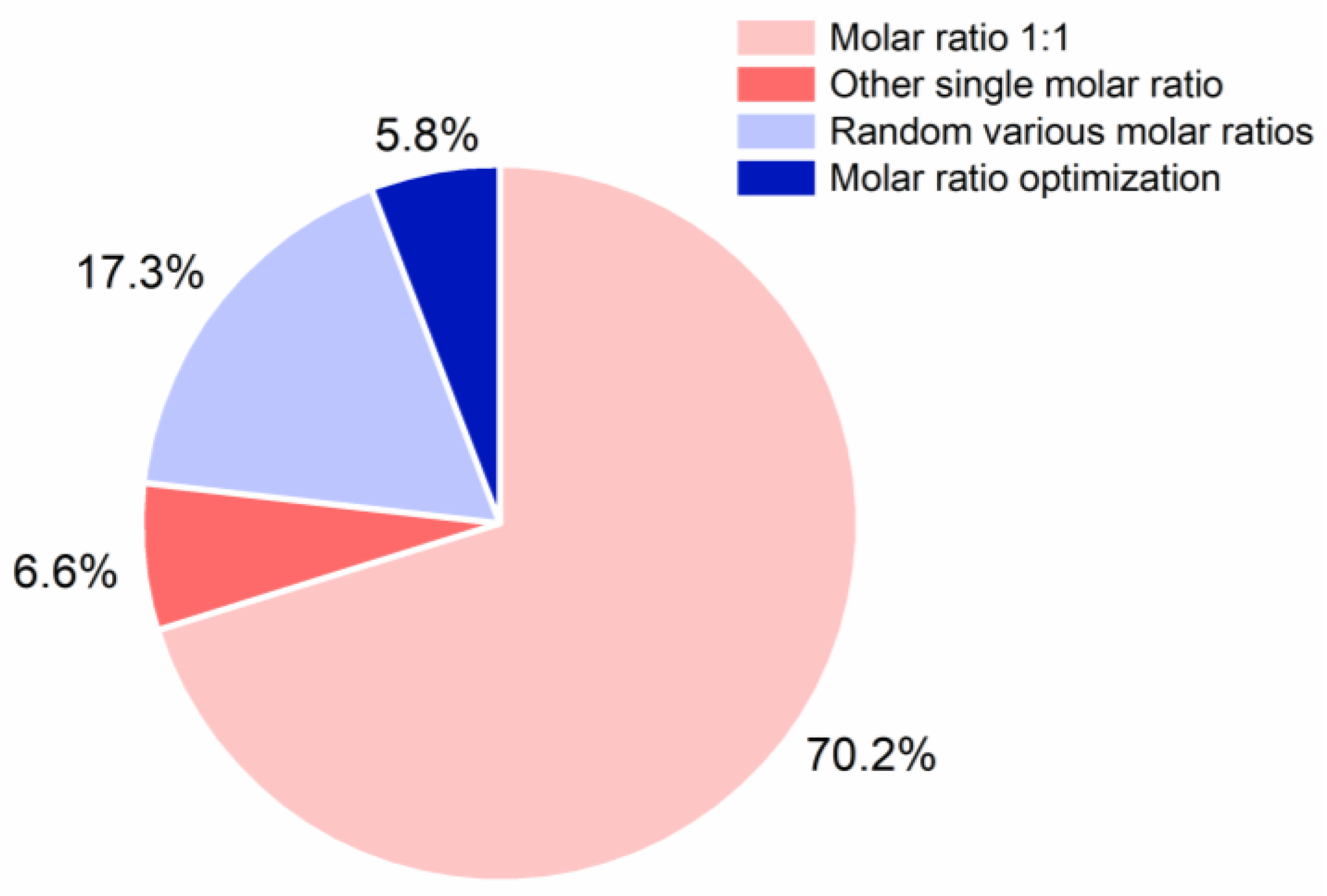
6. Molar ratio Optimization of Investigated CAMS
6.1. Methods for Molar Ratio Optimization
6.1.1. Detection of Endothermic and Exothermic Thermal Events

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amorphous Systems | Preparation Method | Compositions of the Systems | Optimization Methods | The Optimal Molar Ratio(s) | Physical Stability to Confirm the Optimal Molar Ratio | Reference |
|---|---|---|---|---|---|---|
| Atenolol- Urea | Melt-quench | Molar ratios of 1:1, 1:2, 1:4, 1:6, 1:8, 1:10, 1:12 | Thermal analysis by DSC; Precipitation test | Atenolol–Urea 1:4 for CAMS; Atenolol–Urea 1:8 for super-saturation maintenance | N | [44] |
| Carvedilol– Aspartic acid | Spray drying | Molar ratios of 2:1, 1:1, 1:1.25, 1:1.5, 1:1.75, 1:2, 1:2.25, 1:2.5, 1:3, 1:4 | Data fitting methods of Tgs; FTIR-PCA | 1:1.46 (mathematically); 1:1.5 (experimentally) | Y | [72] |
| Carvedilol– Benzoic acid | Spray drying | Molar ratios of 1:4, 1:3, 1:2, 1:1, 1.5:1, 2:1, 4:1 | Determination of the highest Tg | 1.5:1 | Y | [39] |
| Carvedilol– Citric acid | Spray drying | Molar ratios of 1:4, 1:3, 1:2, 1:1, 2:1, 3:1, 4:1 | Determination of the highest Tg | 2:1 | Y | [39] |
| Carvedilol– Glutamic acid | Spray drying | Molar ratios of 2:1, 1:1, 1:1.25, 1:1.5, 1:1.75, 1:2, 1:2.25, 1:2.5, 1:3, 1:4 | Data fitting methods of Tgs; FTIR-PCA | 1:1.43 (mathematically); 1:1.5 (experimentally) | Y | [72] |
| Carvedilol– Malic acid | Spray drying | Molar ratios of 1:4, 1:3, 1:2, 1:1, 2:1, 3:1, 4:1 | Determination of the highest Tg | 2:1 | Y | [39] |
| Carvedilol– Tryptophan | Ball milling | Molar fractions 0.1–0.9, at an interval of 0.1 | Determintion of Tgβ by DMA; thermal analysis by DSC (lack of any endothermic or exothermic events) | The molar fractions of carvedilol were 34–52% (equal to the molar ratio of Carvedilol–Tryptophan from 1:0.92 to 1:1.94). | Y | [73] |
| Ezetimibe– Lovastatin– Soluplus® | Spray drying | Ezetimibe–Lovastatin at the weight ratios of 1:1, 1:2, 1:4. The weight fractions of soluplus® were 50 wt %, 75 wt %, 90 wt %. | Physical stability | Weight ratio of 12.5:12.5:75. | Y | [78] |
| Ezetimide–Simvastatin– Kollidon® VA64 | Melt-quench | Ezetimibe–Simvastatin at the weight ratios of 1:1. The weight fractions of polymer were 5 wt %, 20 wt %, 40 wt %, 60 wt % | The viscoelastic properties measured by oscillatory shear rheology | Minimal 40 wt % polymer required | N (only confirmed CAMS with 40 wt % polymer was stable) | [79] |
| Furosemide– Arginine | Ball milling | Molar fractions of furosemide from 0.09 to 0.9 | Comparison of the experimental Tgs to the theoretical Tgs for the largest deviation | 1:1 | N | [30] |
| Furosemide– Tryptophan | Ball milling | Molar fractions of furosemide from 0.09 to 0.9 | Comparison of the experimental Tgs to the theoretical Tgs for the largest deviation | 1:1 | N | [30] |
| Indomethacin– Arginine | Ball milling | Molar fractions of indomethacin from 0.09 to 0.9 | Comparison of the experimental Tgs to the theoretical Tgs for the largest deviation | 1:1 | N | [30] |
| Indomethacin–Naproxen | Melt-quench | Molar fractions 0.1–0.9, at an interval of 0.1 | Phase diagrams to determine the eutectic point | 1:1.5 | Y | [73] |
| Indomethacin–Tryptophan | Ball milling | Molar fractions of indomethacin from 0.09 to 0.9 | Comparison of the experimental Tgs to the theoretical Tgs for the largest deviation | 1:1 | N | [30] |
| Indomethacin–Tryptophan | Ball milling | Molar fractions 0.1–0.9, at an interval of 0.1 | Determintion of Tgβ by DMA; thermal analysis by DSC (lack of any endothermic or exothermic events) | The molar fractions of indomethacin were 5–25% (equal to the molar ratio of Indomethacin–Tryptophan from 1:3 to 1:19). | Y | [76] |
| Naproxen– Indomethacin | Melt-quench | Molar fractions 0.1–0.9, at an interval of 0.1 | XRP–diffractograms–PCA; FTIR–PCA; Phase diagrams | 1.5:1 | Y | [74] |
| Naproxen– Meglumine | Melt-quench | Molar ratios of 10:1, 2.5:1, 10:7, 1:1, 7:10, 1:2.5, 1:10 | Determination of the highest glass transition temperature; Physical stability | 1:1 | Y | [80] |
| Naproxen–Sodium– Indomethacin | Melt-quench | Molar fractions 0.1–0.9, at an interval of 0.1 | Physical stability | Naproxen–Sodium: 0.1–0.4 (equal to the molar ratio Naproxen–Sodium:Indomethacin from 1:9 to 1:1.5) | Y | [81] |
| Naproxen–Sodium– Naproxen–Indomethacin | Melt-quench | Molar ratio of Naproxen–Sodium:Naproxen fixed at 1:1; Molar fractions of Indomethacin: 0.1–0.9, at an interval of 0.1 | Physical stability | Indomethacin: 0.3–0.9 (equal to the molar ratio Naproxen–Sodium:Naproxen:Indomethacin from 1:1:0.86 to 1:1:1.8) | Y | [81] |
| Nifedipine– Cimetidine | Melt-quench | Molar fractions 0.1–0.9, at an interval of 0.1 | DSC thermograms of both freshly prepared samples and stored sample (increased Tg, and lack of crystallization and melting endotherms) | The molar fractions of cimetidine were 0.3–0.9 (equal to Nifedipine–Cimetidine from 2.3:1 to 1:9). | N (only for the sample at the 1:1 molar ratio) | [77] |
| Nifedipine– Paracetamol | Melt-quench | Molar fractions 0.1–0.9, at an interval of 0.1 | Phase diagrams to determine the eutectic point | 1:1.5 | Y | [73] |
| Nimesulide– Carvedilol | Melt-quench | Molar fractions 0.1–0.9, at an interval of 0.1 | DSC thermograms of both freshly prepared samples and stored sample (increased Tg, and lack of crystallization and melting endotherms) | The molar fractions of carvedilol were 0.3–0.8 (equal to Nimesulide–Carvedilol 2.3:1 to 1:4). | N (only for the sample at the 1:1 molar ratio) | [77] |
| Ofloxacin– Tryptophan | Freeze-drying | Molar ratios of 1:1, 1:2, 1:3, and also weight fractions 0.5–0.95 | Kinetic solubility measurements of drug for freeze-dried samples; Comparison of the experimental Tgs to the theoretical Tgs for the largest deviation | Best solubility was found at the molar ratio of 1:1.76; highest positive deviation in Tg values was also found at a molar ratio of 1:1.76. | N (only for the CAMS at the 1:1.76 molar ratio) | [82] |
| Paracetamol– Antipyrine | Melt-quench | Molar fractions 0.1–0.9, at an interval of 0.1 | Thermal analysis during physical stability | 1:2 | Y | [83] |
| Paracetamol–Celecoxib | Melt-quench | Molar fractions 0.1–0.9, at an interval of 0.1 | Phase diagrams to determine the eutectic point | 1:1 | Y | [73] |
| ROY*– Pyrogallol | Melt-quench | Weight fractions 0–100 wt %, at an interval of 5 wt % | Thermal analysis by DSC (lack of any endothermic or exothermic events) | Pyrogallol content 25–35 wt % (equal to the molar ratio ROY*:Pyrogallol from 1:0.69 to 1:1.11) | N | [75] |
| Simvastatin– Nifedipine | Melt-quench | Molar fractions 0.1–0.9, at an interval of 0.1 | Physical stability; phase diagram | CAMS at the molar ratio of 2:1 to 1:2 were all stable for at least one year (Eutectic composition: 5.375:1) | Y | [84] |
| Ursolic acid– Piperine | Solvent evaporation | 3:1, 2:1, 1.5:1, 1:1 and 1:2 | Determination of the highest Tg; physical stability | 2:1 showed the highest Tg; 1.5:1 was the most stable CAMS | Y | [85] |
| Valsartan– Nifedipine | Melt-quench | Weight ratios of valsartan/nifedipine at 90:10, 80:20, 80:30 (molar 1:1), 60:40, 50:50, 40:60 | Physical stability; in vitro dissolution test | CAMS at all molar ratios were stable; CAMS at the weight fractions of 80:30, 80:20, and 90:10 showed better drug release of both drugs (equal to the molar ratio 1:1, 1:0.67, and 1:0.3) | Y | [86] |
6.1.2. Detection of the Glass Transition Temperatures (Tg)
6.1.3. Relationship between CAMS and the Eutectic Behavior of Crystalline Drug and Co-Former
6.1.4. Application of Multivariate Analysis to Analytical Data
7. Preparation Methods of CAMS
| Drug | Amino Acid | Co- Amorphous (Y/N) | Preparation Method | Notes | Reference |
|---|---|---|---|---|---|
| Carvedilol | Aspartic acid | N | Ball milling | [29,41,54,103] | |
| Carvedilol | Aspartic acid | Y | Spray drying | Dependent on solvent composition | [72,103,104] |
| Carvedilol | Aspartic acid | N | LAG | [103] | |
| Carvedilol | Glutamic acid | N | Ball milling | [29,41,54,103] | |
| Carvedilol | Glutamic acid | Y | Spray drying | Dependent on solvent composition | [72,103] |
| Carvedilol | Glutamic acid | N | LAG | [103] | |
| Indomethacin | Histidine | N | Ball milling | [29,41,54,103] | |
| Indomethacin | Histidine | Y | Spray drying | [105,106] | |
| Indomethacin | Lysine | Y | Ball milling | 60 min | [29,41,54,107,108] |
| Indomethacin | Lysine | N | Ball milling | 60 min at 4 °C | [105] |
| Indomethacin | Lysine | Y | Spray drying | [105,106,107] | |
| Indomethacin | Lysine | N | LAG | Crystalline salt was formed | [108] |
| Mebendazole | Phenylalanine | Y | Ball milling | 60 min | [29,41,54,107] |
| Mebendazole | Phenylalanine | N | Ball milling | Up to 180 min at 4 °C | [109,110] |
| Mebendazole | Proline | Y | Ball milling | Up to 180 min at 4 °C | [110] |
| Mebendazole | Proline | N | Ball milling | 60 min | [29,41,54] |
| Simvastatin | Tryptophan | Y | Ball milling | [29,41,54] | |
| Simvastatin | Tryptophan | N | Spray drying | [111] |
7.1. “Rules of Thumb” for the Preparation Method Ball Milling for Drug–Amino Acid CAMS
7.2. The Use of Peptides Instead of Single Amino Acids as Co-Formers in CAMS
8. Physical Stability of Investigated CAMS
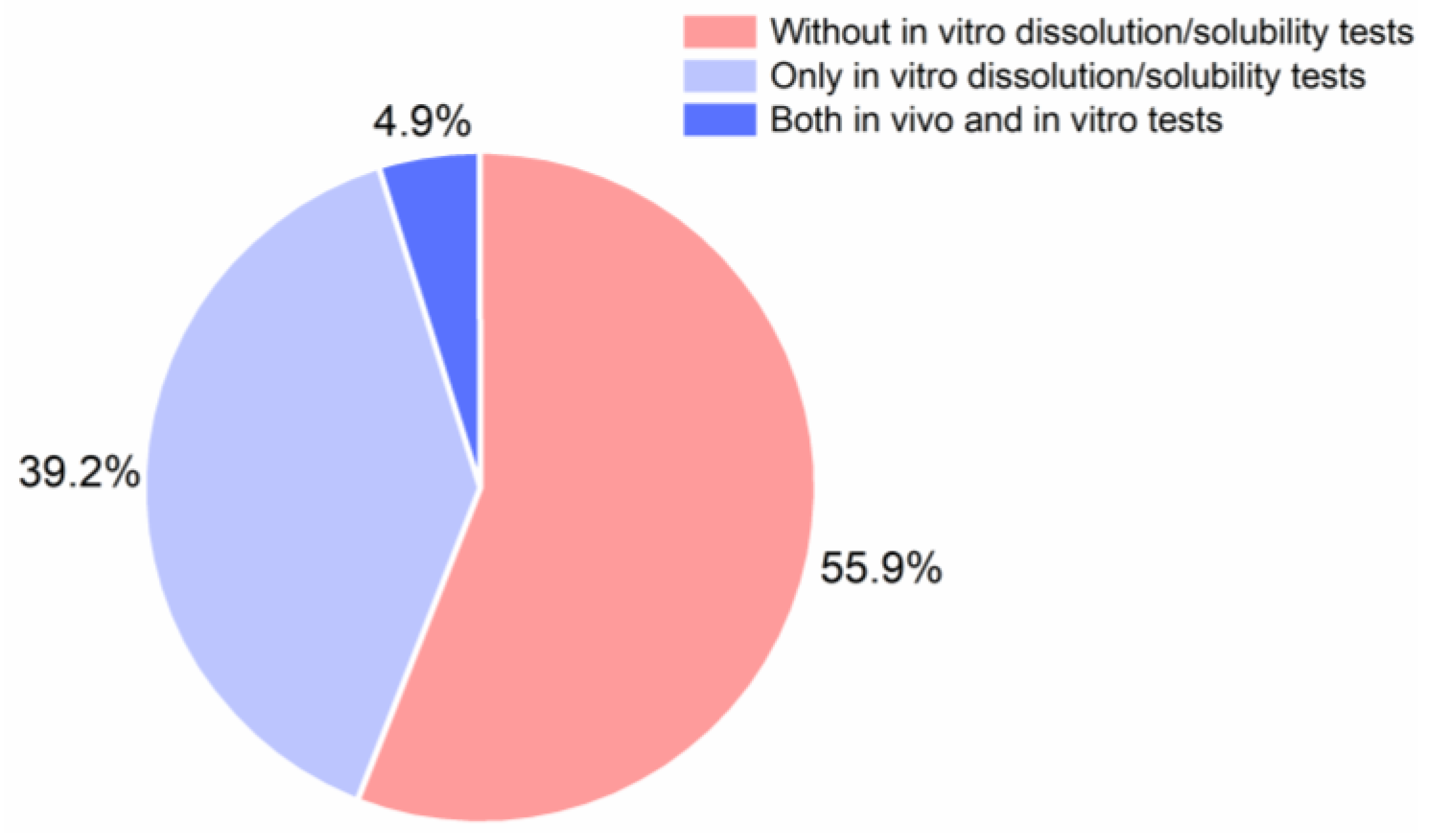
9. In Vitro and In Vivo Performance of CAMS
| Drug | Co-Former | Molar Ratio | In Vitro Study | In Vivo Study | Reference |
|---|---|---|---|---|---|
| Atorvastatin Calcium | Nicotinamide | 1:1 | Intrinsic and powder dissolution rate enhanced compared to physical mixture | Rats: CAMS increased Cmax by 2.25-fold and AUC by 1.72-fold compared to the crystalline drug; | [49] |
| Curcumin | Artemisinin | 1:1 | Enhanced intrinsic dissolution rate compared to crystalline curcumin | Rats: CAMS showed a Cmax of 1005 µg/mL and an AUC of 24.7 µg*h/mL; crystalline CUR could not be detected in the plasma; | [137] |
| Ritonavir | Quercetin | 1:2 | - | Rats: CAMS improved Cmax by 1.26-fold and Tmax decreased by 2 h compared to the crystalline drug; however, no significant enhancement in oral bioavailability was found (AUC); | [132] |
| Talinolol | Naringin | 1:1 | - | Rats: CAMS improved Cmax by 8.6-fold, Tmax decreased by 1.5 h and AUC improved by 5.4-fold compared to the crystalline drug; | [124] |
| Olanzapine | Ascorbic Acid Citric Acid Tartaric Acid | 1:1, 1:2 | Enhanced dissolution rate | Healthy men: CAMS has 115.83% bioavailability compared to a marketed product and showed a faster disintegration; Note: the CAMS was formulated as an oral film with polymeric excipients. | [33] |
| Atenolol | Hydrochlorothiazide | 1:1 | Enhanced intrinsic dissolution rate | Rats: CAMS improved Cmax by 7.3-fold compared to crystalline drug (hydrochlorothiazide), by 2.8-fold compared to the amorphous drug and by 1.7-fold compared to the physical mixture. AUC was increased by 3.4-, 2.6- and 1.4-fold compared to the crystalline drug, the amorphous drug, and the physical mixture. | [138] |
| Loratadine | Citric Acid | 1:1 | Enhanced solubility and dissolution rate compared to the crystalline drug | Rats: CAMS improved Cmax by 2.59-fold compared to the crystalline drug. T½ decreased by 2.5 h. AUC improved by 2.45-fold. Enhanced absorption of loratadine. | [139] |
| Naproxen | Arginine | 1:1 | Enhanced intrinsic dissolution rate compared to the crystalline drug | Rats: CAMS improved Cmax by 2.15-fold and AUC by 1.5-fold compared to the crystalline drug. For the crystalline salt of the CAMS, no increase in bioavailability was seen, even though the in vitro performance was enhanced. | [140] |
| Curcumin | Artemisinin | 1:1 | - | Rats: CAMS showed a Cmax of 1.23 µg/mL, a Tmax of 30 min, and an AUC of 3.68 µg·h/mL. The crystalline drug curcumin could not be detected. | [135] |
| Curcumin | Piperine | 1:1 | Enhanced powder dissolution rate and higher supersaturation compared to crystalline drug | Rats: CAMS improved Cmax by 2.64-fold for curcumin and by 2.41-fold for piperine. The AUC was improved by 2.16-fold and 1.92-fold for the individual drugs. | [133] |
| Docetaxel | Myricetin (natural p-Gp inhibitor) | 1:1 | Enhanced intrinsic and powder dissolution rate compared to crystalline drug | Rats: CAMS improved Cmax by 2.3-fold and AUC by 1.7-fold compared to the physical mixture. Cmax and AUC increased by 1.5- and 2.3-fold respectively compared to the amorphous drug docetaxel. CAMS improved Cmax and AUC by 3.9-fold and 3.13-fold, respectively, compared to the crystalline drug docetaxel. Thus, the bioavailability of docetaxel compared to the crystalline drug is 313%. CAMS improved Cmax and AUC for the crystalline drug myricetin by 2.1-fold and 1.9-fold, respectively. | [141] |
| Docetaxel | Bicalutamide | 1:1 | Enhanced dissolution rate for both drugs, but supersaturation only achieved for docetaxel | Rats: CAMS improved Cmax by 8.8-fold and AUC by 11.8-fold for docetaxel compared to the crystalline drug. Cmax improved by 3.3-fold and AUC by 3.2-fold for bicalutamide compared to the crystalline drug. | [142] |
| Valsartan | Nifedipine | Weight ratio: 80:30 (2.12:1 molar ratio) | Enhanced dissolution rate and supersaturation for both drugs | Rats: CAMS improved Cmax by 3.63-fold and AUC by 1.44-fold compared to the crystalline drug nifedipine. CAMS improved Cmax by 2.2-fold and AUC by 1.4-fold compared to the crystalline drug valsartan. | [86] |
| Ibrutinib | Saccharin | 1:1 | Enhanced dissolution rate and supersaturation | Rats: CAMS increased Cmax by 2.9-fold compared to the crystalline drug. AUC, Tmax, and T½ were not significantly different. | [122] |
| Ibrutinib | Oxalic Acid (and microcrystalline cellulose) | 1:1:1 | Enhanced dissolution rate | Rats: CAMS improved Cmax by 1.49-fold and AUC by 1.48-fold compared to the crystalline drug. | [36] |
| Ursolic acid | Piperine | 1.5:1 | Enhanced dissolution rate and supersaturation | Rats: CAMS improved Cmax by 4.9-fold and AUC by 5.77-fold compared to crystalline ursolic acid. | [85] |
| Sacubitril | Valsartan (additionally with lactose monohydrate or microcrystalline cellulose) | 1:1; weight ratio co-amorphous to excipient 1:3 | Enhanced dissolution rate and supersaturation of the co-amorphous formulations | Rats: CAMS were compared to the marketed formulation Entresto®. A 1.54-fold higher AUC was found for valsartan and a 3.56-fold higher AUC was found for the sacubitril derivate in the CAMS with lactose monohydrate. A 1.39-fold higher AUC was found for valsartan and a 1.25-fold higher AUC was found for the sacubitril derivate in the CAMS with microcrystalline cellulose. However, there was decreased bioavailability for the CAMS with microcrystalline cellulose compared to the binary CAMS. | [121] |
| Atorvastatin | Naringin | 1:1 | Enhanced dissolution rate and supersaturation (but fast precipitation after 30 min) | Rats: Melt-quench CAMS improved Cmax by 1.73-fold compared to the physical mixture. AUC was not found to be significantly different. Solvent evaporated CAMS improved Cmax by 1.73-fold and AUC by 3.3-fold compared to the physical mixture. | [125] |
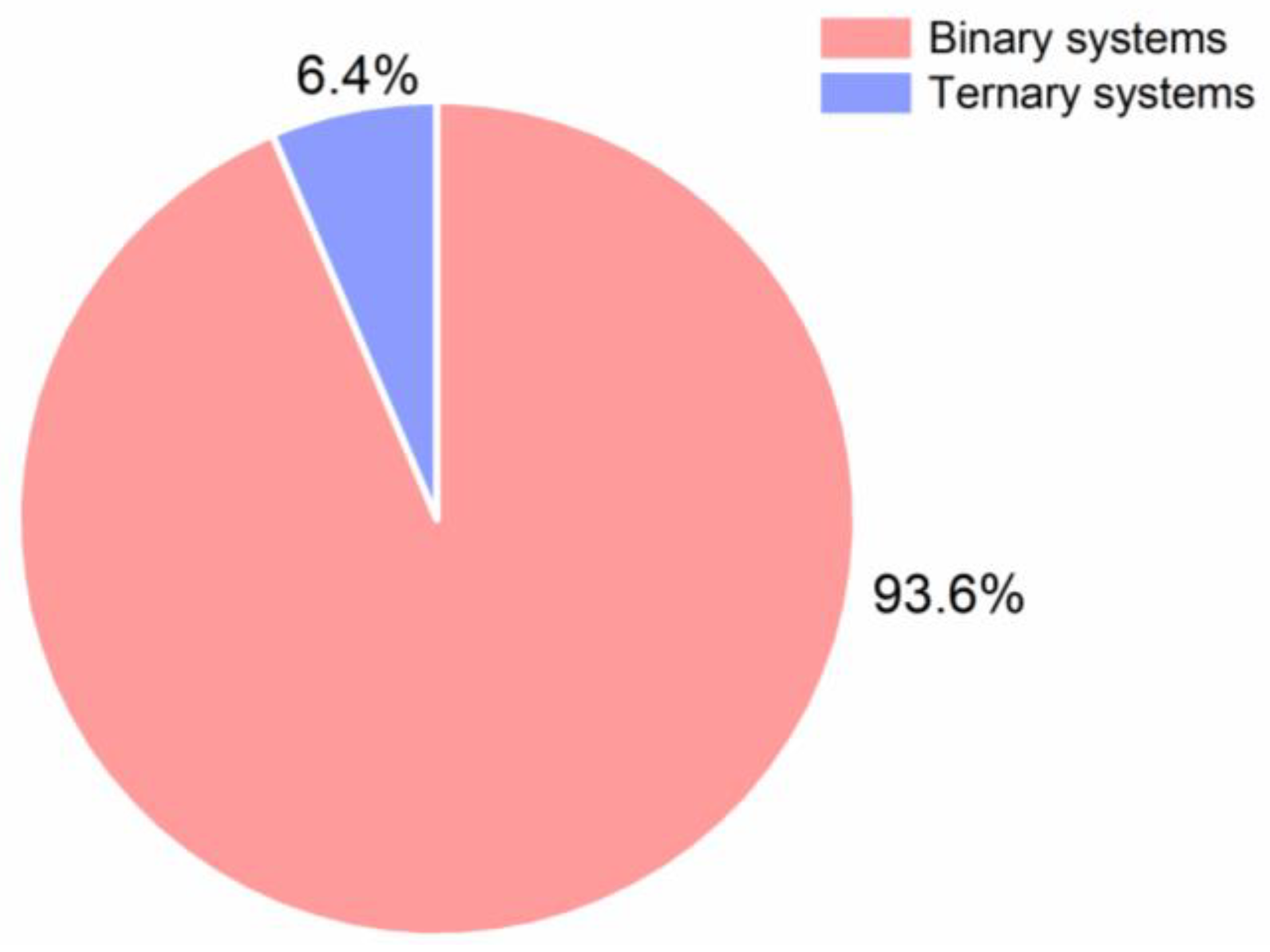
10. Preparation of Ternary CAMS
| Drug | Co-Former | Molar Ratio | In Vitro Study | Outcome | Reference |
|---|---|---|---|---|---|
| Sacubitril | Valsartan (also with lactose monohydrate and microcrystalline cellulose) | 1:1 (with excipient weight ratio: 1:1, 1:2, 1:3, 1:4) | Permeation test | CAMS impaired the permeation of the drugs, i.e., the flux decreased by 27.8% for sacubitril and 31.0% for valsartan. | [121] |
| Hydrochlorothiazide (HCT) | Arginine (L and D form) (and plus PVP) | 1:1 (with PVP in 1:1 weight ratio) | Permeation studies with PAMPA membranes; Permeation studies with MCDKII cellular barriers | Highest cumulative amount of permeated HCT for HCT:L-Arginine (over 2-fold compared to crystalline drug) using PAMPA membranes. Improved permeation of BCS class IV drug HCT even though no specific permeation-enhancing effect for the excipients could be found using MCDKII cellular barriers. | [144] |
| Budesonide | Theophylline | 1:1 | Aerosolization performance by NGI | Higher fine particle fraction compared to nanosuspensions and co-deposition of budesonide and theophylline in the aerodynamic assessment. | [145] |
| Simvastatin | Leucine Tryptophan Lysine | 1:1 | Aerosolization performance by NGI | Simvastatin-Leucine showed the best aerosol performance, followed by CAMS with tryptophan and lysine. | [111] |
| Chloramphenicol | Arginine Cysteine Glycine Leucine | 1:1 | Antimicrobial activity; Oxygen species detection | Chloramphenicol maintained its microbiological activity in CAMS with amino acids, i.e., the amino acids did not interfere with the microbiological activity of chloramphenicol | [146] |
| Ciprofoloxacin | Tartaric acid | 2:1, 1:1, 1:2, 1:3 | Antimicrobial activity | With tartaric acid as a co-former, the CAMS was more potent compared to the drug alone toward P.aeruginosa biofilms, which was described as a synergistic effect of the CAMS. | [147] |
| Ibrutinib | Oxalic acid (also with microcrystalline cellulose) | 1:1 (:1) | Cytotoxicity assay | Ibrutinib–oxalic acid and microcrystalline cellulose as a CAMS reduced the side effects of the drug on the kidney (nephrotoxicity) and showed and improved antitumor effect. | [36] |
| Glibenclamide | Arginine Serine Arginine–sodium lauryl sulfate Serine-sodium lauryl sulfate | 1:1; arginine-sodium lauryl sulfate (1:1:0.083 and 1:1:0.157) serine sodium lauryl sulfate (1:1:0.875 and 1:1:0.154) | Permeation study (PAMPA membranes) | Permeation was increased (AUC) by 7.2-, 5.7-, and 7.0- fold for CAMS with arginine, with arginine–sodium lauryl sulfate (low amounts) and arginine–sodium lauryl sulfate (high amounts), respectively, compared to the amorphous drug. | [143] |
| Azithromycin Tobramycin Ciprofloxacin | N-acetylcysteine | 1:2 1:1.5 1:1 | Aerosolization performance by NGI; Pseudomonas aeruginosa biofilm assay; | All CAMS showed a high fine particle fraction. The CAMS improved or at least maintained the antibiotic susceptibility and the inhibitory properties of N-acetylcysteine against P. aeruginosa biofilms. | [148] |
| Budesonide | Arginine | 1:1 | In vitro lung deposition test; Aerosolization performance by NGI | Aerosolization performance as well as lung deposition of budesonide improved with the co-former arginine. | [149] |
| Ursolic acid | Piperine | 2:1, 1.5:1 | In vitro permeability study across Caco-2 Cell Monolayers | Free piperine significantly increased the permeability of the drug. However, piperine in the CAMS exhibited a much lower level in permeability enhancement compared to its free form arising from the synchronized dissolution characteristic of the preparation. | [85] |
| Piroxicam | Citric Acid | 1:1 | In vitro skin permeation study | The CAMS demonstrated higher skin permeation than piroxicam alone or the physical mixture of the CAMS. | [150] |
| Kanamycin sulfate | Valine Methionine Phenylalanine Tryptophan | 1:1 | Aerosolization performance by NGI | All the CAMS improved the aerosolization performanc compared to the pure drug in the order methionine > tryptophan > phenylalanine > valine. | [88] |
| Glibenclamide | Arginine Serine Quercetine Arginine–sodium lauryl sulfate | 1:1 | Permeability studies conducted with MDCKII-MDR1 cells | The CAMS with arginine-sodium lauryl sulfate exhibited a 9-fold increase in permeating through the MDCKII-MDR1 cell layer as compared to the corresponding physical mixture. Permeability of the CAMS in the order of the co-former: serine < quercetine < arginine < arginine-sodium lauryl sulfate. | [116] |
| Talinolol | Naringin | 1:1 | In vitro single pass perfusion studies conducted on the ileum of Wistar rats | The permeability of talinolol was significantly increased in the presence of naringin due to the p-Gp inhibition effect by naringin. | [124] |
| Atenolol | Urea and PEG400 | various | Skin permeation study | The supersaturated CAMS formulation showed higher permeability for mice skin than that of a supersatured drug formulation, due to the degree of supersaturation. | [44] |
| Curcumin | Piperine | 1:1 | Permeability study with caco-2-cell | The absorptive transport of curcumin was significantly enhanced by 2.67-fold compared to the pure crystalline drug, suggesting the CAMS can promote the intestinal absorption of CUR. | [133] |
| Acyclovir | Citric acid | 1:10 | Skin permeation study | The steady-state permeation flux of the drug in the CAMS was 2.06 µg/cm2/h, much higher compared to the crystalline pure drug (0.02 µg/cm2/h). | [151] |
| Binary CAMS | The Additional Third Component | Type of the Additional Component | Preparation Method(s) | Reasons for the Addition | Outcomes (Compared to the Corresponding Binary CAMS) | Reference |
|---|---|---|---|---|---|---|
| Carbamazepine–Citric acid | Arginine | Small molecule | Ball milling | To design a stable co-amorphous system with an elevated Tg | Significant increase in the Tg value; Improvement of dissolution behavior and physical stability | [153] |
| Carvedilol–Aspartic acid | Eudragit® L 55 | Polymer | Coating and in situ amorphization | As a coating dispersion | - | [100] |
| Carvedilol–Aspartic acid | HPMC | Polymer | Spray drying | To optimize the dissolution behavior | Improvement of dissolution behavior by reducing the initial dissolution rate and maintaining supersaturation for a longer time | [104] |
| Ciprofloxacin–Tartaric acid | Silica-coated silver nanobeads and NaHCO3 | Other | Spray drying | As an external layer for co-amorphous powder coating | Disruptive effect on rheological properties | [147] |
| Ezetimibe–Lovastatin | Soluplus® | Polymer | Spray drying | To improve the poor dissolution characteristic | Improvement of the dissolution behaviors of both drugs | [78] |
| Ezetimibe–Lovastatin | PVP K30 | Polymer | Spray drying | To improve the poor dissolution characteristic | Significant improvement of the dissolution behavior of only one drug | [78] |
| Ezetimibe–Lovastatin | PVP VA64 | Polymer | Spray drying | To improve the poor dissolution characteristic | Significant improvement of the dissolution behavior of only one drug | [78] |
| Ezetimibe–Lovastatin | HPMC | Polymer | Spray drying | To improve the poor dissolution characteristic | Significant improvement of the dissolution behavior of only one drug | [78] |
| Ezetimibe–Simvastatin | Kollidon® VA64 | Polymer | Melt-quench | To verify feasible applications of the developed methods | - | [154] |
| Ezetimibe–Simvastatin | Kollidon® VA64 | Polymer | Melt-quench | To hinder the re-crystallization | Improvement of the physical stability at elevated temperature conditions (T = 373 K) | [79] |
| Flutamide–Bicalutamide | Poly(methyl methacrylate-co-ethyl acrylate) | Polymer | Melt-quench | To stabilize two drugs mixed | Inhomogeneity of the sample | [155] |
| Flutamide–Bicalutamide | PVP | Polymer | Melt-quench | To stabilize two drugs mixed | Sample homogeneity; Inhibition of the re-crystallization | [155] |
| Glibenclamide–Arginine | Sodium lauryl sulfate | Surfactant | Cryo-milling | To act as an absorption enhancer for permeability improvement | Improvement of dissolution and permeability of the drug | [116,143] |
| Glibenclamide–Serine | Sodium lauryl sulfate | Surfactant | Cryo-milling | To act as an absorption enhancer for permeability improvement | No significant improvement on the dissolution and permeability | [143] |
| Hydrochlorothiazide– Arginine | PVP | Polymer | Cryo-milling | Not mentioned | Improvement of drug dissolution behavior; Decrease on the drug permeation behavior | [143] |
| Ibrutinib– Oxalic acid | Microcrystalline cellulose | Polymer | Ball milling | As an effective crystal growth inhibitor | Improvement of solubility and dissolution rate; Improvement of physical stability | [36] |
| Ibuprofen–Arginine | Mannitol+ PVP K30 | Small molecule and polymer | Tableting compaction | As tablet compositions | The addition of PVP increased the initial drug release rate | [67] |
| Ibuprofen–Arginine | Xylitol+ PVP K30 | Small molecule and polymer | Tableting compaction | As tablet compositions | - | [67] |
| Indomethacin–Arginine | Co-povidone | Polymer | HME | To investigate the need for an addition of a polymer in the co-amorphous system preparation by HME | The co-amorphous formulations could be achieved with or without polymer; Enhanced dissolution behavior | [95] |
| Indomethacin–Arginine | Mannitol+ PVP K30 | Small molecule and polymer | Tableting compaction | As tablet compositions | The addition of PVP showed precipitation inhibitory effect | [67] |
| Indomethacin–Arginine | Xylitol+ PVP K30 | Small molecule and polymer | Tableting compaction | As tablet compositions | - | [67] |
| Indomethacin–Arginine | Kollicoat® Protect | Polymer | Coating | To investigate whether polymer coating of co-amorphous formulations is possible without inducing recrystallization | Coating of a co-amorphous formulation is possible without inducing recrystallization; Improvement of the drug release behavior | [97] |
| Indomethacin–Cimetidine | PEO | Polymer | HME | To investigate the effects of the addition of low amounts of polymer on the processability during HME | Inhibition behavior of amorphous–amorphous phase separation; Decrease in melt viscosity | [94] |
| Indomethacin–Citric acid | PVP | Polymer | Solvent evaporation | To prevent self-association between these two small molecules and thus to enhance their mutual miscibility | Enhancement of the mutual miscibility between two small molecules, but the ability is sensitive to PVP concentration | [25] |
| Naproxen–Arginine | Sodium dodecyl sulfate | Surfactant | Freeze-drying | To increase the solubility of drug in the starting solution for freeze-drying; To investigate the influence of the surfactant types | Formation of a heterogeneous system; Improvement of sample physical stability at certain concentration | [57] |
| Naproxen–Arginine | Pluronic F-127 | Surfactant | Freeze-drying | To increase the solubility of drug in the starting solution for freeze-drying; To investigate the influence of the surfactant types | Formation of a homogeneous system | [57] |
| Naproxen–Arginine | Polyoxyethylene (40) stearate | Surfactant | Freeze-drying | To increase the solubility of drug in the starting solution for freeze-drying; To investigate the influence of the surfactant types | Formation of a homogeneous system; Improvement of sample physical stability | [57] |
| Naproxen–Arginine | Tween 20 | Surfactant | Freeze-drying | To increase the solubility of drug in the starting solution for freeze-drying; To investigate the influence of the surfactant types | Formation of a heterogeneous system; Improvement of sample physical stability at certain concentration | [57] |
| Naproxen–Arginine | TPGS 1000 | Surfactant | Freeze-drying | To increase the solubility of drug in the starting solution for freeze-drying; To investigate the influence of the surfactant types | Formation of a heterogeneous system; Improvement of sample physical stability at certain concentration | [57] |
| Naproxen–Arginine | Proline | Small molecule | Ball milling | To achieve an additional improvement of the dissolution rate | Improvement of dissolution rate for Naproxen–Arginine amorphous salt | [156] |
| Naproxen– Indomethacin | Naproxen–sodium | Small molecule | Melt-quench | To optimize the physicochemical properties | Improvement of physical stability | [81] |
| Naproxen– Lysine | Sodium dodecyl sulfate | Surfactant | Freeze-drying | To increase the solubility of drug in the starting solution for freeze-drying; To investigate the influence of the surfactant types | Formation of a homogeneous system | [57] |
| Naproxen– Lysine | Pluronic F-127 | Surfactant | Freeze-drying | To increase the solubility of drug in the starting solution for freeze-drying; To investigate the influence of the surfactant types | Formation of a homogeneous system; Improvement of sample physical stability | [57] |
| Naproxen– Lysine | Polyoxyethylene (40) stearate | Surfactant | Freeze-drying | To increase the solubility of drug in the starting solution for freeze-drying; To investigate the influence of the surfactant types | Formation of a homogeneous system; Improvement of sample physical stability | [57] |
| Naproxen– Lysine | Tween 20 | Surfactant | Freeze-drying | To increase the solubility of drug in the starting solution for freeze-drying; To investigate the influence of the surfactant types | Formation of a homogeneous system; Improvement of sample physical stability | [57] |
| Naproxen– Lysine | TPGS 1000 | Surfactant | Freeze-drying | To increase the solubility of drug in the starting solution for freeze-drying; To investigate the influence of the surfactant types | Formation of a heterogeneous system; Improvement of sample physical stability | [57] |
| Naproxen–Meglumine | Kollidon VA64 | Polymer | Reactive melt extrusion | To combine the advantages of both salts and amorphous solid dispersions for enhancing the solubility and dissolution rates | The presence of a polymer did not interfere with the salt formation; Improvement of the dissolution properties and the physical stability compared to drug–polymer systems | [80] |
| Naproxen– Meglumine | Kollidon K30 | Polymer | Reactive melt extrusion | To combine the advantages of both salts and amorphous solid dispersions for enhancing the solubility and dissolution rates | The presence of a polymer did not interfere with the salt formation; Improvement of the dissolution properties and the physical stability compared to drug–polymer systems | [80] |
| Naproxen–Meglumine | Soluplus | Polymer | Reactive melt extrusion | To combine the advantages of both salts and amorphous solid dispersions for enhancing the solubility and dissolution rates | The presence of a polymer did not interfere with the salt formation; Improvement of the dissolution properties and the physical stability compared to drug–polymer systems | [80] |
| Naproxen–Tryptophan | Proline | Small molecule | Ball milling | To achieve an additional improvement of the dissolution rate | Successful formation of amorphous mixture (while a small remaining degree of crystallinity observed in Naproxen–Tryptophan binary system); Improvement of dissolution rate | [156] |
| Nateglinide–Metformin hydrochloride | Magnesium aluminometasilicate | Small molecule | Spray drying | To improve flowability of spray-dried powder | Improvement of flow properties and enhanced compressibility; Improvement of solubility and dissolution; The formation of spherical microstructured particles; Enhancement of physical stability | [157] |
| Olmesartan medoxomil–Hydrochlorothiazide | HPMC | Polymer | Solvent evaporation | To inhibit the deleterious interactions | Inhibition of co-crystallization but no significant improvement on the dissolution rate | [158] |
| Sacubitril–Valsartan | Lactose monohydrate | Small molecule | Spray drying | As an inert carrier | A slight decrease of solubility of both drugs; Delay of phase transformation; Improvement of in vivo bioavailability; Improvement of powder properties for compressibility | [121] |
| Sacubitril–Valsartan | Microcrystalline cellulose | Polymer | Spray drying | As an inert carrier | A slight decrease of solubility of both drugs; Delay of phase transformation; Decrease on in vivo bioavailability; Improvement of powder properties for compressibility | [121] |
11. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
Appendix C
Appendix D
References
- FDA. The Biopharmaceutical Classification System (BCS) Guidance. Available online: https://www.fda.gov/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cder/ucm128219.htm (accessed on 2 October 2016).
- Jermain, S.V.; Brough, C.; Williams, R.O. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery-An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Hancock Bruno, C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Grohganz, H.; Löbmann, K.; Priemel, P.; Tarp Jensen, K.; Graeser, K.; Strachan, C.; Rades, T. Amorphous drugs and dosage forms. J. Drug Deliv. Sci. Technol. 2013, 23, 403–408. [Google Scholar] [CrossRef]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef]
- Grohganz, H.; Priemel, P.A.; Löbmann, K.; Nielsen, L.H.; Laitinen, R.; Mullertz, A.; Van den Mooter, G.; Rades, T. Refining stability and dissolution rate of amorphous drug formulations. Expert Opin. Drug Deliv. 2014, 11, 977–989. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Riikonen, J.; Xu, W.; Lehto, V.P. Mesoporous systems for poorly soluble drugs-recent trends. Int. J. Pharm. 2018, 536, 178–186. [Google Scholar] [CrossRef]
- Kissi, E.O.; Ruggiero, M.T.; Hempel, N.-J.; Song, Z.; Grohganz, H.; Rades, T.; Löbmann, K. Characterising glass transition temperatures and glass dynamics in mesoporous silica-based amorphous drugs. Phys. Chem. Chem. Phys. 2019, 21, 19686–19694. [Google Scholar] [CrossRef]
- Hempel, N.-J.; Brede, K.; Olesen, N.E.; Genina, N.; Knopp, M.M.; Löbmann, K. A fast and reliable DSC-based method to determine the monomolecular loading capacity of drugs with good glass-forming ability in mesoporous silica. Int. J. Pharm. 2018, 544, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Bavnhøj, C.G.; Knopp, M.M.; Madsen, C.M.; Löbmann, K. The role interplay between mesoporous silica pore volume and surface area and their effect on drug loading capacity. Int. J. Pharm. X 2019, 1, 10008–10012. [Google Scholar] [CrossRef]
- Antonino, R.S.; Ruggiero, M.; Song, Z.; Nascimento, T.L.; Lima, E.M.; Bohr, A.; Knopp, M.M.; Löbmann, K. Impact of drug loading in mesoporous silica-amorphous formulations on the physical stability of drugs with high recrystallization tendency. Int. J. Pharm. X 2019, 1, 100026–100032. [Google Scholar] [CrossRef] [PubMed]
- Veloso, D.F.M.C.; Knopp, M.M.; Löbmann, K. Amorphous drug stabilization using mesoporous materials. Drug Deliv. Trends 2020, 1, 151–166. [Google Scholar]
- Löbmann, K.; Grohganz, H.; Laitinen, R.; Strachan, C.; Rades, T. Amino acids as co-amorphous stabilizers for poorly water soluble drugs—Part 1: Preparation, stability and dissolution enhancement. Eur. J. Pharm. Biopharm. 2013, 85, 873–881. [Google Scholar] [CrossRef]
- Löbmann, K.; Laitinen, R.; Strachan, C.; Rades, T.; Grohganz, H. Amino acids as co-amorphous stabilizers for poorly water-soluble drugs—Part 2: Molecular interactions. Eur. J. Pharm. Biopharm. 2013, 85, 882–888. [Google Scholar] [CrossRef]
- Löbmann, K.; Strachan, C.; Grohganz, H.; Rades, T.; Korhonen, O.; Laitinen, R. Co-amorphous simvastatin and glipizide combinations show improved physical stability without evidence of intermolecular interactions. Eur. J. Pharm. Biopharm. 2012, 81, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Dengale, S.J.; Ranjan, O.P.; Hussen, S.S.; Krishna, B.S.; Musmade, P.B.; Gautham Shenoy, G.; Bhat, K. Preparation and characterization of co-amorphous Ritonavir-Indomethacin systems by solvent evaporation technique: Improved dissolution behavior and physical stability without evidence of intermolecular interactions. Eur. J. Pharm. Sci. 2014, 62, 57–64. [Google Scholar] [CrossRef]
- Fukuoka, E.; Makita, M.; Yamamura, S. Glassy state of pharmaceuticals. III: Thermal properties and stability of glassy pharmaceuticals and their binary glass systems. Chem. Pharma. Bull. 1989, 37, 1047–1050. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, S.; Gotoh, H.; Sakamoto, Y.; Momose, Y. Physicochemical properties of amorphous precipitates of cimetidine–indomethacin binary system. Eur. J. Pharm. Biopharm. 2000, 49, 259–265. [Google Scholar] [CrossRef]
- Yamamura, S.; Momose, Y.; Takahashi, K.; Nagatani, S. Solid-state interaction between cimetidine and naproxen. Drug Stab. 1996, 1, 173–178. [Google Scholar]
- Lu, Q.; Zografi, G. Phase behavior of binary and ternary amorphous mixtures containing indomethacin, citric acid, and PVP. Pharm. Res. 1998, 15, 1202–1206. [Google Scholar] [CrossRef]
- Chieng, N.; Aaltonen, J.; Saville, D.; Rades, T. Physical characterization and stability of amorphous indomethacin and ranitidine hydrochloride binary systems prepared by mechanical activation. Eur. J. Pharm. Biopharm. 2009, 71, 47–54. [Google Scholar] [CrossRef]
- Korhonen, O.; Pajula, K.; Laitinen, R. Rational excipient selection for co-amorphous formulations. Expert Opin. Drug Deliv. 2017, 14, 551–569. [Google Scholar] [CrossRef]
- Laitinen, R.; Löbmann, K.; Grohganz, H.; Strachan, C.; Rades, T. Amino acids as co-amorphous excipients for simvastatin and glibenclamide: Physical properties and stability. Mol. Pharm. 2014, 11, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Kasten, G.; Grohganz, H.; Rades, T.; Löbmann, K. Development of a screening method for co-amorphous formulations of drugs and amino acids. Eur. J. Pharm. Sci. 2016, 95, 28–35. [Google Scholar] [CrossRef]
- Jensen, K.T.; Larsen, F.H.; Löbmann, K.; Rades, T.; Grohganz, H. Influence of variation in molar ratio on co-amorphous drug-amino acid systems. Eur. J. Pharm. Biopharm. 2016, 107, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Ali, A.A.; Maghrabi, I.A. Clozapine-carboxylic acid plasticized co-amorphous dispersions: Preparation, characterization and solution stability evaluation. Acta. Pharm. 2015, 65, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Gniado, K.; Löbmann, K.; Rades, T.; Erxleben, A. The influence of co-formers on the dissolution rates of co-amorphous sulfamerazine/excipient systems. Int. J. Pharm. 2016, 504, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, E.M.; Ali, A.M.; Salem, H.F.; Abdelrahman, A.A. In vitro/in vivo evaluation of an optimized fast dissolving oral film containing olanzapine co-amorphous dispersion with selected carboxylic acids. Drug Deliv. 2016, 23, 3088–3100. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Kojima, T.; Karashima, M.; Ikeda, Y. Physicochemical evaluation and developability assessment of co-amorphouses of low soluble drugs and comparison to the co-crystals. Chem. Pharm. Bull. 2016, 1, 1739–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.H.; Lim, C.; Kiyonga, A.N.; Chung, I.H.; Lee, I.K.; Mo, K.; Park, M.; Youn, W.; Choi, W.R.; Suh, Y.G.; et al. Co-amorphous screening for the solubility enhancement of poorly water-soluble mirabegron and investigation of their intermolecular interactions and dissolution behaviors. Pharmaceutics 2018, 10, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Suo, Z.; Peng, X.; Gan, N.; Zhao, L.; Tang, P.; Wei, X.; Li, H. Microcrystalline cellulose as an effective crystal growth inhibitor for the ternary Ibrutinib formulation. Carbohydr. Polym. 2020, 229, 115476–115484. [Google Scholar] [CrossRef]
- Fung, M.; Be Rzins, K.R.; Suryanarayanan, R. Physical stability and dissolution behavior of ketoconazole-organic acid coamorphous systems. Mol. Pharm. 2018, 15, 1862–1869. [Google Scholar] [CrossRef]
- Fung, M.H.; DeVault, M.; Kuwata, K.T.; Suryanarayanan, R. Drug-excipient interactions: Effect on molecular mobility and physical stability of ketoconazole-organic acid coamorphous systems. Mol. Pharm. 2018, 15, 1052–1061. [Google Scholar] [CrossRef]
- Wu, W.; Ueda, H.; Löbmann, K.; Rades, T.; Grohganz, H. Organic acids as co-formers for co-amorphous systems-Influence of variation in molar ratio on the physicochemical properties of the co-amorphous systems. Eur. J. Pharm. Biopharm. 2018, 131, 25–32. [Google Scholar] [CrossRef]
- Macfhionnghaile, P.; Hu, Y.; Gniado, K.; Curran, S.; McArdle, P.; Erxleben, A. Effects of ball-milling and cryomilling on sulfamerazine polymorphs: A quantitative study. J. Pharm. Sci. 2014, 103, 1766–1778. [Google Scholar] [CrossRef] [Green Version]
- Chambers, L.I.; Grohganz, H.; Palmelund, H.; Löbmann, K.; Rades, T.; Musa, O.M.; Steed, J.W. Predictive identification of co-formers in co-amorphous systems. Eur. J. Pharm. Sci. 2021, 157, 105636–105643. [Google Scholar] [CrossRef]
- Hu, Y.; Gniado, K.; Erxleben, A.; McArdle, P. Mechanochemical reaction of sulfathiazole with carboxylic acids: Formation of a cocrystal, a salt, and coamorphous solids. Cryst. Growth Des. 2014, 14, 803–813. [Google Scholar] [CrossRef]
- Wu, W.; Wang, Y.; Löbmann, K.; Grohganz, H.; Rades, T. Transformations between co-amorphous and co-crystal systems and their influence on the formation and physical stability of co-amorphous systems. Mol. Pharm. 2019, 16, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, Y.; Ueda, H.; Miyano, T.; Kamiya, N.; Goto, M. New insight into transdermal drug delivery with supersaturated formulation based on co-amorphous system. Int. J. Pharm. 2019, 569, 118582–118588. [Google Scholar] [CrossRef]
- Ahuja, N.; Katare, O.P.; Singh, B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur. J. Pharm. Biopharm. 2007, 65, 26–38. [Google Scholar] [CrossRef]
- Ali, A.M.; Al-Remawi, M.M. Freeze dried quetiapine-nicotinamide binary solid dispersions: A new strategy for improving physicochemical properties and ex vivo diffusion. J. Pharm. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Tawfeek, H.M.; Chavan, T.; Kunda, N.K. Effect of spray drying on amorphization of indomethacin nicotinamide cocrystals; optimization, characterization, and stability study. AAPS Pharm. Sci. Tech. 2020, 21, 181–193. [Google Scholar] [CrossRef]
- Bi, Y.; Xiao, D.; Ren, S.; Bi, S.; Wang, J.; Li, F. The binary system of ibuprofen-nicotinamide under nanoscale confinement: From cocrystal to coamorphous state. J. Pharm. Sci. 2017, 106, 3150–3155. [Google Scholar] [CrossRef]
- Shayanfar, A.; Jouyban, A. Drug–drug coamorphous systems: Characterization and physicochemical properties of coamorphous atorvastatin with carvedilol and glibenclamide. J. Pharm. Innov. 2013, 8, 218–228. [Google Scholar] [CrossRef]
- Pajula, K.; Taskinen, M.; Lehto, V.-P.; Ketolainen, J.; Korhonen, O. Predicting the formation and stability of amorphous small molecule binary mixtures from computationally determined Flory− Huggins interaction parameter and phase diagram. Mol. Pharm. 2010, 7, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Muranushi, N.; Sakuma, S.; Ida, Y.; Endoh, T.; Kadota, K.; Tozuka, Y. A strategy for co-former selection to design stable co-amorphous formations based on physicochemical properties of non-steroidal inflammatory drugs. Pharm. Res. 2016, 33, 1018–1029. [Google Scholar] [CrossRef]
- Mizoguchi, R.; Waraya, H.; Hirakura, Y. Application of co-amorphous technology for improving the physicochemical properties of amorphous formulations. Mol. Pharm. 2019, 16, 2142–2152. [Google Scholar] [CrossRef] [PubMed]
- Pajula, K.; Wittoek, L.; Lehto, V.P.; Ketolainen, J.; Korhonen, O. Phase separation in coamorphous systems: In silico prediction and the experimental challenge of detection. Mol. Pharm. 2014, 11, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Meng-Lund, H.; Kasten, G.; Jensen, K.T.; Poso, A.; Pantsar, T.; Rades, T.; Rantanen, J.; Grohganz, H. The use of molecular descriptors in the development of co-amorphous formulations. Eur. J. Pharm. Sci. 2018, 119, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Pajula, K.; Hyyrylainen, J.; Koistinen, A.; Leskinen, J.T.T.; Korhonen, O. Detection of amorphous-amorphous phase separation in small molecular co-amorphous mixtures with SEM-EDS. Eur. J. Pharm. Biopharm. 2020, 150, 43–49. [Google Scholar] [CrossRef]
- Veith, H.; Wiechert, F.; Luebbert, C.; Sadowski, G. Combining crystalline and polymeric excipients in API solid dispersions-Opportunity or risk? Eur. J. Pharm. Biopharm. 2021, 158, 323–335. [Google Scholar] [CrossRef]
- Wostry, M.; Plappert, H.; Grohganz, H. Preparation of co-amorphous systems by freeze-drying. Pharmaceutics 2020, 12, 941. [Google Scholar] [CrossRef]
- Greenhalgh, D.J.; Williams, A.C.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. [Google Scholar] [CrossRef]
- Marsac, P.J.; Shamblin, S.L.; Taylor, L.S. Theoretical and practical approaches for prediction of drug-polymer miscibility and solubility. Pharm. Res. 2006, 23, 2417–2426. [Google Scholar] [CrossRef]
- Thakral, S.; Thakral, N.K. Prediction of drug–polymer miscibility through the use of solubility parameter based Flory–Huggins interaction parameter and the experimental validation: PEG as model polymer. J. Pharm. Sci. 2013, 102, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Kitak, T.; Dumicic, A.; Planinsek, O.; Sibanc, R.; Srcic, S. Determination of solubility parameters of ibuprofen and ibuprofen lysinate. Molecules 2015, 20, 21549–21568. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Alzghoul, A.; Kaialy, W. Data mining of solubility parameters for computational prediction of drug-excipient miscibility. Drug Dev. Ind. Pharm. 2014, 40, 904–909. [Google Scholar] [CrossRef]
- Calahan, J.L.; Zanon, R.L.; Alvarez-Nunez, F.; Munson, E.J. Isothermal microcalorimetry to investigate the phase separation for amorphous solid dispersions of AMG 517 with HPMC-AS. Mol. Pharm. 2013, 10, 1949–1957. [Google Scholar] [CrossRef]
- Padilla, A.M.; Chou, S.G.; Luthra, S.; Pikal, M.J. The study of amorphous phase separation in a model polymer phase-separating system using Raman microscopy and a low-temperature stage: Effect of cooling rate and nucleation temperature. J. Pharm. Sci. 2011, 100, 1362–1376. [Google Scholar] [CrossRef]
- Padilla, A.M.; Ivanisevic, I.; Yang, Y.; Engers, D.; Bogner, R.H.; Pikal, M.J. The study of phase separation in amorphous freeze-dried systems. Part I: Raman mapping and computational analysis of XRPD data in model polymer systems. J. Pharm. Sci. 2011, 100, 206–222. [Google Scholar] [CrossRef]
- Van Eerdenbrugh, B.; Lo, M.; Kjoller, K.; Marcott, C.; Taylor, L.S. Nanoscale mid-infrared imaging of phase separation in a drug-polymer blend. J. Pharm. Sci. 2012, 101, 2066–2073. [Google Scholar] [CrossRef] [PubMed]
- Ojarinta, R.; Saarinen, J.; Strachan, C.J.; Korhonen, O.; Laitinen, R. Preparation and characterization of multi-component tablets containing co-amorphous salts: Combining multimodal non-linear optical imaging with established analytical methods. Eur. J. Pharm. Biopharm. 2018, 132, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpelainen, T.; Pajula, K.; Ervasti, T.; Uurasjarvi, E.; Koistinen, A.; Korhonen, O. Raman imaging of amorphous-amorphous phase separation in small molecule co-amorphous systems. Eur. J. Pharm. Biopharm. 2020, 155, 49–54. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Alzghoul, A.; Kaialy, W.; Mahlin, D.; Bergstrom, C.A. Computational predictions of glass-forming ability and crystallization tendency of drug molecules. Mol. Pharm. 2014, 11, 3123–3132. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.H.; Wermuth, C.G. Pharmaceutical Salts: Properties, Selection and Use; John Wiley & Sons: Hoboken, NJ, USA, 2002. [Google Scholar]
- Han, J.; Wei, Y.; Lu, Y.; Wang, R.; Zhang, J.; Gao, Y.; Qian, S. Co-amorphous systems for the delivery of poorly water-soluble drugs: Recent advances and an update. Expert Opin. Drug Deliv. 2020, 17, 1411–1435. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rades, T.; Grohganz, H. Determination of the optimal molar ratio in amino acid-based coamorphous systems. Mol. Pharm. 2020, 17, 1335–1342. [Google Scholar] [CrossRef]
- Kissi, E.O.; Khorami, K.; Rades, T. Determination of stable co-amorphous drug-drug ratios from the eutectic behavior of crystalline physical mixtures. Pharmaceutics 2019, 11, 628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, A.; Grohganz, H.; Löbmann, K.; Rades, T.; Leopold, C.S. Influence of the cooling rate and the blend ratio on the physical stability of co-amorphous naproxen/indomethacin. Eur. J. Pharm. Biopharm. 2016, 109, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Corner, P.A.; Harburn, J.J.; Steed, J.W.; McCabe, J.F.; Berry, D.J. Stabilisation of an amorphous form of ROY through a predicted co-former interaction. Chem. Commun. 2016, 52, 6537–6540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kissi, E.; Kasten, G.; Löbmann, K.; Rades, T.; Grohganz, H. The role of glass transition temperatures in co-amorphous drug-amino acid formulations. Mol. Pharm. 2018, 15, 4247–4256. [Google Scholar] [CrossRef]
- Martinez, L.M.; Videa, M.; Sosa, N.G.; Ramirez, J.H.; Castro, S. Long-term stability of new co-amorphous drug binary systems: Study of glass transitions as a function of composition and shelf time. Molecules 2016, 21, 1712. [Google Scholar] [CrossRef] [Green Version]
- Riekes, M.K.; Engelen, A.; Appeltans, B.; Rombaut, P.; Stulzer, H.K.; Van den Mooter, G. New perspectives for fixed dose combinations of poorly water-soluble compounds: A case study with ezetimibe and lovastatin. Pharm. Res. 2016, 33, 1259–1275. [Google Scholar] [CrossRef]
- Knapik-Kowalczuk, J.; Chmiel, K.; Jurkiewicz, K.; Correia, N.T.; Sawicki, W.; Paluch, M. Physical stability and viscoelastic properties of co-amorphous ezetimibe/simvastatin system. Pharmaceuticals 2019, 12, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhou, L.; Zhang, F. Reactive melt extrusion to improve the dissolution performance and physical stability of naproxen amorphous solid dispersions. Mol. Pharm. 2017, 14, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.; Grohganz, H.; Löbmann, K.; Rades, T.; Leopold, C.S. Improvement of the physicochemical properties of co-amorphous naproxen-indomethacin by naproxen-sodium. Int. J. Pharm. 2017, 526, 88–94. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, H.; Babu, S.; Garad, S. Co-amorphous formation of high-dose zwitterionic compounds with amino acids to improve solubility and enable parenteral delivery. Mol. Pharm. 2018, 15, 97–107. [Google Scholar] [CrossRef]
- Martinez, L.M.; Videa, M.; Lopez-Silva, G.A.; de Los Reyes, C.A.; Cruz-Angeles, J.; Gonzalez, N. Stabilization of amorphous paracetamol based systems using traditional and novel strategies. Int. J. Pharm. 2014, 477, 294–305. [Google Scholar] [CrossRef]
- Martinez-Jimenez, C.; Cruz-Angeles, J.; Videa, M.; Martinez, L.M. Co-amorphous simvastatin-nifedipine with enhanced solubility for possible use in combination therapy of hypertension and hypercholesterolemia. Molecules 2018, 23, 2161. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Kan, Z.; Shan, F.; Zang, J.; Zhou, J. Triple strategies to improve oral bioavailability by fabricating coamorphous forms of ursolic acid with piperine: Enhancing water-solubility, permeability, and inhibiting cytochrome p450 isozymes. Mol. Pharm. 2020, 17, 4443–4462. [Google Scholar] [CrossRef]
- Lodagekar, A.; Chavan, R.B.; Mannava, M.K.C.; Yadav, B.; Chella, N.; Nangia, A.K.; Shastri, N.R. Co amorphous valsartan nifedipine system: Preparation, characterization, in vitro and in vivo evaluation. Eur. J. Pharm. Sci. 2019, 139, 105048–105056. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Jin Seo, H.; Hong, S.H.; Ha, E.S.; Lee, S.; Kim, J.S.; Baek, I.H.; Kim, M.S.; Hwang, S.J. Characterization and therapeutic efficacy evaluation of glimepiride and L-arginine co-amorphous formulation prepared by supercritical antisolvent process: Influence of molar ratio and preparation methods. Int. J. Pharm. 2020, 581, 119232–119244. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, B.R.; Berzins, K.; Fraser-Miller, S.J.; Gordon, K.C.; Das, S.C. Co-amorphization of kanamycin with amino acids improves aerosolization. Pharmaceutics 2020, 12, 715. [Google Scholar] [CrossRef]
- Ueda, H.; Peter Botker, J.; Edinger, M.; Löbmann, K.; Grohganz, H.; Mullertz, A.; Rades, T.; Ostergaard, J. Formulation of co-amorphous systems from naproxen and naproxen sodium and in situ monitoring of physicochemical state changes during dissolution testing by Raman spectroscopy. Int. J. Pharm. 2020, 587, 119662–119671. [Google Scholar] [CrossRef]
- Otsuka, Y.; Kuwashima, W.; Tanaka, Y.; Yamaki, Y.; Shimada, Y.; Goto, S. Effects of heat treatment on indomethacin-cimetidine mixture; investigation of drug-drug interaction using singular value decomposition in ftir spectroscopy. J. Pharm. Sci. 2021, 110, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Rades, T.; Gordon, K.C.; Graeser, K. Molecular structure, properties, and states of matter. In Remington: Essentials of Pharmaceutics; Pharmaceutical Press: London, UK, 2013; pp. 177–206. [Google Scholar]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating cocrystals: A review of pharmaceutical cocrystal preparation routes and applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Wickstrom, H.; Palo, M.; Rijckaert, K.; Kolakovic, R.; Nyman, J.O.; Maattanen, A.; Ihalainen, P.; Peltonen, J.; Genina, N.; de Beer, T.; et al. Improvement of dissolution rate of indomethacin by inkjet printing. Eur. J. Pharm. Sci. 2015, 75, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Arnfast, L.; Kamruzzaman, M.; Löbmann, K.; Aho, J.; Baldursdottir, S.; Rades, T.; Rantanen, J. Melt extrusion of high-dose co-amorphous drug-drug combinations: Theme: Formulation and manufacturing of solid dosage forms. Pharm. Res. 2017, 34, 2689–2697. [Google Scholar] [CrossRef]
- Lenz, E.; Löbmann, K.; Rades, T.; Knop, K.; Kleinebudde, P. Hot melt extrusion and spray drying of co-amorphous indomethacin-arginine with polymers. J. Pharm. Sci. 2017, 106, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bounartzi, M.; Panagopoulou, A.; Kantiranis, N.; Malamataris, S.; Nikolakakis, I. Effect of plasticiser type on the hot melt extrusion of venlafaxine hydrochloride. J. Pharm. Pharmacol. 2014, 66, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. Solid state properties and drug release behavior of co-amorphous indomethacin-arginine tablets coated with Kollicoat® Protect. Eur. J. Pharm. Biopharm. 2017, 119, 150–160. [Google Scholar] [CrossRef]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. Undesired co-amorphisation of indomethacin and arginine during combined storage at high humidity conditions. Int. J. Pharm. 2018, 544, 172–180. [Google Scholar] [CrossRef]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. In situ co-amorphisation of arginine with indomethacin or furosemide during immersion in an acidic medium—A proof of concept study. Eur. J. Pharm. Biopharm. 2018, 133, 151–160. [Google Scholar] [CrossRef]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. In situ co-amorphisation in coated tablets-the combination of carvedilol with aspartic acid during immersion in an acidic medium. Int. J. Pharm. 2019, 558, 357–366. [Google Scholar] [CrossRef]
- Lim, A.W.; Löbmann, K.; Grohganz, H.; Rades, T.; Chieng, N. Investigation of physical properties and stability of indomethacin-cimetidine and naproxen-cimetidine co-amorphous systems prepared by quench cooling, coprecipitation and ball milling. J. Pharm. Pharmacol. 2016, 68, 36–45. [Google Scholar] [CrossRef]
- Russo, M.G.; Sancho, M.I.; Silva, L.M.; Baldoni, H.A.; Venancio, T.; Ellena, J.; Narda, G.E. Looking for the interactions between omeprazole and amoxicillin in a disordered phase. An experimental and theoretical study. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2016, 156, 70–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, J.; Löbmann, K.; Grohganz, H.; Rades, T. Influence of preparation technique on co-amorphization of carvedilol with acidic amino acids. Int. J. Pharm. 2018, 552, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Grohganz, H.; Rades, T. Influence of polymer addition on the amorphization, dissolution and physical stability of co-amorphous systems. Int. J. Pharm. 2020, 588, 119768–119776. [Google Scholar] [CrossRef]
- Mishra, J.; Rades, T.; Löbmann, K.; Grohganz, H. Influence of solvent composition on the performance of spray-dried co-amorphous formulations. Pharmaceutics 2018, 10, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, K.T.; Blaabjerg, L.I.; Lenz, E.; Bohr, A.; Grohganz, H.; Kleinebudde, P.; Rades, T.; Löbmann, K. Preparation and characterization of spray-dried co-amorphous drug-amino acid salts. J. Pharm. Pharmacol. 2016, 68, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Kasten, G.; Duarte, I.; Paisana, M.; Löbmann, K.; Rades, T.; Grohganz, H. Process optimization and upscaling of spray-dried drug-amino acid co-amorphous formulations. Pharmaceutics 2019, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasten, G.; Nouri, K.; Grohganz, H.; Rades, T.; Löbmann, K. Performance comparison between crystalline and co-amorphous salts of indomethacin-lysine. Int. J. Pharm. 2017, 533, 138–144. [Google Scholar] [CrossRef]
- Wu, W.; Löbmann, K.; Schnitzkewitz, J.; Knuhtsen, A.; Pedersen, D.S.; Grohganz, H.; Rades, T. Aspartame as a co-former in co-amorphous systems. Int. J. Pharm. 2018, 549, 380–387. [Google Scholar] [CrossRef]
- Wu, W.; Löbmann, K.; Schnitzkewitz, J.; Knuhtsen, A.; Pedersen, D.S.; Rades, T.; Grohganz, H. Dipeptides as co-formers in co-amorphous systems. Eur. J. Pharm. Biopharm. 2019, 134, 68–76. [Google Scholar] [CrossRef]
- Lu, W.; Rades, T.; Rantanen, J.; Chan, H.K.; Yang, M. Amino acids as stabilizers for spray-dried simvastatin powder for inhalation. Int. J. Pharm. 2019, 572, 118724–118735. [Google Scholar] [CrossRef]
- Shi, Q.; Moinuddin, S.M.; Cai, T. Advances in coamorphous drug delivery systems. Acta Pharm. Sin. B 2019, 9, 19–35. [Google Scholar] [CrossRef]
- Graeser, K.A.; Strachan, C.J.; Patterson, J.E.; Gordon, K.C.; Rades, T. Physicochemical properties and stability of two differently prepared amorphous forms of simvastatin. Cryst. Growth Des. 2008, 8, 128–135. [Google Scholar] [CrossRef]
- Ke, P.; Hasegawa, S.; Al-Obaidi, H.; Buckton, G. Investigation of preparation methods on surface/bulk structural relaxation and glass fragility of amorphous solid dispersions. Int. J. Pharm. 2012, 422, 170–178. [Google Scholar] [CrossRef]
- Heikkinen, A.; DeClerck, L.; Löbmann, K.; Grohganz, H.; Rades, T.; Laitinen, R. Dissolution properties of co-amorphous drug-amino acid formulations in buffer and biorelevant media. Pharmazie 2015, 70, 452–457. [Google Scholar]
- Sormunen, H.; Ruponen, M.; Laitinen, R. The effect of co-amorphization of glibenclamide on its dissolution properties and permeability through an MDCKII-MDR1 cell layer. Int. J. Pharm. 2019, 570, 118653–118663. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Löbmann, K.; Rades, T.; Grohganz, H. On the role of salt formation and structural similarity of co-formers in co-amorphous drug delivery systems. Int. J. Pharm. 2018, 535, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.M.; Navale, T.S.; Jones, S.D.; Bates, F.S.; Reineke, T.M. Deconstructing hpmcas: Excipient design to tailor polymer–drug interactions for oral drug delivery. ACS Biomater. Sci. Eng. 2015, 1, 978–990. [Google Scholar] [CrossRef]
- Wu, W.; Grohganz, H.; Rades, T.; Löbmann, K. Comparison of co-former performance in co-amorphous formulations: Single amino acids, amino acid physical mixtures, amino acid salts and dipeptides as co-formers. Eur. J. Pharm. Sci. 2020, 156, 105582–105589. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, Q.; Wang, J.R.; Lin, K.L.; Mei, X. Amino acids as co-amorphous excipients for tackling the poor aqueous solubility of valsartan. Pharm. Dev. Technol. 2017, 22, 69–76. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Y.; Du, X.; Guan, R.; He, Z.; Liu, H. Combining co-amorphous-based spray drying with inert carriers to achieve improved bioavailability and excellent downstream manufacturability. Pharmaceutics 2020, 12, 1063. [Google Scholar] [CrossRef]
- Shi, X.; Song, S.; Ding, Z.; Fan, B.; Huang, W.; Xu, T. Improving the solubility, dissolution, and bioavailability of ibrutinib by preparing it in a coamorphous state with saccharin. J. Pharm. Sci. 2019, 108, 3020–3028. [Google Scholar] [CrossRef] [PubMed]
- Hoppu, P.; Jouppila, K.; Rantanen, J.; Schantz, S.; Juppo, A.M. Characterisation of blends of paracetamol and citric acid. J. Pharm. Pharmacol. 2007, 59, 373–381. [Google Scholar] [CrossRef]
- Teja, A.; Musmade, P.B.; Khade, A.B.; Dengale, S.J. Simultaneous improvement of solubility and permeability by fabricating binary glassy materials of Talinolol with Naringin: Solid state characterization, in-vivo in-situ evaluation. Eur. J. Pharm. Sci. 2015, 78, 234–244. [Google Scholar] [CrossRef]
- Nair, A.; Varma, R.; Gourishetti, K.; Bhat, K.; Dengale, S. Influence of preparation methods on physicochemical and pharmacokinetic properties of co-amorphous formulations: The case of co-amorphous atorvastatin: Naringin. J. Pharm. Innov. 2019, 15, 365–379. [Google Scholar] [CrossRef]
- Qian, S.; Li, Z.; Heng, W.; Liang, S.; Ma, D.; Gao, Y.; Zhang, J.; Wei, Y. Charge-assisted intermolecular hydrogen bond formed in coamorphous system is important to relieve the pH-dependent solubility behavior of lurasidone hydrochloride. RSC Adv. 2016, 6, 106396–106412. [Google Scholar] [CrossRef]
- Hoppu, P.; Hietala, S.; Schantz, S.; Juppo, A.M. Rheology and molecular mobility of amorphous blends of citric acid and paracetamol. Eur. J. Pharm. Biopharm. 2009, 71, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Theil, F.; Anantharaman, S.; Kyeremateng, S.O.; van Lishaut, H.; Dreis-Kuhne, S.H.; Rosenberg, J.; Magerlein, M.; Woehrle, G.H. Frozen in time: Kinetically stabilized amorphous solid dispersions of nifedipine stable after a quarter century of storage. Mol. Pharm. 2017, 14, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Aaltonen, J.; Tian, F.; Saville, D.J.; Rades, T. Influence of particle size and preparation methods on the physical and chemical stability of amorphous simvastatin. Eur. J. Pharm. Biopharm. 2009, 71, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, R.; Löbmann, K.; Grohganz, H.; Priemel, P.; Strachan, C.J.; Rades, T. Supersaturating drug delivery systems: The potential of co-amorphous drug formulations. Int. J. Pharm. 2017, 532, 1–12. [Google Scholar] [CrossRef]
- Gao, Y.; Liao, J.; Qi, X.; Zhang, J. Coamorphous repaglinide-saccharin with enhanced dissolution. Int. J. Pharm. 2013, 450, 290–295. [Google Scholar] [CrossRef]
- Dengale, S.J.; Hussen, S.S.; Krishna, B.S.; Musmade, P.B.; Gautham Shenoy, G.; Bhat, K. Fabrication, solid state characterization and bioavailability assessment of stable binary amorphous phases of Ritonavir with Quercetin. Eur. J. Pharm. Biopharm. 2015, 89, 329–338. [Google Scholar] [CrossRef]
- Wang, R.; Han, J.; Jiang, A.; Huang, R.; Fu, T.; Wang, L.; Zheng, Q.; Li, W.; Li, J. Involvement of metabolism-permeability in enhancing the oral bioavailability of curcumin in excipient-free solid dispersions co-formed with piperine. Int. J. Pharm. 2019, 561, 9–18. [Google Scholar] [CrossRef]
- Haneef, J.; Chadha, R. Drug-drug multicomponent solid forms: Cocrystal, coamorphous and eutectic of three poorly soluble antihypertensive drugs using mechanochemical approach. AAPS Pharm. Sci. Tech. 2017, 18, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Mannava, M.K.C.; Suresh, K.; Kumar Bommaka, M.; Bhavani Konga, D.; Nangia, A. Curcumin-artemisinin coamorphous solid: Xenograft model preclinical study. Pharmaceutics 2018, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Sai Krishna Anand, V.; Sakhare, S.D.; Navya Sree, K.S.; Nair, A.R.; Raghava Varma, K.; Gourishetti, K.; Dengale, S.J. The relevance of co-amorphous formulations to develop supersaturated dosage forms: In-vitro, and ex-vivo investigation of Ritonavir-Lopinavir co-amorphous materials. Eur. J. Pharm. Sci. 2018, 123, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Mannava, M.K.C.; Nangia, A. A novel curcumin–artemisinin coamorphous solid: Physical properties and pharmacokinetic profile. RSC Adv. 2014, 4, 58357–58361. [Google Scholar] [CrossRef]
- Moinuddin, S.M.; Ruan, S.; Huang, Y.; Gao, Q.; Shi, Q.; Cai, B.; Cai, T. Facile formation of co-amorphous atenolol and hydrochlorothiazide mixtures via cryogenic-milling: Enhanced physical stability, dissolution and pharmacokinetic profile. Int. J. Pharm. 2017, 532, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chang, R.; Zhao, Y.; Zhang, J.; Zhang, T.; Fu, Q.; Chang, C.; Zeng, A. Coamorphous loratadine-citric acid system with enhanced physical stability and bioavailability. AAPS Pharm. Sci.Tech. 2017, 18, 2541–2550. [Google Scholar] [CrossRef]
- Kasten, G.; Lobo, L.; Dengale, S.; Grohganz, H.; Rades, T.; Löbmann, K. In vitro and in vivo comparison between crystalline and co-amorphous salts of naproxen-arginine. Eur. J. Pharm. Biopharm. 2018, 132, 192–199. [Google Scholar] [CrossRef]
- Wei, Y.; Zhou, S.; Hao, T.; Zhang, J.; Gao, Y.; Qian, S. Further enhanced dissolution and oral bioavailability of docetaxel by coamorphization with a natural P-gp inhibitor myricetin. Eur. J. Pharm. Sci. 2019, 129, 21–30. [Google Scholar] [CrossRef]
- Bohr, A.; Nascimento, T.L.; Harmankaya, N.; Weisser, J.J.; Wang, Y.; Grohganz, H.; Rades, T.; Löbmann, K. Efflux inhibitor bicalutamide increases oral bioavailability of the poorly soluble efflux substrate docetaxel in co-amorphous anti-cancer combination therapy. Molecules 2019, 24, 266. [Google Scholar] [CrossRef] [Green Version]
- Ruponen, M.; Visti, M.; Ojarinta, R.; Laitinen, R. Permeability of glibenclamide through a PAMPA membrane: The effect of co-amorphization. Eur. J. Pharm. Biopharm. 2018, 129, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Ruponen, M.; Rusanen, H.; Laitinen, R. Dissolution and permeability properties of co-amorphous formulations of hydrochlorothiazide. J. Pharm. Sci. 2020, 109, 2252–2261. [Google Scholar] [CrossRef]
- Leng, D.; Kissi, E.O.; Löbmann, K.; Thanki, K.; Fattal, E.; Rades, T.; Foged, C.; Yang, M. Design of inhalable solid dosage forms of budesonide and theophylline for pulmonary combination therapy. AAPS Pharm. Sci. Tech. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sterren, V.B.; Aiassa, V.; Garnero, C.; Linck, Y.G.; Chattah, A.K.; Monti, G.A.; Longhi, M.R.; Zoppi, A. Preparation of chloramphenicol/amino acid combinations exhibiting enhanced dissolution rates and reduced drug-induced oxidative stress. AAPS Pharm. Sci. Tech. 2017, 18, 2910–2918. [Google Scholar] [CrossRef]
- Mohammed, A.; Zurek, J.; Madueke, S.; Al-Kassimy, H.; Yaqoob, M.; Houacine, C.; Ferraz, A.; Kalgudi, R.; Zariwala, M.G.; Hawkins, N.; et al. Generation of high dose inhalable effervescent dispersions against pseudomonas aeruginosa biofilms. Pharm. Res. 2020, 37, 1–14. [Google Scholar] [CrossRef]
- Lababidi, N.; Ofosu Kissi, E.; Elgaher, W.A.M.; Sigal, V.; Haupenthal, J.; Schwarz, B.C.; Hirsch, A.K.H.; Rades, T.; Schneider, M. Spray-drying of inhalable, multifunctional formulations for the treatment of biofilms formed in cystic fibrosis. J. Control Release 2019, 314, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Rades, T.; Rantanen, J.; Yang, M. Inhalable co-amorphous budesonide-arginine dry powders prepared by spray drying. Int. J. Pharm. 2019, 565, 1–8. [Google Scholar] [CrossRef]
- Hirakawa, Y.; Ueda, H.; Takata, Y.; Minamihata, K.; Wakabayashi, R.; Kamiya, N.; Goto, M. Co-amorphous formation of piroxicam-citric acid to generate supersaturation and improve skin permeation. Eur. J. Pharm. Sci. 2021, 158, 105667–105673. [Google Scholar] [CrossRef]
- Masuda, T.; Yoshihashi, Y.; Yonemochi, E.; Fujii, K.; Uekusa, H.; Terada, K. Cocrystallization and amorphization induced by drug-excipient interaction improves the physical properties of acyclovir. Int. J. Pharm. 2012, 422, 160–169. [Google Scholar] [CrossRef]
- Fung, M.H.; Suryanarayanan, R. Effect of organic acids on molecular mobility, physical stability, and dissolution of ternary ketoconazole spray-dried dispersions. Mol. Pharm. 2019, 16, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Wu, W.; Löbmann, K.; Grohganz, H.; Mullertz, A.; Rades, T. Application of a salt coformer in a co-amorphous drug system dramatically enhances the glass transition temperature: A case study of the ternary system carbamazepine, citric acid, and l-arginine. Mol. Pharm. 2018, 15, 2036–2044. [Google Scholar] [CrossRef]
- Phan, A.D.; Knapik-Kowalczuk, J.; Paluch, M.; Hoang, T.X.; Wakabayashi, K. Theoretical model for the structural relaxation time in coamorphous drugs. Mol. Pharm. 2019, 16, 2992–2998. [Google Scholar] [CrossRef] [PubMed]
- Pacult, J.; Rams-Baron, M.; Chmiel, K.; Jurkiewicz, K.; Antosik, A.; Szafraniec, J.; Kurek, M.; Jachowicz, R.; Paluch, M. How can we improve the physical stability of co-amorphous system containing flutamide and bicalutamide? The case of ternary amorphous solid dispersions. Eur. J. Pharm. Sci. 2019, 136, 104947–104955. [Google Scholar] [CrossRef]
- Jensen, K.T.; Lobmann, K.; Rades, T.; Grohganz, H. Improving co-amorphous drug formulations by the addition of the highly water soluble amino acid, proline. Pharmaceutics 2014, 6, 416–435. [Google Scholar] [CrossRef] [PubMed]
- Wairkar, S.; Gaud, R. Development and characterization of microstructured, spray-dried co-amorphous mixture of antidiabetic agents stabilized by silicate. AAPS Pharm. Sci. Tech. 2019, 20, 1–10. [Google Scholar] [CrossRef]
- Abdelquader, M.M.; Essa, E.A.; El Maghraby, G.M. Inhibition of co-crystallization of olmesartan medoxomil and hydrochlorothiazide for enhanced dissolution rate in their fixed dose combination. AAPS Pharm. Sci. Tech. 2018, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]










| Drug | Amino Acid | Co- Amorphous (Y/N) | Preparation Method | Molar Ratio | Reference |
|---|---|---|---|---|---|
| Carvedilol | Aspartic acid | Y | Spray drying | 1:1 [103], 2:1 to 1:4 [72] | [72,103] |
| Carvedilol | Aspartic acid | Y | Spray drying | With HPMC; 1:1, 1:1.5, 1:2 | [104] |
| Carvedilol | Glutamic acid | Y | Spray drying | 1:1 [103], 2:1 to 1:4 [72] | [72,103] |
| Drug | Amino Acid | Co- Amorphous (Y/N) | Preparation Method | Molar Ratio | Reference |
|---|---|---|---|---|---|
| Glibenclamide | Serine | Y | Cryo- milling | 1:1 | [28,115,116] |
| Glibenclamide | Threonine | Y | Cryo- milling | 1:1 | [28,115] |
| Drug | Amino Acid | Co- Amorphous (Y/N) | Preparation Method | Molar Ratio | Reference |
|---|---|---|---|---|---|
| Ibrutinib | Arginine | N | LAG | 1:1 | [36] |
| Cimetidine | Arginine | Y | Ball milling | 1:1 | [117] |
| Glimepiride | Arginine | N | Ball milling, Melt-quench, Solvent evaporation | 1:1 | [87] * |
| Mebendazole | Arginine | Y, but two-phase system | Ball milling | 1:1 | [117] |
| Ibrutinib | Histidine | N | LAG | 1:2 | [36] |
| Indomethacin | Histidine | N | Ball milling | 1:1 | [103] |
| Glibenclamide | Lysine | N | Cryo-milling | 1:1 | [28] |
| Indomethacin | Lysine | N | Ball milling | 1:1 | [103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Grohganz, H.; Löbmann, K.; Rades, T.; Hempel, N.-J. Co-Amorphous Drug Formulations in Numbers: Recent Advances in Co-Amorphous Drug Formulations with Focus on Co-Formability, Molar Ratio, Preparation Methods, Physical Stability, In Vitro and In Vivo Performance, and New Formulation Strategies. Pharmaceutics 2021, 13, 389. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13030389
Liu J, Grohganz H, Löbmann K, Rades T, Hempel N-J. Co-Amorphous Drug Formulations in Numbers: Recent Advances in Co-Amorphous Drug Formulations with Focus on Co-Formability, Molar Ratio, Preparation Methods, Physical Stability, In Vitro and In Vivo Performance, and New Formulation Strategies. Pharmaceutics. 2021; 13(3):389. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13030389
Chicago/Turabian StyleLiu, Jingwen, Holger Grohganz, Korbinian Löbmann, Thomas Rades, and Nele-Johanna Hempel. 2021. "Co-Amorphous Drug Formulations in Numbers: Recent Advances in Co-Amorphous Drug Formulations with Focus on Co-Formability, Molar Ratio, Preparation Methods, Physical Stability, In Vitro and In Vivo Performance, and New Formulation Strategies" Pharmaceutics 13, no. 3: 389. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13030389