
Targeting Mitochondrial Oncometabolites: A New Approach to Overcome Drug Resistance in Cancer

 and
and
Abstract
:1. Introduction: Mitochondria in Cancer
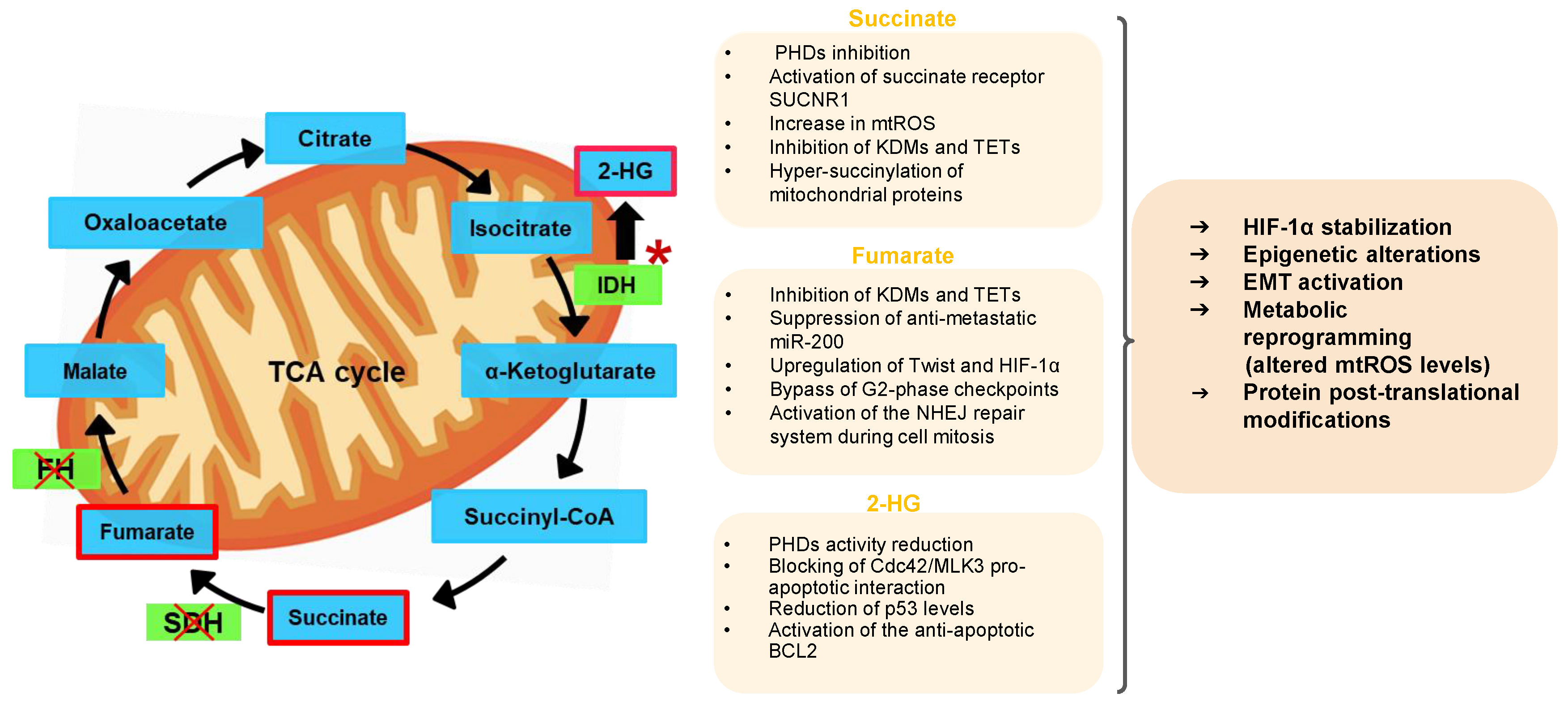
2. Mitochondrial Oncometabolites and Cancer Biology
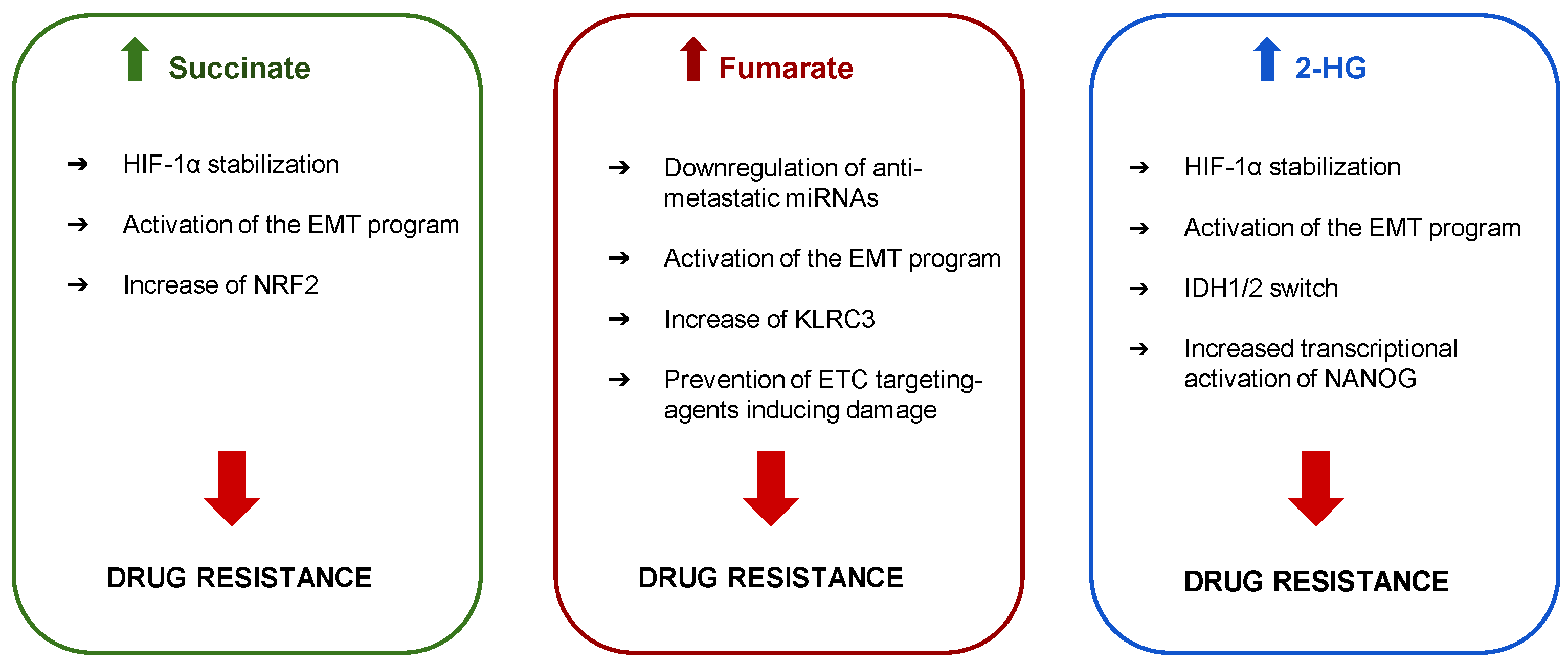
3. Mitochondrial Oncometabolites and Drug Resistance
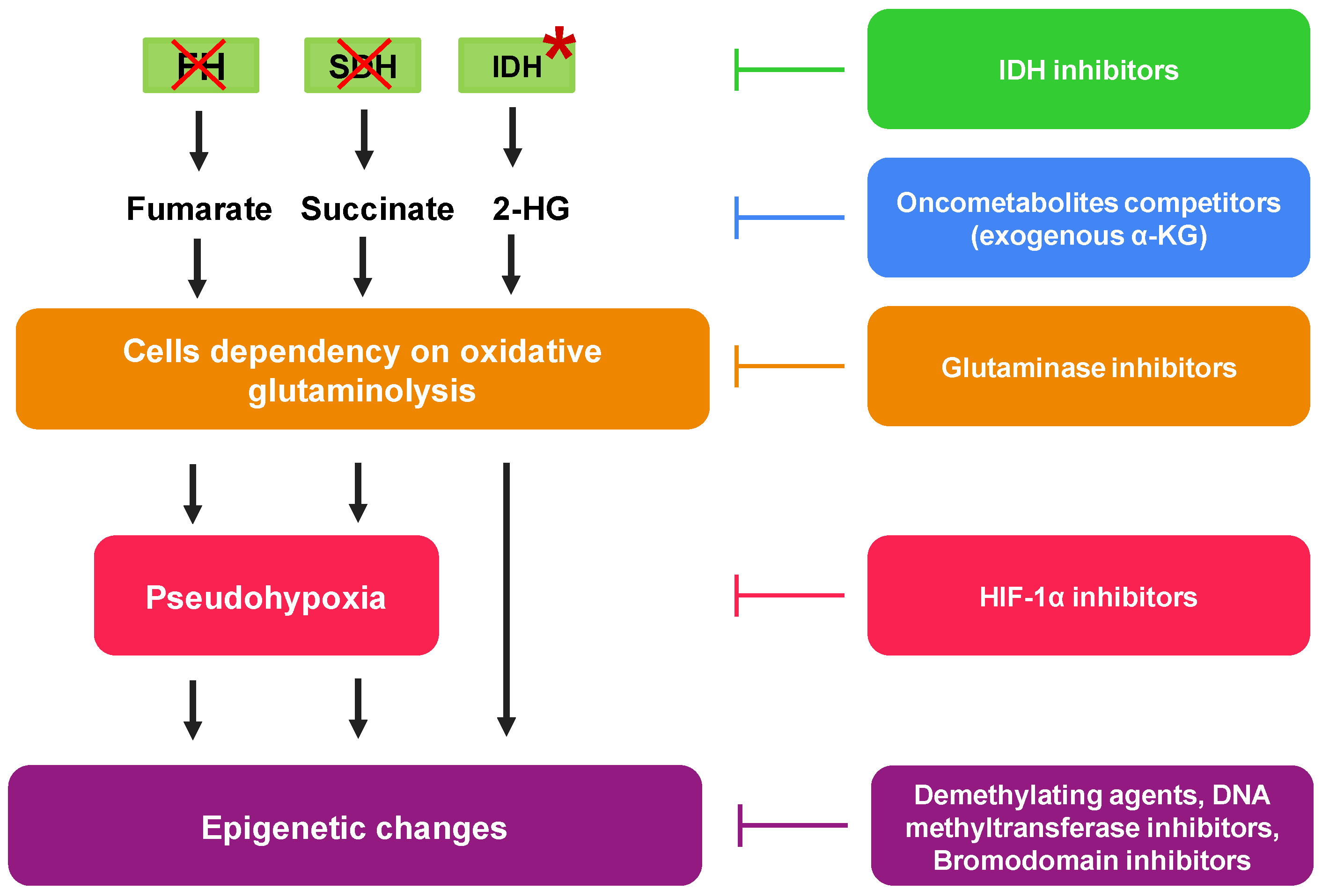
4. Pharmacological Approaches to Reduce Mitochondrial Oncometabolites

{kind=link}
{kind=link}
{kind=link}
| Targeted Oncometabolite | Mutated Gene | Drugs |
|---|---|---|
| Succinate | SDH | Compound 968 and CB-839 [37] |
| Fumarate | FH | JQ1 [37] Exogenous αKG [67,68,69] HIF-1α inhibitors [36] |
| 2-HG | IDH | Venetoclax [33,53]. Enasidenib/AG-221 [33,70] Ivosidenib/AG-120 [33,64] Vorasidenib [71] ML309 [72] AGI-5198 [72] GSK864 [72] Azacytidine [75,76] Decitabine [75,76] Temozolomide [77,78] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokil, A.; Sancho, P. Mitochondrial determinants of chemoresistance. Cancer Drug Resist. 2019, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C.; et al. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 2019, 25, 101076. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Choudhary, D.; Mikhalyuk, A.; Trammel, C.; Shanmugam, S.; Abbott, E.; Pilbeam, C.C.; Taylor, J.A. The role of pyruvate dehydrogenase kinase-4 (PDK4) in bladder cancer and chemoresistance. Mol. Cancer Ther. 2018, 17, 2004–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Yu, A.Q. The functional role of peroxiredoxin 3 in reactive oxygen species, apoptosis, and chemoresistance of cancer cells. J. Cancer Res. Clin. Oncol. 2015, 141, 2071–2077. [Google Scholar] [CrossRef]
- Alexa-Stratulat, T.; Pešić, M.; Gašparović, A.Č.; Trougakos, I.P.; Riganti, C. What sustains the multidrug resistance phenotype beyond ABC efflux transporters? Looking beyond the tip of the iceberg. Drug Resist. Updat. 2019, 46. [Google Scholar] [CrossRef]
- Kapoor, I.; Bodo, J.; Hill, B.T.; Hsi, E.D.; Almasan, A. Targeting BCL-2 in B-cell malignancies and overcoming therapeutic resistance. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef]
- Van Gisbergen, M.W.; Voets, A.M.; Starmans, M.H.; De Coo, I.F.; Yadak, R.; Hoffmann, R.F.; Boutros, P.C.; Smeets, H.J.; Dubois, L.; Lambin, P. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat. Res. Rev. Mutat. Res. 2015, 764, 16–30. [Google Scholar] [CrossRef]
- Aminuddin, A.; Ng, P.Y.; Leong, C.O.; Chua, E.W. Mitochondrial DNA alterations may influence the cisplatin responsiveness of oral squamous cell carcinoma. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Pozza, E.D.; Ambrosini, G.; Torrens-Mas, M.; Butera, G.; Mullappilly, N.; Pacchiana, R.; Palmieri, M.; Donadelli, M. Oncometabolites in cancer aggressiveness and tumour repopulation. Biol. Rev. Camb. Philos. Soc. 2019, 94, 1530–1546. [Google Scholar] [CrossRef] [PubMed]
- Aldera, A.P.; Govender, D. Gene of the month: SDH. J. Clin. Pathol. 2018, 71, 95–97. [Google Scholar] [CrossRef]
- Neppala, P.; Banerjee, S.; Fanta, P.T.; Yerba, M.; Porras, K.A.; Burgoyne, A.M.; Sicklick, J.K. Current management of succinate dehydrogenase–deficient gastrointestinal stromal tumors. Cancer Metastasis Rev. 2019, 38, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.E.; Wang, F.; Yu, F.; Zeng, Z.L.; Wang, Y.; Lu, Y.X.; Jin, Y.; Wang, D.S.; Qiu, M.Z.; Pu, H.Y.; et al. Suppression of fumarate hydratase activity increases the efficacy of cisplatin-mediated chemotherapy in gastric cancer. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Dalla Pozza, E.; Dando, I.; Pacchiana, R.; Liboi, E.; Scupoli, M.T.; Donadelli, M.; Palmieri, M. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin. Cell Dev. Biol. 2020, 9, 4–14. [Google Scholar] [CrossRef]
- Hoekstra, A.S.; Bayley, J.P. The role of complex II in disease. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Feng, F.; Guo, Q.H.; Wang, Y.P.; Zhao, R. Role of succinate dehydrogenase deficiency and oncometabolites in gastrointestinal stromal tumors. World J. Gastroenterol. 2020, 26, 5074–5089. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Wu, G. SDHB deficiency promotes TGFβ-mediated invasion and metastasis of colorectal cancer through transcriptional repression complex SNAIL1-SMAD3/4. Transl. Oncol. 2016, 9, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.; Sciacovelli, M.; Frezza, C. Fumarate hydratase in cancer: A multifaceted tumour suppressor. Semin. Cell Dev. Biol. 2020, 98, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.M.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.M.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer the multiple leiomyoma consortium. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.S.; Linehan, W.M. Hereditary leiomyomatosis and renal cell carcinoma. Int. J. Nephrol. Renov. Dis. 2014, 7, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Vega, L.J.; Buffet, A.; De Cubas, A.A.; Cascón, A.; Menara, M.; Khalifa, E.; Amar, L.; Azriel, S.; Bourdeau, I.; Chabre, O.; et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 2014, 23, 2440–2446. [Google Scholar] [CrossRef] [Green Version]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G.; Godley, L.A.; Koivunen, P. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Mir, A.; Glaser, B.; Chuang, G.S.; Horev, L.; Waldman, A.; Engler, D.E.; Gordon, D.; Spelman, L.J.; Hatzibougias, I.; Green, J.; et al. Germline fumarate hydratase mutations in families with multiple cutaneous and uterine leiomyomata. J. Invest. Dermatol. 2003, 121, 741–744. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Ohshima, K.; Nojima, S.; Tahara, S.; Kurashige, M.; Hori, Y.; Okuzaki, D.; Wada, N.; Ikeda, J.I.; Morii, E. Adenylosuccinate lyase enhances aggressiveness of endometrial cancer by increasing killer cell lectin-like receptor C3 expression by fumarate. Lab. Investig. 2018, 98, 449–461. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Yan, B.; Liu, S.; Jia, J.; Lai, W.; Xin, X.; Tang, C.E.; Luo, D.; Tan, T.; Jiang, Y.; et al. Chromatin remodeling factor LSH drives cancer progression by suppressing the activity of fumarate hydratase. Cancer Res. 2016, 76, 5743–5755. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Zhao, W.; Shi, M.; Zhang, J.; Chen, A.; Ma, H.; Suleman, M.; Lin, F.; Zhou, L.; Wang, J.; et al. IDH1 Arg-132 mutant promotes tumor formation through down-regulating P53. J. Biol. Chem. 2018, 293, 9747–9758. [Google Scholar] [CrossRef] [Green Version]
- Ježek, P. 2-Hydroxyglutarate in cancer cells. Antioxid. Redox Signal. 2020, 33, 903–926. [Google Scholar] [CrossRef] [Green Version]
- Tinoco, G.; Wilky, B.A.; Paz-Mejia, A.; Rosenberg, A.; Trent, J.C. The biology and management of cartilaginous tumors: A role for targeting isocitrate dehydrogenase. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e648–e655. [Google Scholar] [CrossRef]
- Amaya, M.L.; Pollyea, D.A. Targeting the IDH2 pathway in acute myeloid leukemia. Clin. Cancer Res. 2018, 24, 4931–4936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smestad, J.; Erber, L.; Chen, Y.; Maher, L.J. Chromatin succinylation correlates with active gene expression and is perturbed by defective TCA cycle metabolism. Iscience 2018, 2, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belisario, D.C.; Kopecka, J.; Pasino, M.; Akman, M.; Smaele, E.D.; Donadelli, M.; Riganti, C. Hypoxia dictates metabolic rewiring of tumors: Implications for chemoresistance. Cells 2020, 9, 2598. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitazawa, S.; Ebara, S.; Ando, A.; Baba, Y.; Satomi, Y.; Soga, T.; Hara, T. Succinate dehydrogenase B-deficient cancer cells are highly sensitive to bromodomain and extra-terminal inhibitors. Oncotarget 2017, 8, 28922–28938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Y.; Huang, T.W.; Hsieh, Y.T.; Wang, Y.F.; Yen, C.C.; Lee, G.L.; Yeh, C.C.; Peng, Y.J.; Kuo, Y.Y.; Wen, H.T.; et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. Cell 2020, 77, 213–227.e5. [Google Scholar] [CrossRef] [PubMed]
- Ristic, B.; Bhutia, Y.; Ganapathy, V. Cell-surface G-protein-coupled receptors for tumor-associated metabolites: A direct link to mitochondrial dysfunction in cancer. Biochim. Biophys. Acta 2017, 1868, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratcliffe, P.J. Oxygen sensing and hypoxia signalling pathways in animals: The implications of physiology for cancer. J. Physiol. 2013, 591, 2027–2042. [Google Scholar] [CrossRef]
- Aggarwal, R.K.; Zou, Y.; Luchtel, R.A.; Pradhan, K.; Ashai, N.; Ramachandra, N.; Albanese, J.M.; Yang, J.-I.; Wang, X.; Aluri, S.; et al. Functional succinate dehydrogenase deficiency is a pathognomonic adverse feature of clear cell renal cancer. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cervera, A.M.; Bayley, J.P.; Devilee, P.; McCreath, K.J. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol. Cancer 2009, 8, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loriot, C.; Domingues, M.; Berger, A.; Menara, M.; Ruel, M.; Morin, A.; Castro-Vega, L.J.; Letouzé, É.; Martinelli, C.; Bemelmans, A.P.; et al. Deciphering the molecular basis of invasiveness in sdhbdeficient cells. Oncotarget 2015, 6, 32955–32965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef]
- Johnson, T.I.; Costa, A.S.H.; Ferguson, A.N.; Frezza, C. Fumarate hydratase loss promotes mitotic entry in the presence of DNA damage after ionising radiation. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Wentzel, J.F.; Lewies, A.; Bronkhorst, A.J.; Van Dyk, E.; Du Plessis, L.H.; Pretorius, P.J. Exposure to high levels of fumarate and succinate leads to apoptotic cytotoxicity and altered global DNA methylation profiles in vitro. Biochimie 2017, 135, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Ricciardiello, F.; Gang, Y.; Palorini, R.; Li, Q.; Giampà, M.; Zhao, F.; You, L.; La Ferla, B.; De Vitto, H.; Guan, W.; et al. Hexosamine pathway inhibition overcomes pancreatic cancer resistance to gemcitabine through unfolded protein response and EGFR-Akt pathway modulation. Oncogene 2020, 39, 4103–4117. [Google Scholar] [CrossRef]
- Wang, T.; Yu, Q.; Li, J.; Hu, B.; Zhao, Q.; Ma, C.; Huang, W.; Zhuo, L.; Fang, H.; Liao, L.; et al. O-GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. Nat. Cell Biol. 2017, 19, 833–843. [Google Scholar] [CrossRef]
- Chen, T.; Wang, T.; Liang, W.; Zhao, Q.; Yu, Q.; Ma, C.M.; Zhuo, L.; Guo, D.; Zheng, K.; Zhou, C.; et al. PAK4 phosphorylates fumarase and blocks TGFβ-induced cell growth arrest in lung cancer cells. Cancer Res. 2019, 79, 1383–1397. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Zhang, J.; Xia, J.; Zhao, W.; Wu, Y.; Shi, M.; Luo, L.; Zhou, H.; Chen, A.; Ma, H.; et al. IDH1 mutation promotes tumorigenesis by inhibiting JNK activation and apoptosis induced by serum starvation. Cell Rep. 2017, 19, 389–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.S.; Daniel, T.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.-J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreedhar, A.; Wiese, E.K.; Hitosugi, T. Enzymatic and metabolic regulation of lysine succinylation. Genes Dis. 2020, 7, 166–171. [Google Scholar] [CrossRef]
- Kinch, L.; Grishin, N.V.; Brugarolas, J. Succination of keap1 and activation of Nrf2-dependent antioxidant pathways in FH-deficient papillary renal cell carcinoma type 2. Cancer Cell 2011, 20, 418–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; He, X.; Ye, D.; Lin, Y.; Yu, H.; Yao, C.; Huang, L.; Zhang, J.; Wang, F.; Xu, S.; et al. NADP(+)-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol. Cell 2015, 60, 661–675. [Google Scholar] [CrossRef]
- Timmers, H.J.; Gimenez-Roqueplo, A.P.; Mannelli, M.; Pacak, K. Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr. Relat. Cancer 2009, 16, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheray, M.; Bessette, B.; Lacroix, A.; Mélin, C.; Jawhari, S.; Pinet, S.; Deluche, E.; Clavère, P.; Durand, K.; Sanchez-Prieto, R.; et al. KLRC3, a natural killer receptor gene, is a key factor involved in glioblastoma tumourigenesis and aggressiveness. J. Cell Mol. Med. 2017, 21, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Greer, Y.E.; Porat-Shliom, N.; Nagashima, K.; Stuelten, C.; Crooks, D.; Koparde, V.N.; Gilbert, S.F.; Islam, C.; Ubaldini, A.; Ji, Y.; et al. ONC201 kills breast cancer cells in vitro by targeting mitochondria. Oncotarget 2018, 9, 18454–18479. [Google Scholar] [CrossRef] [Green Version]
- Pruss, M.; Dwucet, A.; Tanriover, M.; Hlavac, M.; Kast, R.E.; Debatin, K.M.; Wirtz, C.R.; Halatsch, M.E.; Siegelin, M.D.; Westhoff, M.A.; et al. Dual metabolic reprogramming by ONC201/TIC10 and 2-deoxyglucose induces energy depletion and synergistic anti-cancer activity in glioblastoma. Br. J. Cancer 2020, 122, 1146–1157. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, K.; Terai, S.; Takami, T.; Yamamoto, N.; Yamasaki, T.; Matsumoto, T.; Yamaguchi, K.; Owada, Y.; Nishina, H.; Noma, T.; et al. Modulation of anti-cancer drug sensitivity through the regulation of mitochondrial activity by adenylate kinase 4. J. Exp. Clin. Cancer Res. 2016, 35, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Ariën, S.; Albanese, S.K.; Patel, M.; Famulare, C.; Fabian, M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef]
- Harding, J.J.; Lowery, M.A.; Shih, A.H.; Schvartzman, J.M.; Hou, S.; Famulare, C.; Patel, M.; Roshal, M.; Do, R.K.; Zehir, A.; et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov. 2018, 8, 1540–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, M.; Sperry, J.; Braas, D.; Yan, W.; Le, T.M.; Mottahedeh, J.; Ludwig, K.; Eskin, A.; Qin, Y.; Levy, R.; et al. Metabolic characterization of isocitrate dehydrogenase (IDH) mutant and IDH wildtype gliomaspheres uncovers cell type-specific vulnerabilities. Cancer Metab. 2018, 6, 1–15. [Google Scholar] [CrossRef]
- Kim, G.H.; Choi, S.Y.; Oh, T.I.; Kan, S.Y.; Kang, H.; Lee, S.; Oh, T.; Ko, H.M.; Lim, J.H. IDH1R132H causes resistance to HDAC inhibitors by increasing NANOG in glioblastoma cells. Int. J. Mol. Sci. 2019, 20, 2679. [Google Scholar] [CrossRef] [Green Version]
- Brière, J.J.; Favier, J.; Bénit, P.; El Ghouzzi, V.; Lorenzato, A.; Rabier, D.; Di Renzo, M.F.; Gimenez-Roqueplo, A.P.; Rustin, P. Mitochondrial succinate is instrumental for HIF1α nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum. Mol. Genet. 2005, 14, 3263–3269. [Google Scholar] [CrossRef] [Green Version]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.; Obara, N.; Ema, M.; Horie, M.; Naka, A.; Takahashi, S.; Imagawa, S. Antitumor effects of 2-Oxoglutarate through inhibition of angiogenesis in a murine tumor model. Cancer Sci. 2009, 100, 1639–1647. [Google Scholar] [CrossRef] [Green Version]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.; Narayaraswamy, R.; Tong, S.; Wang, F.; et al. Vorasidenib (AG-881): A first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Matteo, D.A.; Wells, G.; Luna, L.; Grunseth, A.; Zagnitko, O.; Scott, D.; Hoang, A.; Luthra, A.; Swairjo, M.; Schiffer, J.; et al. Inhibitor potency varies widely among tumor-relevant human isocitrate dehydrogenase 1 mutants. Biochem. J. 2019, 475, 3221–3238. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Wu, T.; Miao, L.; Mei, Y.; Wu, M. MiR-181a regulates lipid metabolism via IDH1. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Sasayama, T.; Tanaka, K.; Nakamizo, S.; Nishihara, M.; Mizukawa, K.; Kohta, M.; Koyama, J.; Miyake, S.; Taniguchi, M.; et al. MicroRNA-183 upregulates HIF-1α by targeting isocitrate dehydrogenase 2 (IDH2) in glioma cells. J. Neurooncol. 2013, 111, 273–283. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs. conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Ravandi, F.; Wilson, W.; Estey, E. Decitabine in older adults with acute myeloid leukemia: Why was the dream broken? J. Clin. Oncol. 2013, 31, 1795–1796. [Google Scholar] [CrossRef]
- Hadoux, J.; Favier, J.; Scoazec, J.-Y.; Leboulleux, S.; Ghuzlan, A.A.; Caramella, C.; Deandreis, D.; Borget, I.; Loriot, C.; Chougnet, C.; et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 2014, 135, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Songtao, Q.; Lei, Y.; Si, G.; Yanqing, D.; Huixia, H.; Xuelin, Z.; Lanxiao, W.; Fei, Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef]
- Su, R.; Lei, D.; Chenying, L.; Nachtergaele, S.; Wunderlich, M.; YQing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R-2HG exhibits anti-tumor activity by targeting FTO/M6 A/MYC/CEBPA signaling. Cell 2018, 172, 90–105.e23. [Google Scholar] [CrossRef] [Green Version]
- Dong, B.; Qiu, Z.; Wu, Y. Tackle epithelial-mesenchymal transition with epigenetic drugs in cancer. Front. Pharmacol. 2020, 11, 596239. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godel, M.; Ortone, G.; Anobile, D.P.; Pasino, M.; Randazzo, G.; Riganti, C.; Kopecka, J. Targeting Mitochondrial Oncometabolites: A New Approach to Overcome Drug Resistance in Cancer. Pharmaceutics 2021, 13, 762. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13050762
Godel M, Ortone G, Anobile DP, Pasino M, Randazzo G, Riganti C, Kopecka J. Targeting Mitochondrial Oncometabolites: A New Approach to Overcome Drug Resistance in Cancer. Pharmaceutics. 2021; 13(5):762. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13050762
Chicago/Turabian StyleGodel, Martina, Giacomo Ortone, Dario Pasquale Anobile, Martina Pasino, Giulio Randazzo, Chiara Riganti, and Joanna Kopecka. 2021. "Targeting Mitochondrial Oncometabolites: A New Approach to Overcome Drug Resistance in Cancer" Pharmaceutics 13, no. 5: 762. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13050762