
A Computational Investigation of In Vivo Cytosolic Protein Delivery for Cancer Therapy

 ,
,
Abstract
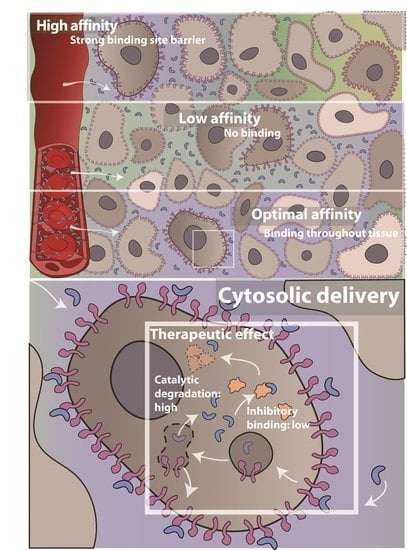
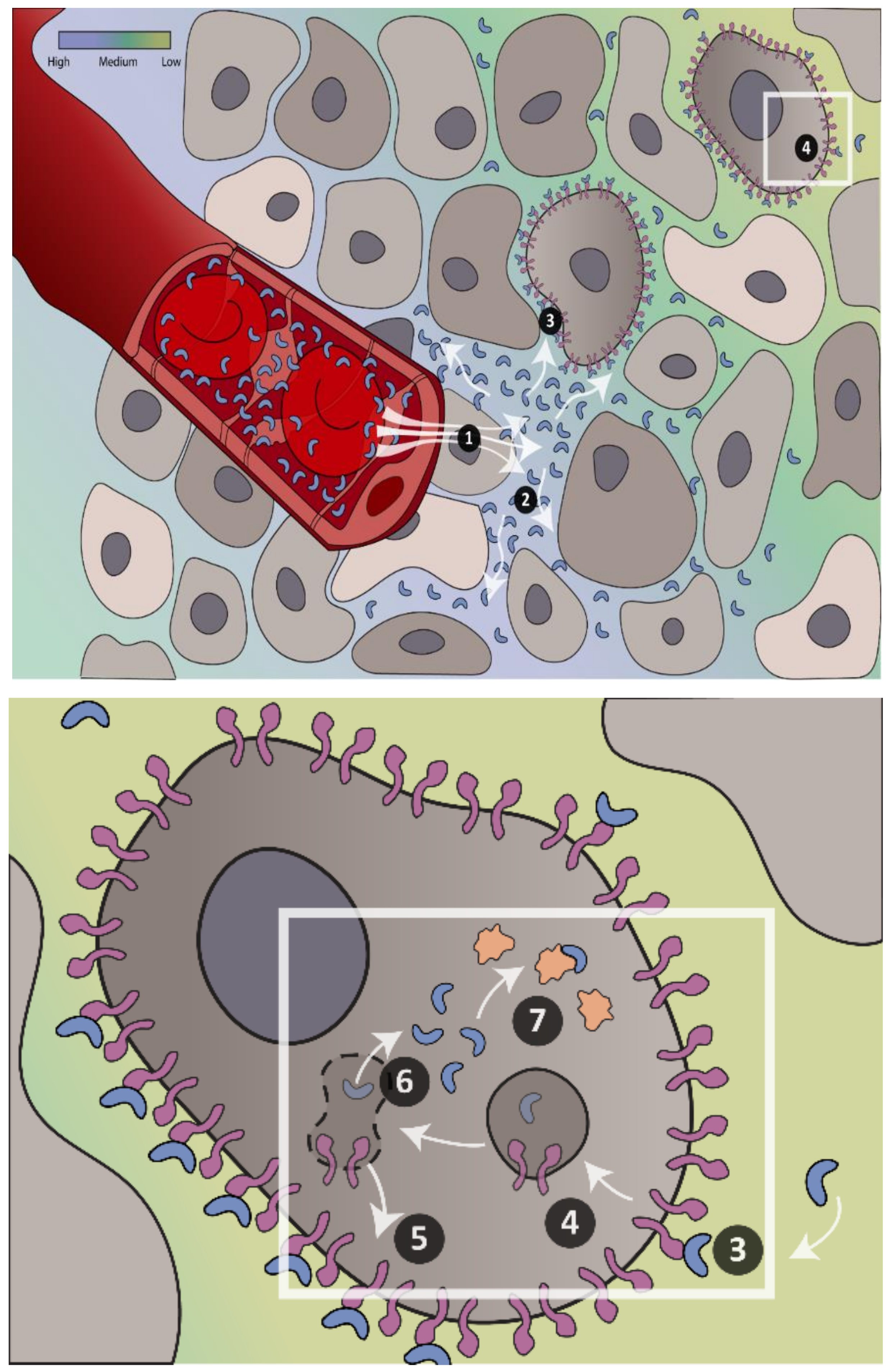
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
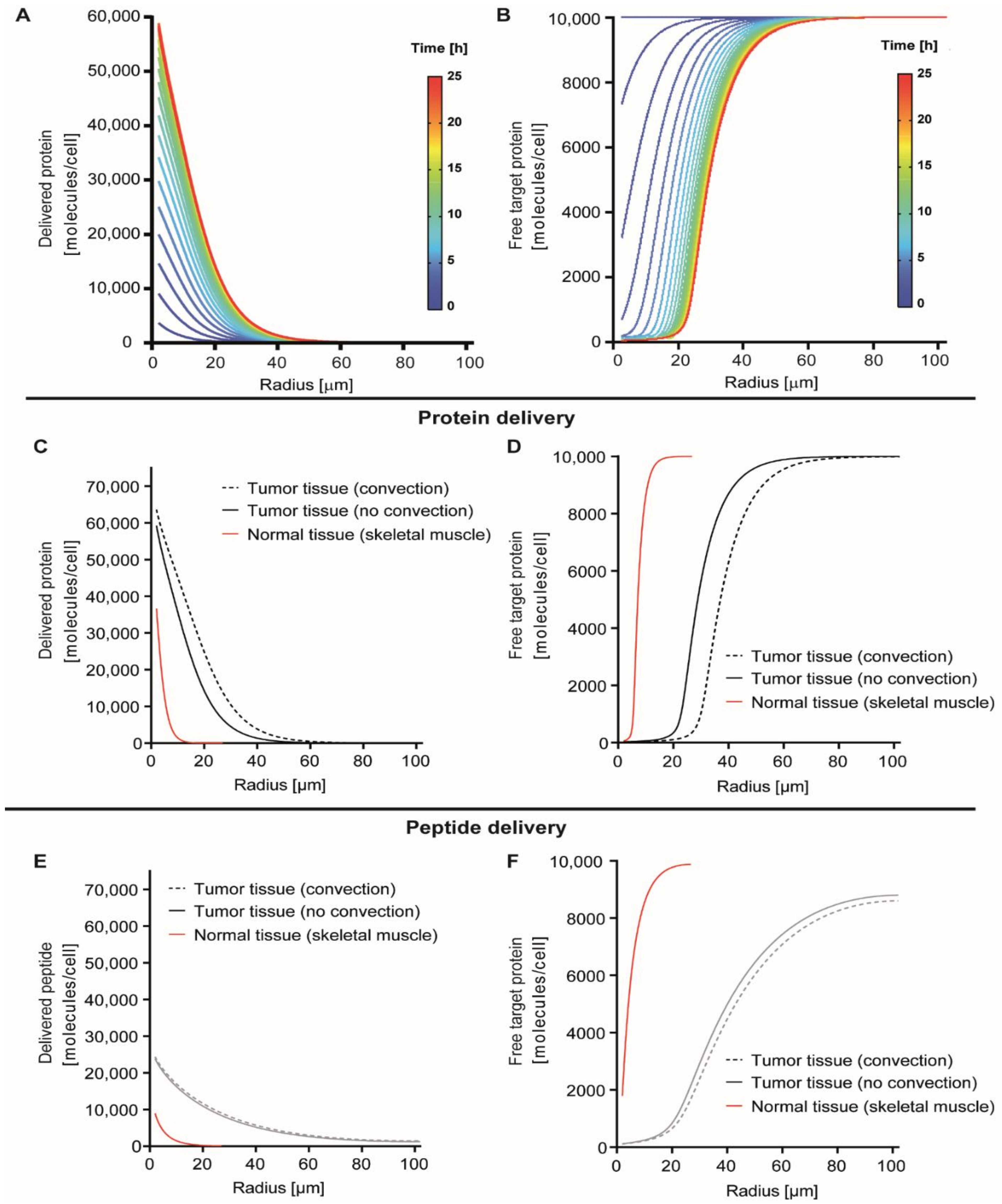
3.1. Modeling Protein Delivery
3.2. Simulation of Delivery and Therapeutic Effects In Vivo
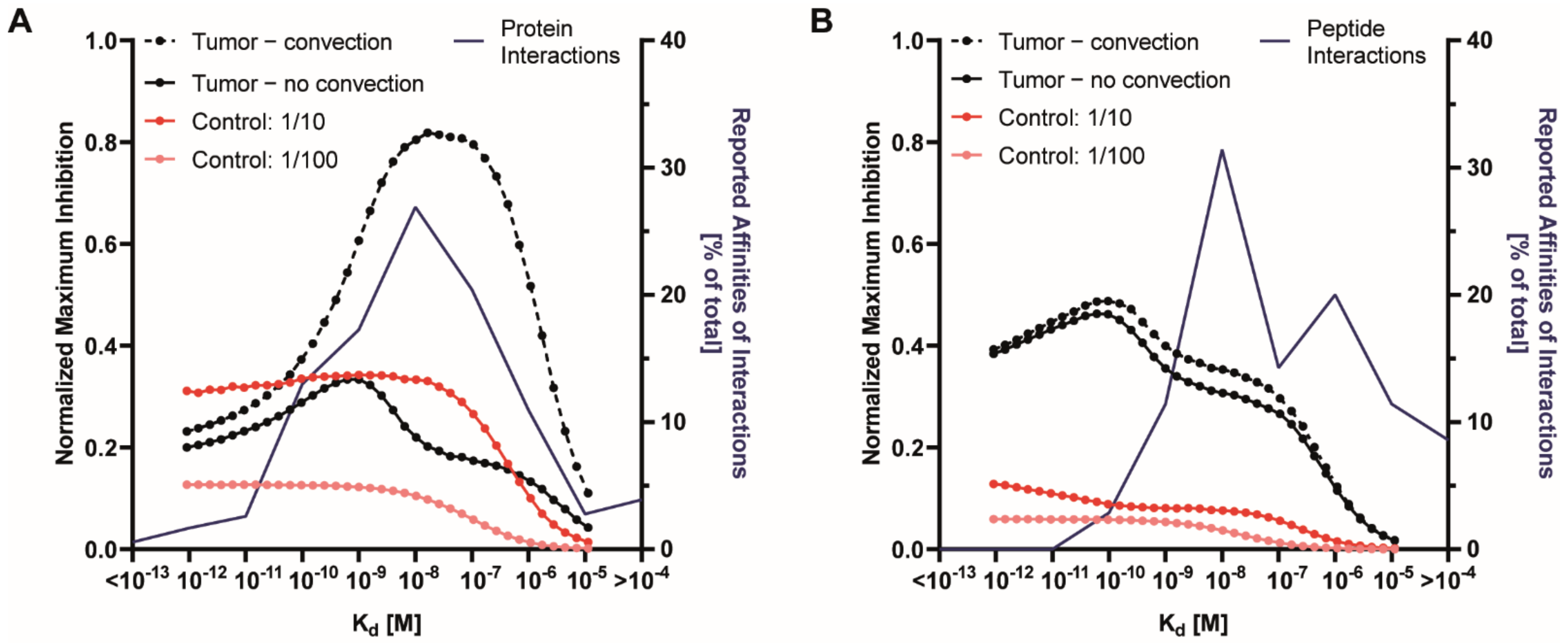
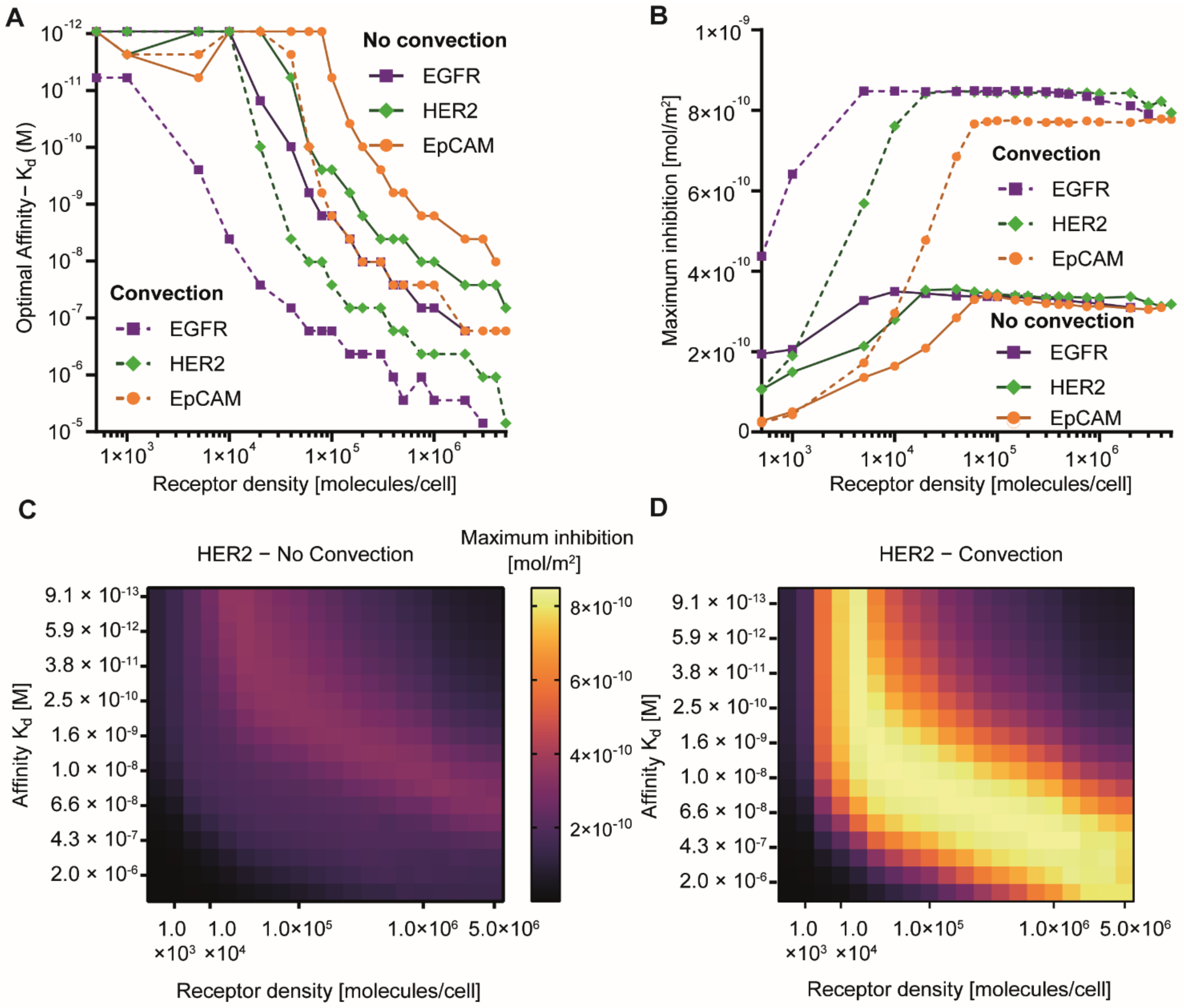
3.3. Effect of Receptor Affinity on Peptide and Protein Delivery
3.4. Interplay Between Receptor Affinity, Receptor Density and Internalization Rate
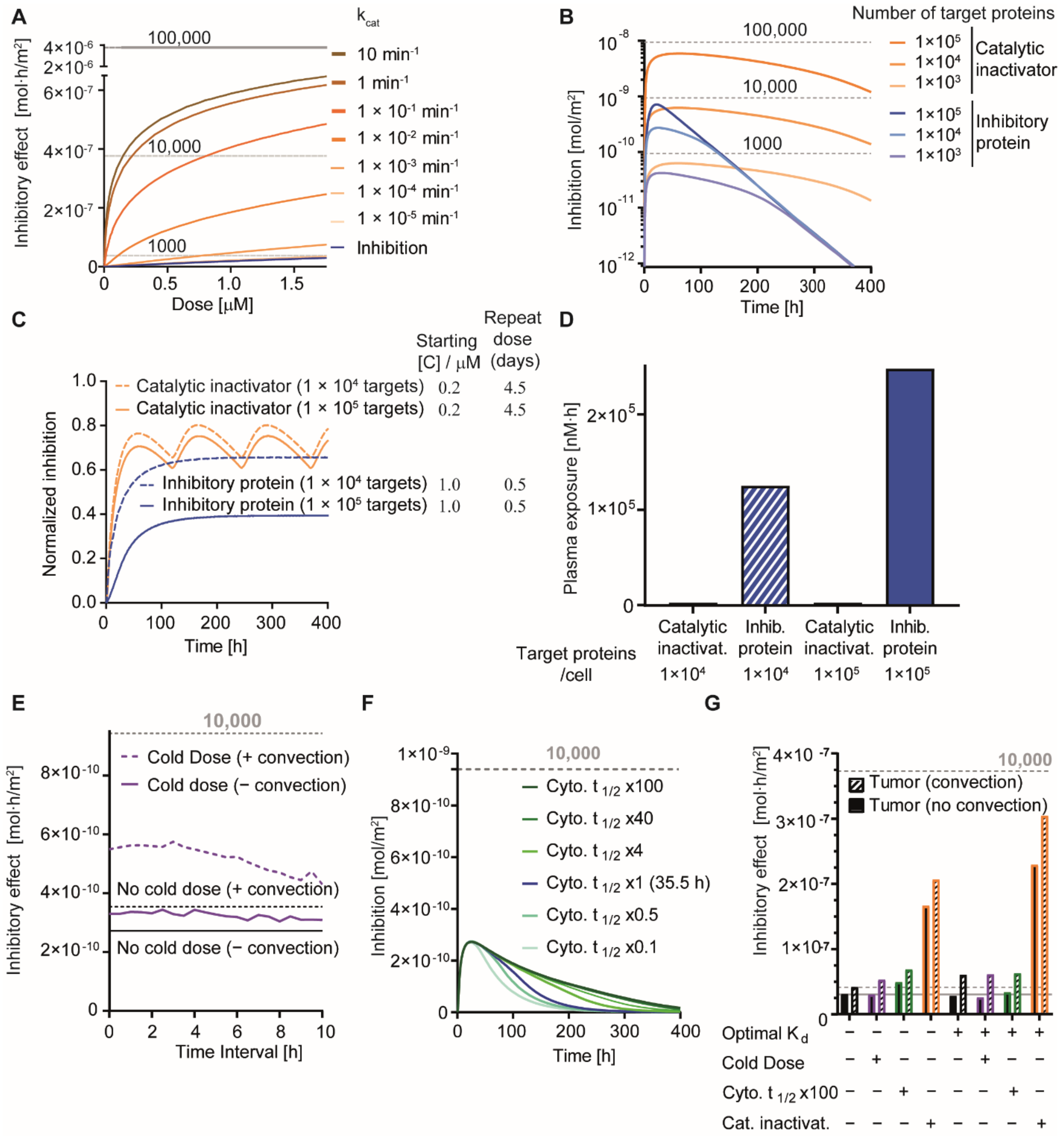
3.5. Effect of Cold Dosing, Targeted Protein Degradation and Degradation-Resistant Proteins on the Binding-Site Barrier
4. Discussion
Limitations and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Lin, A.Y.; Dinner, S.N. Moxetumomab pasudotox for hairy cell leukemia: Preclinical development to FDA approval. Blood Adv. 2019, 3, 2905–2910. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.; Pemmaraju, N. Tagraxofusp for Blastic Plasmacytoid Dendritic Cell Neoplasm. Hematol. Oncol. Clin. N. Am. 2020, 34, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Foss, F.M. DAB(389)IL-2 (ONTAK): A novel fusion toxin therapy for lymphoma. Clin. Lymphoma 2000, 1, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Z.S.; Liu, X.L.; Hui, Q.; Lu, S.Y.; Qu, L.L.; Li, Y.S.; Zhou, Y.; Ren, H.L.; Hu, P. Clinical targeting recombinant immunotoxins for cancer therapy. OncoTargets Ther. 2017, 10, 3645–3665. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Kwaku Dad, A.B.; Beloor, J.; Gopalappa, R.; Lee, S.K.; Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014, 24, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Vidimar, V.; Beilhartz, G.L.; Park, M.; Biancucci, M.; Kieffer, M.B.; Gius, D.R.; Melnyk, R.A.; Satchell, K.J.F. An engineered chimeric toxin that cleaves activated mutant and wild-type RAS inhibits tumor growth. Proc. Natl. Acad. Sci. USA 2020, 117, 16938–16948. [Google Scholar] [CrossRef]
- Chopra, R.; Sadok, A.; Collins, I. A critical evaluation of the approaches to targeted protein degradation for drug discovery. Drug Discov. Today Technol. 2019, 31, 5–13. [Google Scholar] [CrossRef]
- Jost, C.; Plückthun, A. Engineered proteins with desired specificity: DARPins, other alternative scaffolds and bispecific IgGs. Curr. Opin. Struct. Biol. 2014, 27, 102–112. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef]
- Beilhartz, G.L.; Sugiman-Marangos, S.N.; Melnyk, R.A. Repurposing bacterial toxins for intracellular delivery of therapeutic proteins. Biochem. Pharm. 2017, 142, 13–20. [Google Scholar] [CrossRef]
- Stewart, M.P.; Sharei, A.; Ding, X.; Sahay, G.; Langer, R.; Jensen, K.F. In vitro and ex vivo strategies for intracellular delivery. Nature 2016, 538, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Yuvienco, C.; Montclare, J.K. Protein based therapeutic delivery agents: Contemporary developments and challenges. Biomaterials 2017, 134, 91–116. [Google Scholar] [CrossRef]
- Zhang, Y.; Roise, J.J.; Lee, K.; Li, J.; Murthy, N. Recent developments in intracellular protein delivery. Curr. Opin. Biotechnol. 2018, 52, 25–31. [Google Scholar] [CrossRef]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Verdurmen, W.P.R.; Mazlami, M.; Plückthun, A. A quantitative comparison of cytosolic delivery via different protein uptake systems. Sci. Rep. 2017, 7, 13194. [Google Scholar] [CrossRef] [Green Version]
- Thurber, G.M.; Zajic, S.C.; Wittrup, K.D. Theoretic criteria for antibody penetration into solid tumors and micrometastases. J. Nucl. Med. 2007, 48, 995–999. [Google Scholar] [CrossRef] [Green Version]
- D’Esposito, A.; Sweeney, P.W.; Ali, M.; Saleh, M.; Ramasawmy, R.; Roberts, T.A.; Agliardi, G.; Desjardins, A.; Lythgoe, M.F.; Pedley, R.B.; et al. Computational fluid dynamics with imaging of cleared tissue and of in vivo perfusion predicts drug uptake and treatment responses in tumours. Nat. Biomed. Eng. 2018, 2, 773–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefan, N.; Martin-Killias, P.; Wyss-Stoeckle, S.; Honegger, A.; Zangemeister-Wittke, U.; Plückthun, A. DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J. Mol. Biol. 2011, 413, 826–843. [Google Scholar] [CrossRef] [PubMed]
- Chernyavska, M.; Schmid, M.; Freitag, P.C.; Palacio-Castañeda, V.; Piruska, A.; Huck, W.T.S.; Plückthun, A.; Verdurmen, W.P.R. Unravelling Receptor and RGD Motif Dependence of Retargeted Adenoviral Vectors using Advanced Tumor Model Systems. Sci. Rep. 2019, 9, 18568. [Google Scholar] [CrossRef] [PubMed]
- Leveque, D.; Wisniewski, S.; Jehl, F. Pharmacokinetics of therapeutic monoclonal antibodies used in oncology. Anticancer Res. 2005, 25, 2327–2343. [Google Scholar] [PubMed]
- Verdurmen, W.P.; Luginbuhl, M.; Honegger, A.; Plückthun, A. Efficient cell-specific uptake of binding proteins into the cytoplasm through engineered modular transport systems. J. Control. Release 2015, 200, 13–22. [Google Scholar] [CrossRef]
- Buclin, T.; Cosma Rochat, M.; Burckhardt, P.; Azria, M.; Attinger, M. Bioavailability and biological efficacy of a new oral formulation of salmon calcitonin in healthy volunteers. J. Bone Miner. Res. 2002, 17, 1478–1485. [Google Scholar] [CrossRef]
- Tang, L.; Persky, A.M.; Hochhaus, G.; Meibohm, B. Pharmacokinetic aspects of biotechnology products. J. Pharm. Sci. 2004, 93, 2184–2204. [Google Scholar] [CrossRef]
- De Godoy, L.M.; Olsen, J.V.; Cox, J.; Nielsen, M.L.; Hubner, N.C.; Frohlich, F.; Walther, T.C.; Mann, M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 2008, 455, 1251–1254. [Google Scholar] [CrossRef]
- Juweid, M.; Neumann, R.; Paik, C.; Perez-Bacete, M.J.; Sato, J.; van Osdol, W.; Weinstein, J.N. Micropharmacology of monoclonal antibodies in solid tumors: Direct experimental evidence for a binding site barrier. Cancer Res. 1992, 52, 5144–5153. [Google Scholar]
- Thurber, G.M.; Wittrup, K.D. Quantitative spatiotemporal analysis of antibody fragment diffusion and endocytic consumption in tumor spheroids. Cancer Res. 2008, 68, 3334–3341. [Google Scholar] [CrossRef] [Green Version]
- Norval, L.W.; Kramer, S.D.; Gao, M.; Herz, T.; Li, J.; Rath, C.; Wohrle, J.; Gunther, S.; Roth, G. KOFFI and Anabel 2.0-a new binding kinetics database and its integration in an open-source binding analysis software. Database 2019, 2019. [Google Scholar] [CrossRef]
- Kim, H.Y.; Wang, X.; Wahlberg, B.; Edwards, W.B. Discovery of hapten-specific scFv from a phage display library and applications for HER2-positive tumor imaging. Bioconjug. Chem. 2014, 25, 1311–1322. [Google Scholar] [CrossRef]
- Hendriks, B.S.; Opresko, L.K.; Wiley, H.S.; Lauffenburger, D. Quantitative analysis of HER2-mediated effects on HER2 and epidermal growth factor receptor endocytosis: Distribution of homo- and heterodimers depends on relative HER2 levels. J. Biol. Chem. 2003, 278, 23343–23351. [Google Scholar] [CrossRef] [Green Version]
- Hazin, J.; Moldenhauer, G.; Altevogt, P.; Brady, N.R. A novel method for measuring cellular antibody uptake using imaging flow cytometry reveals distinct uptake rates for two different monoclonal antibodies targeting L1. J. Immunol. Methods 2015, 423, 70–77. [Google Scholar] [CrossRef]
- Knox, S.J.; Goris, M.L.; Trisler, K.; Negrin, R.; Davis, T.; Liles, T.M.; Grillo-Lopez, A.; Chinn, P.; Varns, C.; Ning, S.C.; et al. Yttrium-90-labeled anti-CD20 monoclonal antibody therapy of recurrent B-cell lymphoma. Clin. Cancer Res. 1996, 2, 457–470. [Google Scholar]
- Prinssen, H.M.; Molthoff, C.F.; Verheijen, R.H.; Broadhead, T.J.; Kenemans, P.; Roos, J.C.; Davies, Q.; van Hof, A.C.; Frier, M.; den Hollander, W.; et al. Biodistribution of 111In-labelled engineered human antibody CTM01 (hCTM01) in ovarian cancer patients: Influence of prior administration of unlabelled hCTM01. Cancer Immunol. Immunother. 1998, 47, 39–46. [Google Scholar] [CrossRef]
- Cilliers, C.; Menezes, B.; Nessler, I.; Linderman, J.; Thurber, G.M. Improved Tumor Penetration and Single-Cell Targeting of Antibody-Drug Conjugates Increases Anticancer Efficacy and Host Survival. Cancer Res. 2018, 78, 758–768. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.P.; Guo, L.; Verma, A.; Wong, G.G.; Thurber, G.M.; Shah, D.K. Antibody Coadministration as a Strategy to Overcome Binding-Site Barrier for ADCs: A Quantitative Investigation. AAPS J. 2020, 22, 28. [Google Scholar] [CrossRef]
- Loftis, A.R.; Santos, M.S.; Truex, N.L.; Biancucci, M.; Satchell, K.J.F.; Pentelute, B.L. Anthrax Protective Antigen Retargeted with Single-Chain Variable Fragments Delivers Enzymes to Pancreatic Cancer Cells. ChemBioChem 2020, 21, 2772–2776. [Google Scholar] [CrossRef]
- Schmit, N.E.; Neopane, K.; Hantschel, O. Targeted Protein Degradation through Cytosolic Delivery of Monobody Binders Using Bacterial Toxins. ACS Chem. Biol. 2019, 14, 916–924. [Google Scholar] [CrossRef]
- Rabideau, A.E.; Liao, X.; Pentelute, B.L. Delivery of mirror image polypeptides into cells. Chem. Sci. 2015, 6, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Verdurmen, W.P.; Bovee-Geurts, P.H.; Wadhwani, P.; Ulrich, A.S.; Hallbrink, M.; van Kuppevelt, T.H.; Brock, R. Preferential uptake of L- versus D-amino acid cell-penetrating peptides in a cell type-dependent manner. Chem. Biol. 2011, 18, 1000–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirmule, N.; Jawa, V.; Meibohm, B. Immunogenicity to therapeutic proteins: Impact on PK/PD and efficacy. AAPS J. 2012, 14, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Freiman, R.N.; Tjian, R. Regulating the regulators: Lysine modifications make their mark. Cell 2003, 112, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Ofenbauer, A.; Tursun, B. Strategies for in vivo reprogramming. Curr. Opin. Cell Biol. 2019, 61, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Burlina, F.; Sagan, S.; Bolbach, G.; Chassaing, G. Quantification of the cellular uptake of cell-penetrating peptides by MALDI-TOF mass spectrometry. Angew. Chem. Int. Ed. Engl. 2005, 44, 4244–4247. [Google Scholar] [CrossRef]
- Illien, F.; Rodriguez, N.; Amoura, M.; Joliot, A.; Pallerla, M.; Cribier, S.; Burlina, F.; Sagan, S. Quantitative fluorescence spectroscopy and flow cytometry analyses of cell-penetrating peptides internalization pathways: Optimization, pitfalls, comparison with mass spectrometry quantification. Sci. Rep. 2016, 6, 36938. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Choi, D.K.; Park, S.W.; Shin, S.M.; Bae, J.; Kim, D.M.; Yoo, T.H.; Kim, Y.S. Quantitative assessment of cellular uptake and cytosolic access of antibody in living cells by an enhanced split GFP complementation assay. Biochem. Biophys. Res. Commun. 2015, 467, 771–777. [Google Scholar] [CrossRef]
- LaRochelle, J.R.; Cobb, G.B.; Steinauer, A.; Rhoades, E.; Schepartz, A. Fluorescence correlation spectroscopy reveals highly efficient cytosolic delivery of certain penta-arg proteins and stapled peptides. J. Am. Chem. Soc. 2015, 137, 2536–2541. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Rabideau, A.E.; Pentelute, B.L. Delivery of antibody mimics into mammalian cells via anthrax toxin protective antigen. ChemBioChem 2014, 15, 2458–2466. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.; de la Torre, B.G.; Radis-Baptista, G.; Santos, N.C.; Andreu, D. Efficient cellular delivery of beta-galactosidase mediated by NrTPs, a new family of cell-penetrating peptides. Bioconjugate Chem. 2011, 22, 2339–2344. [Google Scholar] [CrossRef]
- Taylor, M.; Banerjee, T.; VanBennekom, N.; Teter, K. Detection of toxin translocation into the host cytosol by surface plasmon resonance. J. Vis. Exp. 2012, e3686. [Google Scholar] [CrossRef]
- Waizenegger, T.; Fischer, R.; Brock, R. Intracellular concentration measurements in adherent cells: A comparison of import efficiencies of cell-permeable peptides. Biol. Chem. 2002, 383, 291–299. [Google Scholar] [CrossRef]
- Wissner, R.F.; Steinauer, A.; Knox, S.L.; Thompson, A.D.; Schepartz, A. Fluorescence Correlation Spectroscopy Reveals Efficient Cytosolic Delivery of Protein Cargo by Cell-Permeant Miniature Proteins. ACS Cent. Sci. 2018, 4, 1379–1393. [Google Scholar] [CrossRef]
- Becker, L.; Verdurmen, W.P.R.; Plückthun, A. Reengineering anthrax toxin protective antigen for improved receptor-specific protein delivery. BMC Biol. 2020, 18, 100. [Google Scholar] [CrossRef]
- Bartelink, I.H.; Jones, E.F.; Shahidi-Latham, S.K.; Lee, P.R.E.; Zheng, Y.; Vicini, P.; van ‘t Veer, L.; Wolf, D.; Iagaru, A.; Kroetz, D.L.; et al. Tumor Drug Penetration Measurements Could Be the Neglected Piece of the Personalized Cancer Treatment Puzzle. Clin. Pharmacol. Ther. 2019, 106, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Factors determining antibody distribution in tumors. Trends Pharmacol. Sci. 2008, 29, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [Green Version]
- Pluen, A.; Boucher, Y.; Ramanujan, S.; McKee, T.D.; Gohongi, T.; di Tomaso, E.; Brown, E.B.; Izumi, Y.; Campbell, R.B.; Berk, D.A.; et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: Cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 4628–4633. [Google Scholar] [CrossRef] [Green Version]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar]
- Rhoden, J.J.; Wittrup, K.D. Dose dependence of intratumoral perivascular distribution of monoclonal antibodies. J. Pharm. Sci. 2012, 101, 860–867. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Kwon, I.K.; Lee, S.C.; Han, B.; Park, K. Analysis on the current status of targeted drug delivery to tumors. J. Control. Release 2012, 164, 108–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biancucci, M.; Rabideau, A.E.; Lu, Z.; Loftis, A.R.; Pentelute, B.L.; Satchell, K.J.F. Substrate Recognition of MARTX Ras/Rap1-Specific Endopeptidase. Biochemistry 2017, 56, 2747–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 2016, 55, 807–810. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- He, Y.; Zhang, X.; Chang, J.; Kim, H.N.; Zhang, P.; Wang, Y.; Khan, S.; Liu, X.; Zhang, X.; Lv, D.; et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat. Commun. 2020, 11, 1996. [Google Scholar] [CrossRef]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S.; et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 2020, 11, 1545. [Google Scholar] [CrossRef] [Green Version]
- Pignolo, R.J.; Passos, J.F.; Khosla, S.; Tchkonia, T.; Kirkland, J.L. Reducing Senescent Cell Burden in Aging and Disease. Trends Mol. Med. 2020, 26, 630–638. [Google Scholar] [CrossRef]
- Takakura, Y.; Fujita, T.; Hashida, M.; Sezaki, H. Disposition characteristics of macromolecules in tumor-bearing mice. Pharm. Res. 1990, 7, 339–346. [Google Scholar] [CrossRef]
- Bajpayee, A.G.; Grodzinsky, A.J. Cartilage-targeting drug delivery: Can electrostatic interactions help? Nat. Rev. Rheumatol. 2017, 13, 183–193. [Google Scholar] [CrossRef]
- Wiig, H.; Gyenge, C.C.; Tenstad, O. The interstitial distribution of macromolecules in rat tumours is influenced by the negatively charged matrix components. J. Physiol. 2005, 567, 557–567. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef]
- Steiner, D.; Merz, F.W.; Sonderegger, I.; Gulotti-Georgieva, M.; Villemagne, D.; Phillips, D.J.; Forrer, P.; Stumpp, M.T.; Zitt, C.; Binz, H.K. Half-life extension using serum albumin-binding DARPin(R) domains. Protein Eng. Des. Sel. 2017, 30, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.Y.; Park, J.H.; Shim, H.E.; Choi, D.S.; Lee, D.E.; Song, J.J.; Kim, H.S. Prolonged half-life of small-sized therapeutic protein using serum albumin-specific protein binder. J. Control. Release 2019, 315, 31–39. [Google Scholar] [CrossRef]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [Green Version]
- Paterson, J.; Webster, C.I. Exploiting transferrin receptor for delivering drugs across the blood-brain barrier. Drug Discov. Today Technol. 2016, 20, 49–52. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, C.; Dumas, S.; Palacio-Castañeda, V.; Descroix, S.; Brock, R.; Verdurmen, W.P.R. A Computational Investigation of In Vivo Cytosolic Protein Delivery for Cancer Therapy. Pharmaceutics 2021, 13, 562. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13040562
Torres C, Dumas S, Palacio-Castañeda V, Descroix S, Brock R, Verdurmen WPR. A Computational Investigation of In Vivo Cytosolic Protein Delivery for Cancer Therapy. Pharmaceutics. 2021; 13(4):562. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13040562
Chicago/Turabian StyleTorres, Camilo, Simon Dumas, Valentina Palacio-Castañeda, Stéphanie Descroix, Roland Brock, and Wouter P. R. Verdurmen. 2021. "A Computational Investigation of In Vivo Cytosolic Protein Delivery for Cancer Therapy" Pharmaceutics 13, no. 4: 562. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13040562