
Systemically Administered Homing Peptide Targets Dystrophic Lesions and Delivers Transforming Growth Factor-β (TGFβ) Inhibitor to Attenuate Murine Muscular Dystrophy Pathology

 and
and
Abstract
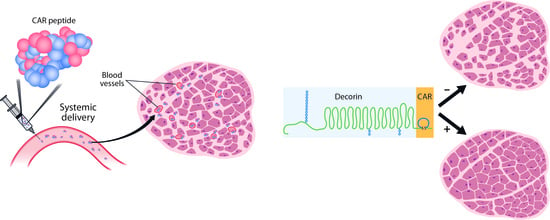
:
1. Introduction
2. Materials and Methods
2.1. Peptide Production
2.2. Recombinant Protein Production
2.3. Characterization of Recombinant Decorins
2.4. Heparan Sulfate Binding Analysis
2.5. Animals
2.6. Peptide Targeting
2.7. Treatment Schedules
2.8. Immunofluorescence
2.9. Histology
2.10. Evans Blue Dye (EBD) Uptake
2.11. Hydroxyproline (HOP) Assay
2.12. Gastrocnemius and Soleus Muscle Function Testing
2.13. Plethysmography
2.14. Statistics
3. Results
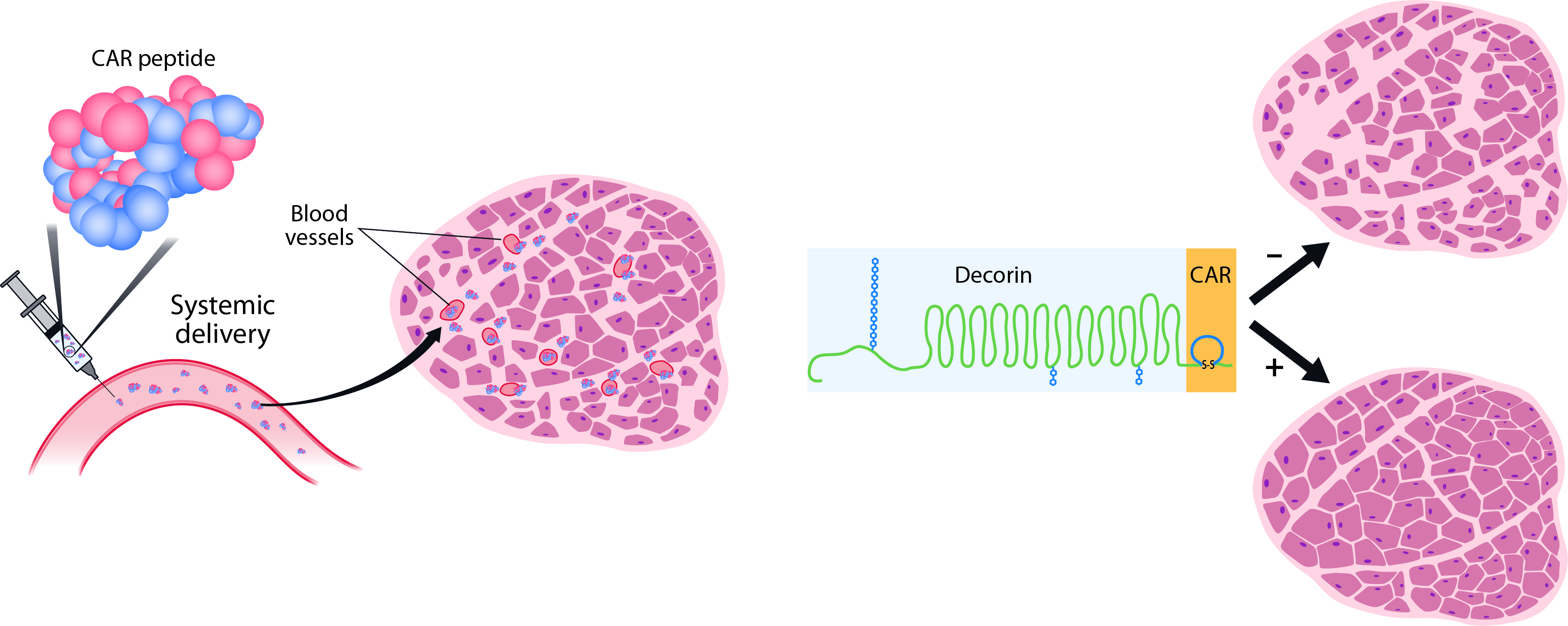
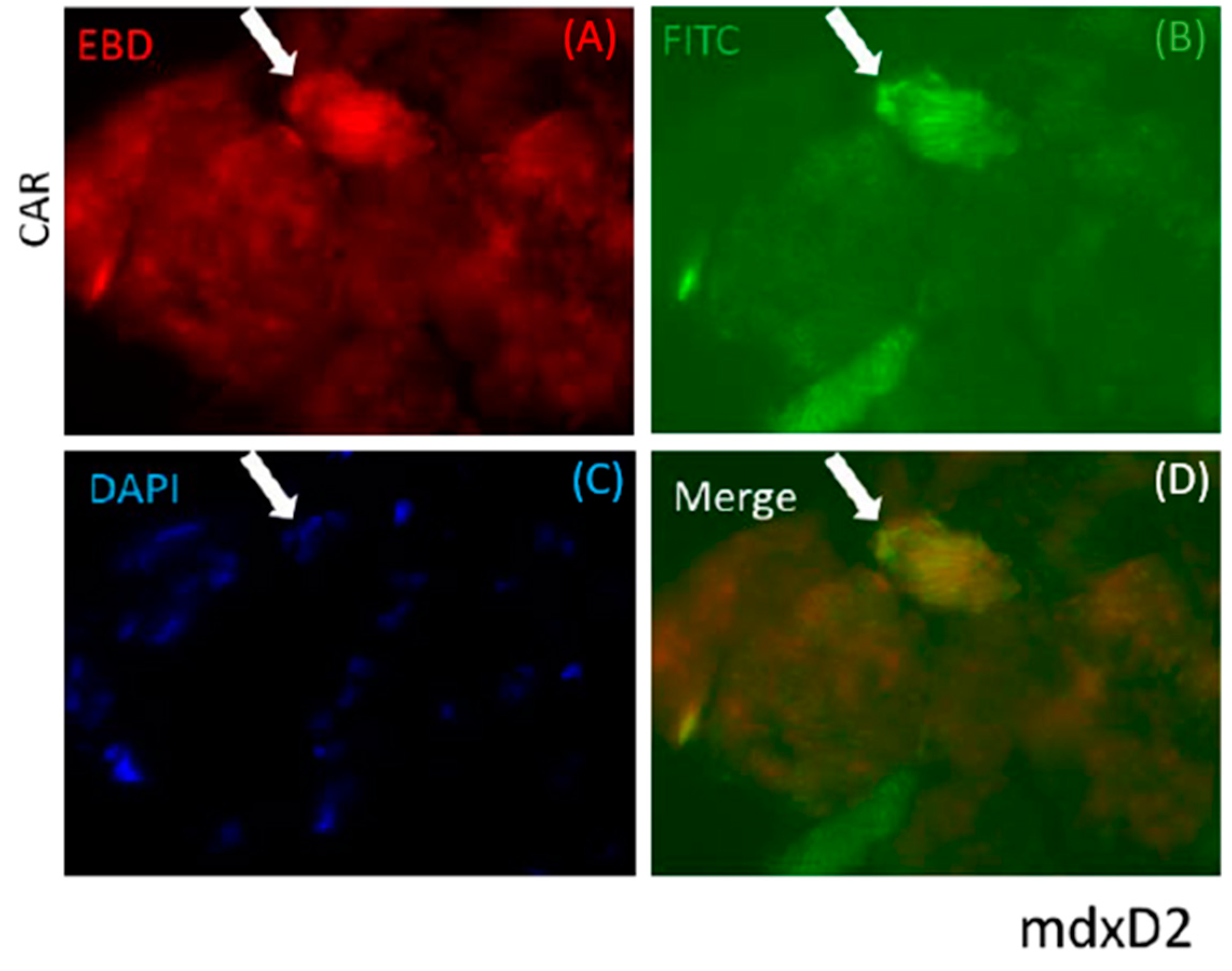
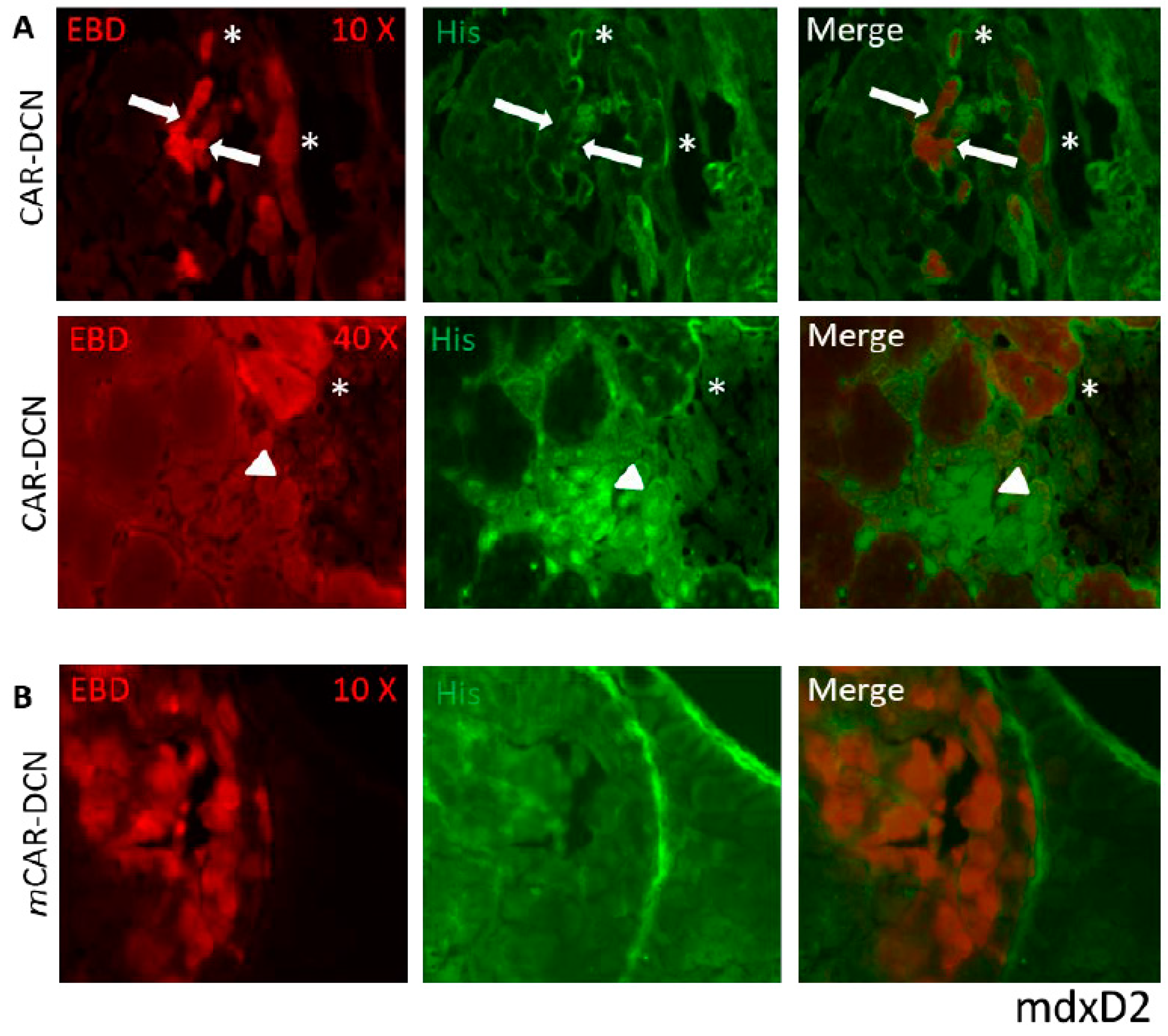
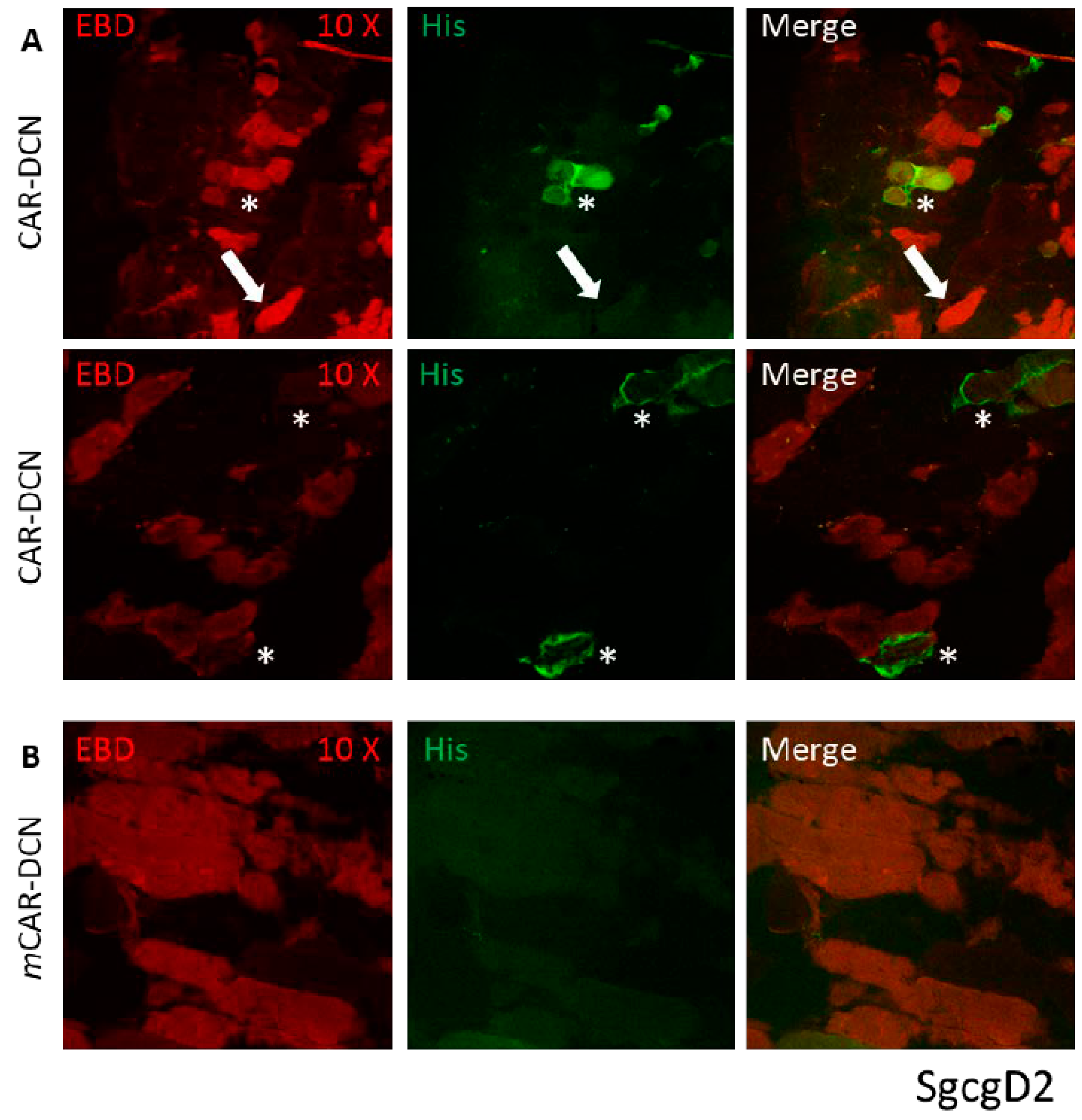
3.1. CAR Peptide Targets Muscular Dystrophy Lesions in Skeletal Muscle
3.2. Decorin Fused to CAR Peptide Maintains Its Biological Activity
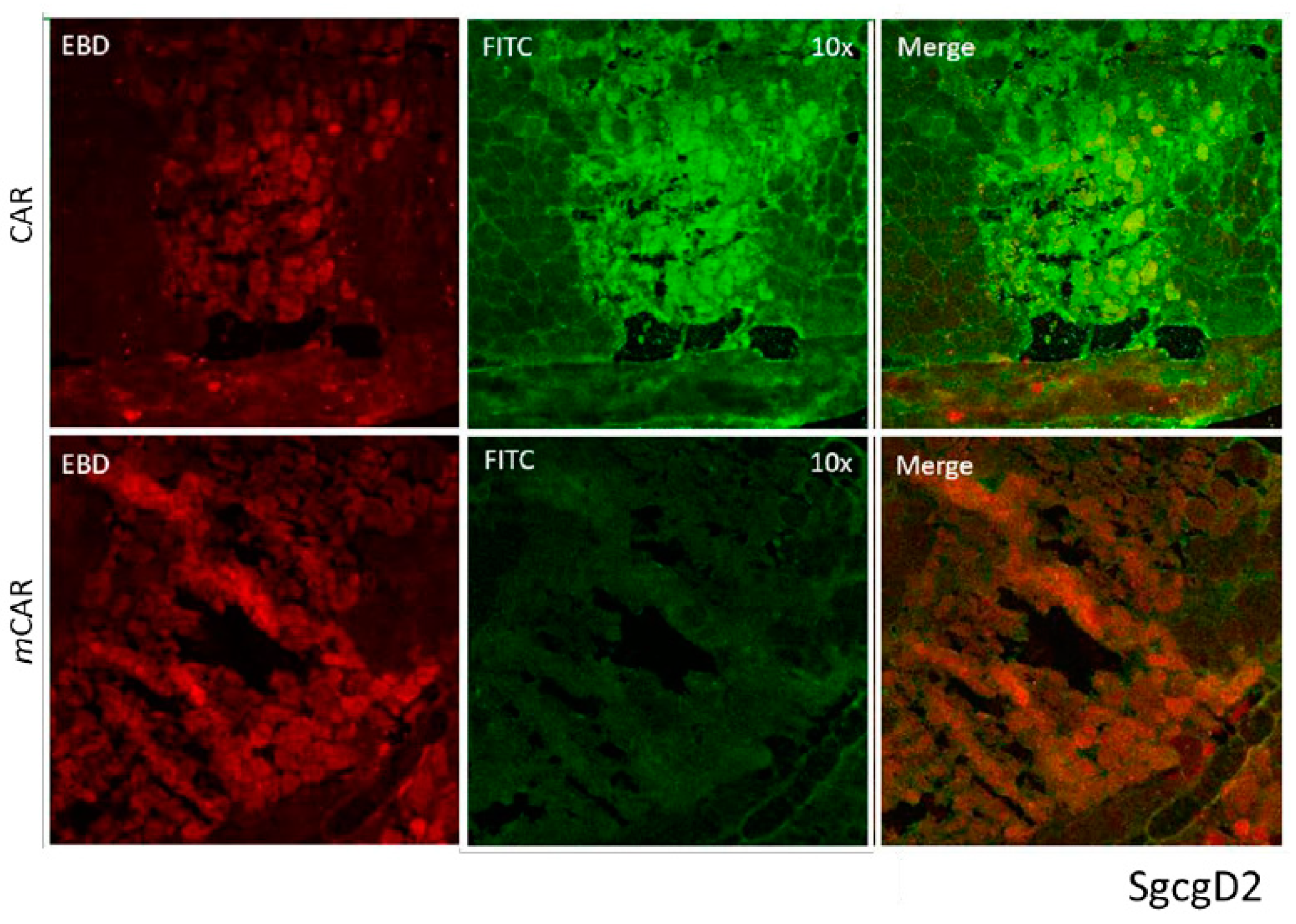
3.3. CAR-DCN Homes to Muscular Dystrophy Lesions
3.4. Treatment of Muscular Dystrophy with Recombinant DCNs
3.5. Targeted CAR-DCN Reduces TGFβ1 Signaling in Muscular Dystrophy
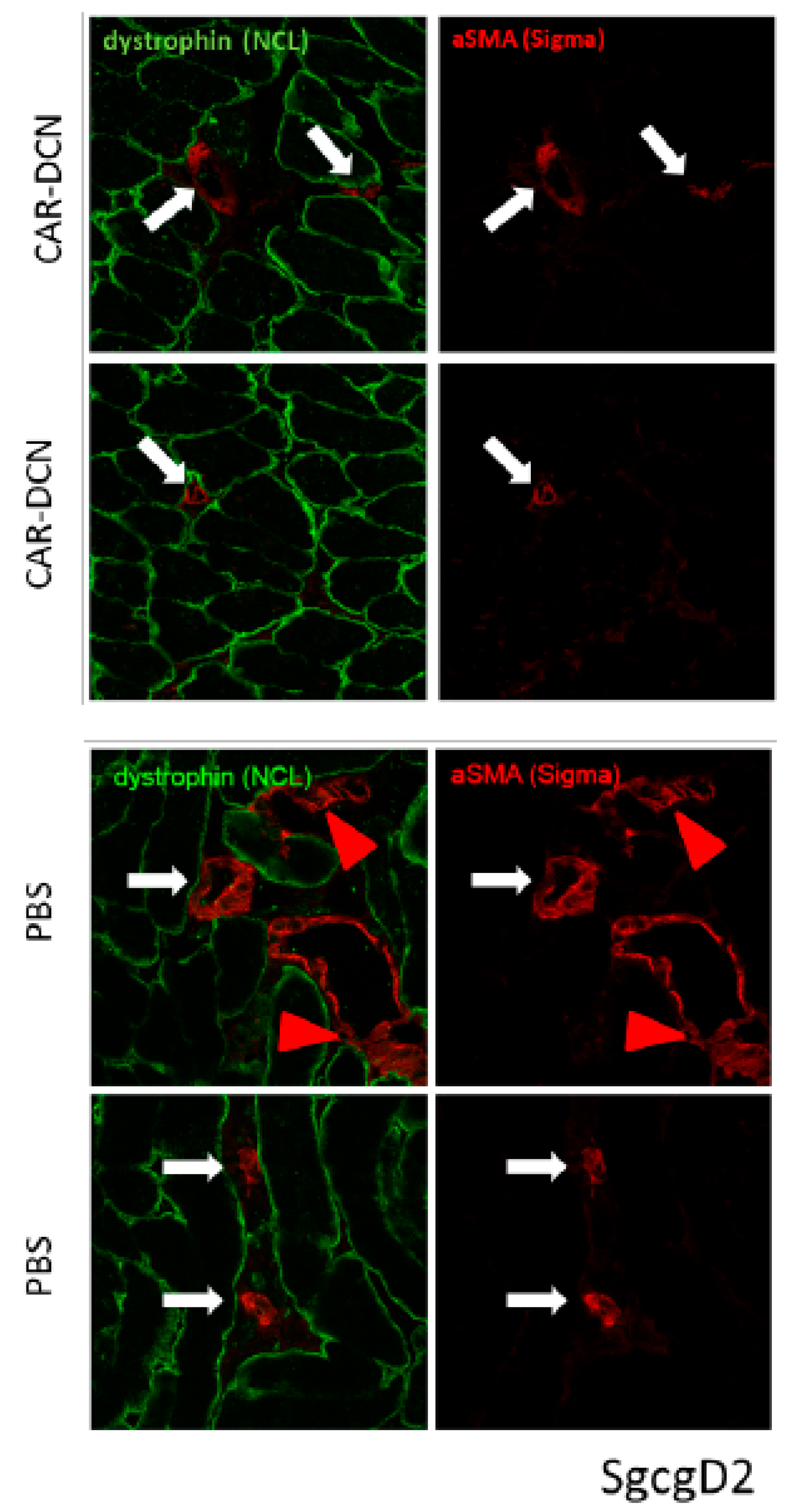
3.6. Targeted CAR-DCN Reduces α-Smooth Muscle Actin (αSMA) Staining in Muscular Dystrophy
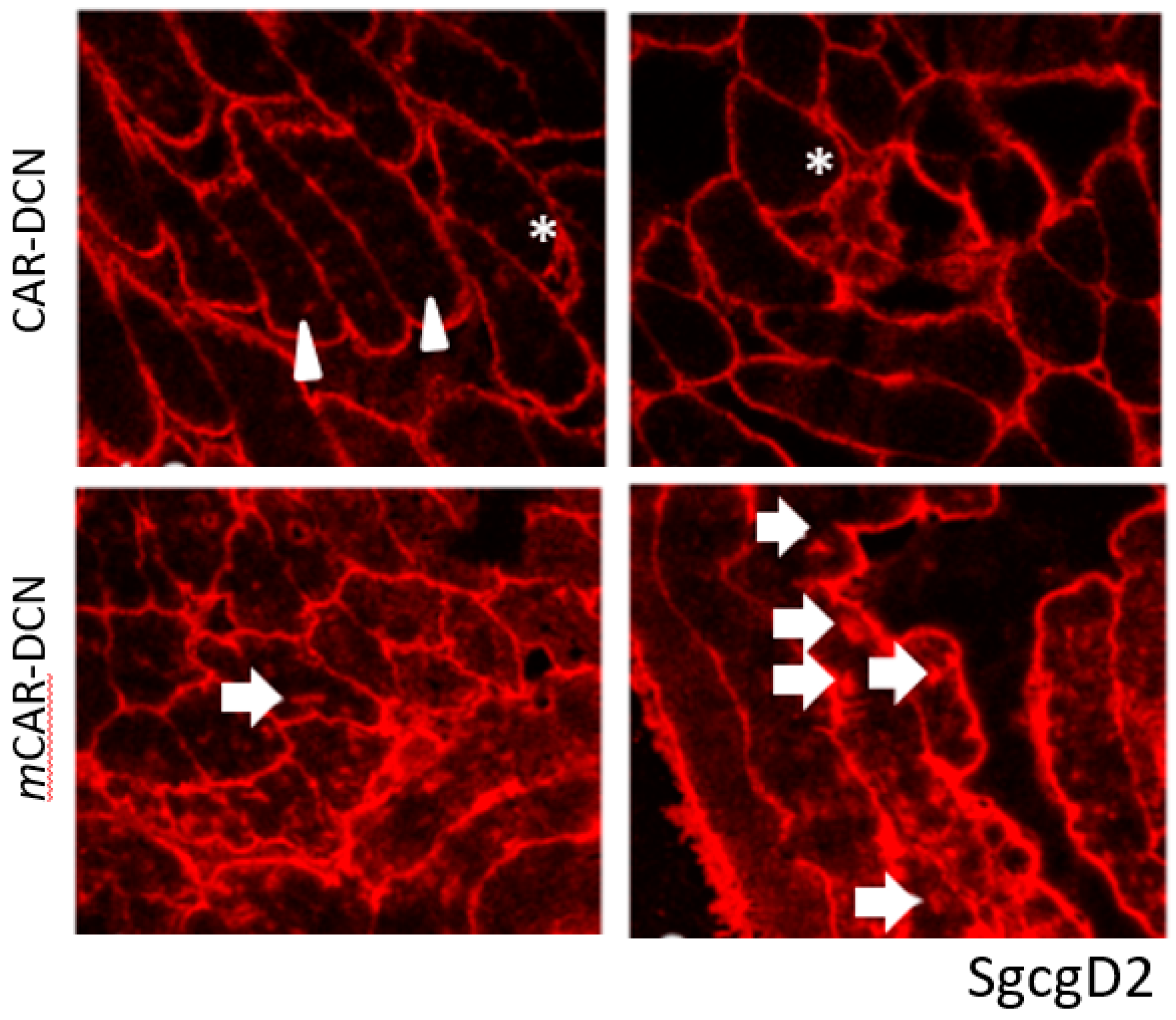
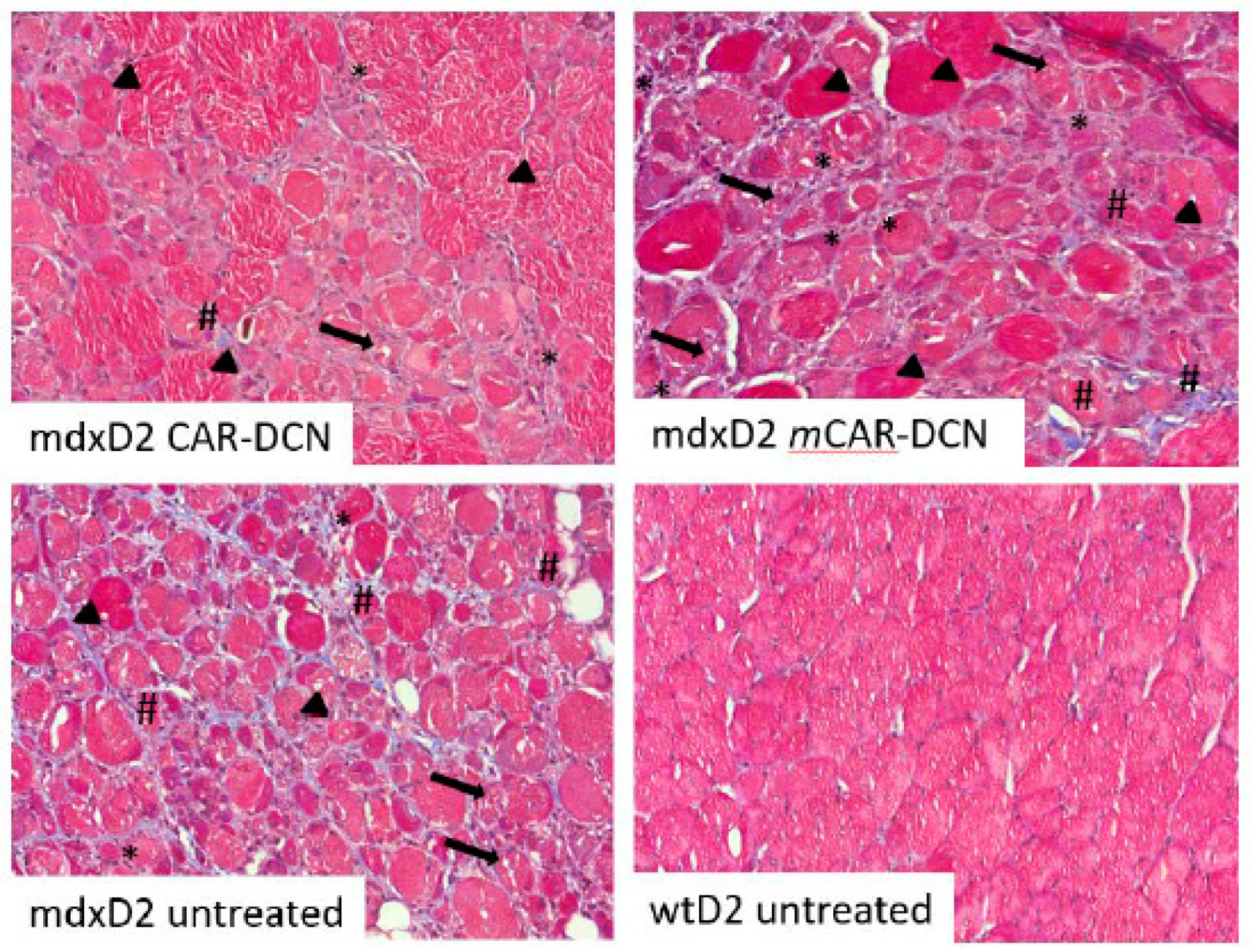
3.7. Skeletal Muscle Morphology Is Improved by CAR-DCN Treatment in Muscular Dystrophy
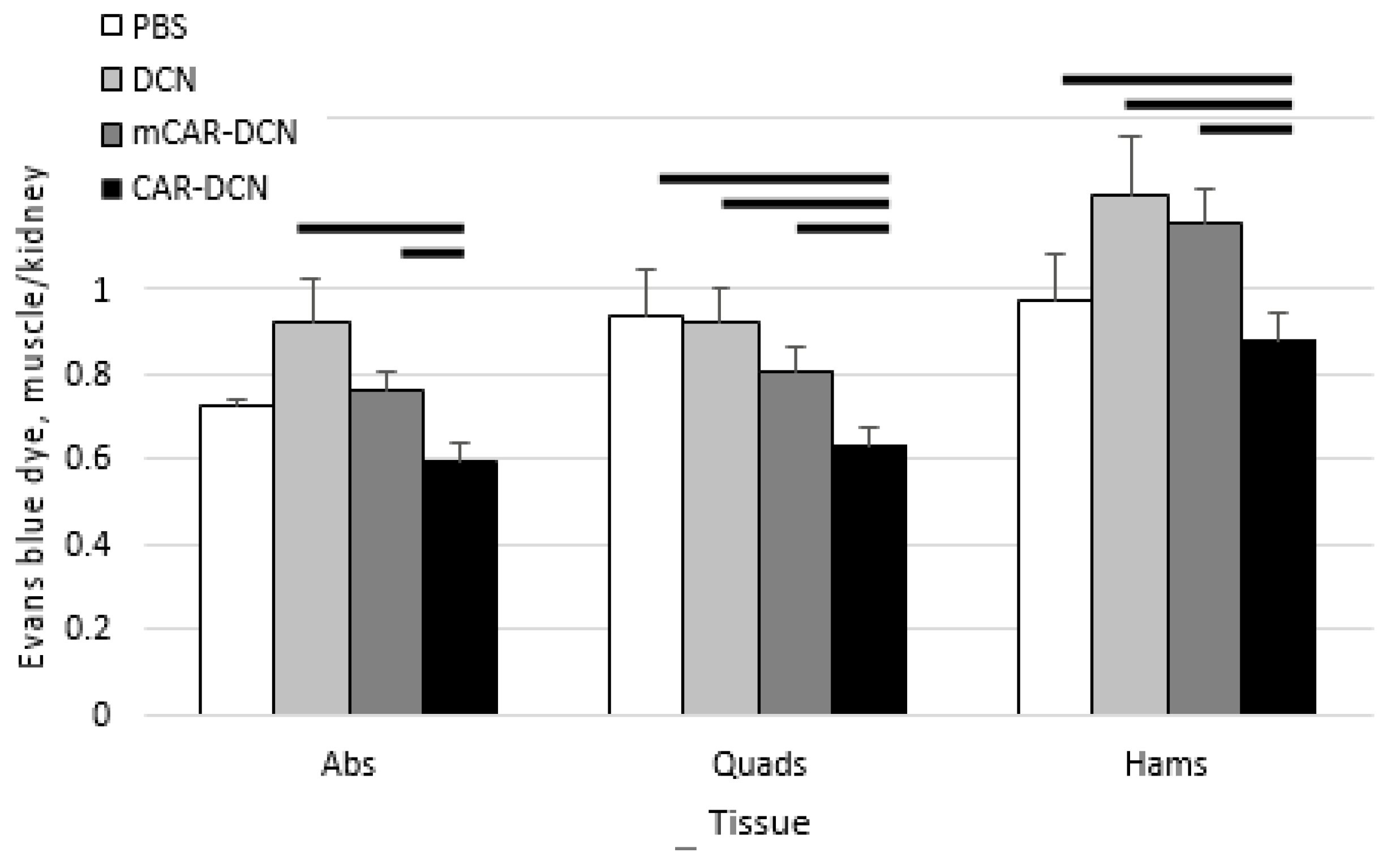
3.8. Pathological Sarcolemmal Permeability Is Significantly Reduced by CAR-DCN Treatment
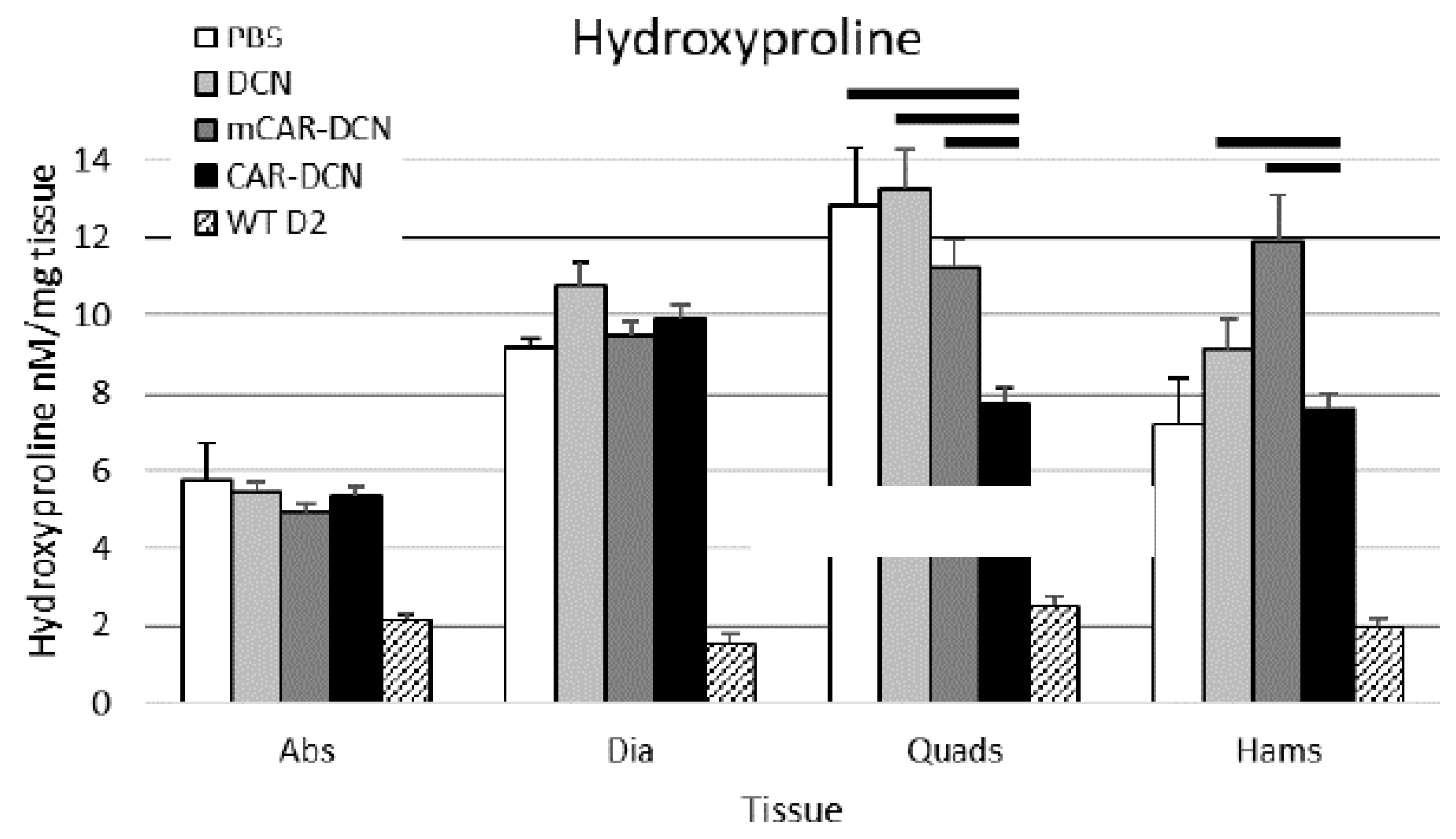
3.9. Muscular Collagen Deposition Is Decreased with CAR-DCN Treatment
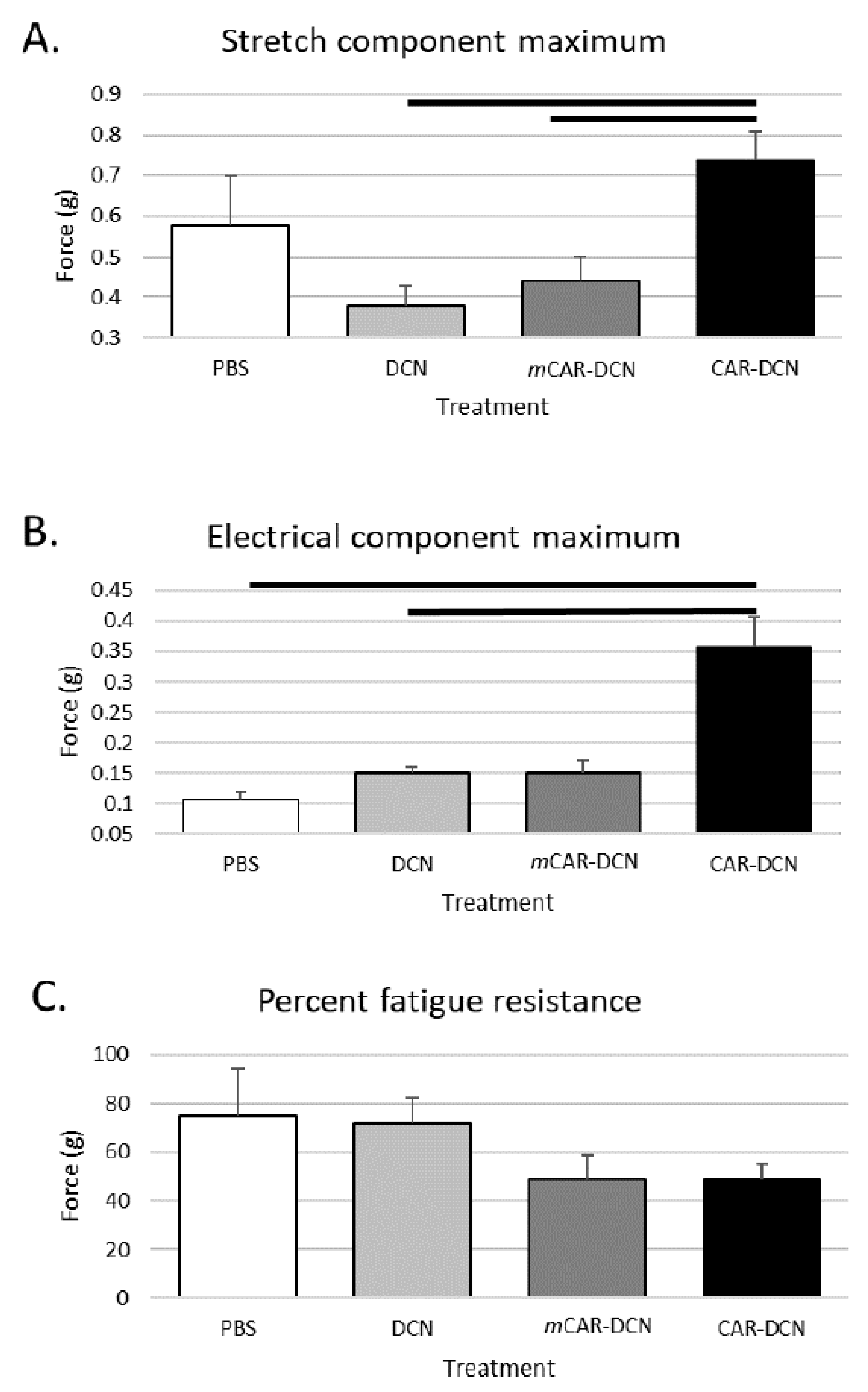
3.10. CAR-DCN Treatment Enhances Skeletal Muscle Function
3.11. No Respiratory Defect Is Detected in Young Muscular Dystrophy Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Munoz-Canoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.W.; Holley-Cuthrell, J.; Gonzalez-Vega, M.; Mull, A.J.; Heydemann, A. Biochemical and Functional Comparisons of mdx and Sgcg(−/−) Muscular Dystrophy Mouse Models. BioMed Res. Int. 2015, 2015, 131436. [Google Scholar] [CrossRef] [Green Version]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Burks, T.N.; Cohn, R.D. Role of TGF-β signaling in inherited and acquired myopathies. Skelet. Muscle 2011, 1, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceco, E.; McNally, E.M. Modifying muscular dystrophy through transforming growth factor-β. FEBS J. 2013, 280, 4198–4209. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.A.; Hunter, R.B.; Quigley, L.A.; Girgenrath, S.; Weber, W.D.; McCullough, J.A.; Dinardo, C.J.; Keefe, K.A.; Ceci, L.; Clayton, N.P.; et al. Inhibiting TGF-β activity improves respiratory function in mdx mice. Am. J. Pathol. 2011, 178, 2611–2621. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor β 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Ren, J.; Ten Dijke, P. Targeting TGFbeta signal transduction for cancer therapy. Signal Transduct Target Ther. 2021, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Lamar, K.M.; Bogdanovich, S.; Gardner, B.B.; Gao, Q.Q.; Miller, T.; Earley, J.U.; Hadhazy, M.; Vo, A.H.; Wren, L.; Molkentin, J.D.; et al. Overexpression of Latent TGFβ Binding Protein 4 in Muscle Ameliorates Muscular Dystrophy through Myostatin and TGFbeta. PLoS Genet. 2016, 12, e1006019. [Google Scholar] [CrossRef] [Green Version]
- Cuende, J.; Lienart, S.; Dedobbeleer, O.; van der Woning, B.; De Boeck, G.; Stockis, J.; Huygens, C.; Colau, D.; Somja, J.; Delvenne, P.; et al. Monoclonal antibodies against GARP/TGF-β1 complexes inhibit the immunosuppressive activity of human regulatory T cells In Vivo. Sci. Transl. Med. 2015, 7, 284ra256. [Google Scholar] [CrossRef]
- Pasqualini, R.; Ruoslahti, E. Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380, 364–366. [Google Scholar] [CrossRef]
- Ruoslahti, E. Vascular zip codes in angiogenesis and metastasis. Biochem. Soc. Trans. 2004, 32 Pt 3, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Järvinen, T.A.H.; Pemmari, T. Systemically Administered, Target-Specific, Multi-Functional Therapeutic Recombinant Proteins in Regenerative Medicine. Nanomaterials 2020, 10, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Järvinen, T.A.; Ruoslahti, E. Molecular changes in the vasculature of injured tissues. Am. J. Pathol. 2007, 171, 702–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urakami, T.; Järvinen, T.A.; Toba, M.; Sawada, J.; Ambalavanan, N.; Mann, D.; McMurtry, I.; Oka, M.; Ruoslahti, E.; Komatsu, M. Peptide-directed highly selective targeting of pulmonary arterial hypertension. Am. J. Pathol. 2011, 178, 2489–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toba, M.; Alzoubi, A.; O’Neill, K.; Abe, K.; Urakami, T.; Komatsu, M.; Alvarez, D.; Järvinen, T.A.; Mann, D.; Ruoslahti, E.; et al. A novel vascular homing peptide strategy to selectively enhance pulmonary drug efficacy in pulmonary arterial hypertension. Am. J. Pathol. 2014, 184, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Russo, V.; Zeglinski, M.R.; Sellers, S.L.; Wu, Z.; Oram, C.; Santacruz, S.; Merkulova, Y.; Turner, C.; Tauh, K.; et al. Recombinant Decorin Fusion Protein Attenuates Murine Abdominal Aortic Aneurysm Formation and Rupture. Sci. Rep. 2017, 7, 15857. [Google Scholar] [CrossRef]
- Rashid, J.; Nahar, K.; Raut, S.; Keshavarz, A.; Ahsan, F. Fasudil and DETA NONOate, Loaded in a Peptide-Modified Liposomal Carrier, Slow PAH Progression upon Pulmonary Delivery. Mol. Pharm. 2018, 15, 1755–1765. [Google Scholar] [CrossRef]
- Gupta, N.; Rashid, J.; Nozik-Grayck, E.; McMurtry, I.F.; Stenmark, K.R.; Ahsan, F. Cocktail of Superoxide Dismutase and Fasudil Encapsulated in Targeted Liposomes Slows PAH Progression at a Reduced Dosing Frequency. Mol. Pharm. 2017, 14, 830–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Al-Saikhan, F.I.; Patel, B.; Rashid, J.; Ahsan, F. Fasudil and SOD packaged in peptide-studded-liposomes: Properties, pharmacokinetics and ex-vivo targeting to isolated perfused rat lungs. Int. J. Pharm. 2015, 488, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Patel, B.; Nahar, K.; Ahsan, F. Cell permeable peptide conjugated nanoerythrosomes of fasudil prolong pulmonary arterial vasodilation in PAH rats. Eur. J. Pharm. Biopharm. 2014, 88, 1046–1055. [Google Scholar] [CrossRef] [Green Version]
- Nahar, K.; Absar, S.; Gupta, N.; Kotamraju, V.R.; McMurtry, I.F.; Oka, M.; Komatsu, M.; Nozik-Grayck, E.; Ahsan, F. Peptide-coated liposomal fasudil enhances site specific vasodilation in pulmonary arterial hypertension. Mol. Pharm. 2014, 11, 4374–4384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Järvinen, T.A.; Ruoslahti, E. Target-seeking antifibrotic compound enhances wound healing and suppresses scar formation in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 21671–21676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshavarz, A.; Alobaida, A.; McMurtry, I.F.; Nozik-Grayck, E.; Stenmark, K.R.; Ahsan, F. CAR, A Homing Peptide, Prolongs Pulmonary Preferential Vasodilation by Increasing Pulmonary Retention and Reducing Systemic Absorption of Liposomal Fasudil. Mol. Pharm. 2019, 16, 3414–3429. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, L.; Huang, H.; Yu, Y.; Wang, J.; Yu, Y.; Li, K.; Li, Y.; Tian, T.; Chen, F. CAR (CARSKNKDC) Peptide Modified ReNcell-Derived Extracellular Vesicles as a Novel Therapeutic Agent for Targeted Pulmonary Hypertension Therapy. Hypertension 2020, 76, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Border, W.A.; Noble, N.A.; Yamamoto, T.; Harper, J.R.; Yamaguchi, Y.; Pierschbacher, M.D.; Ruoslahti, E. Natural inhibitor of transforming growth factor-β protects against scarring in experimental kidney disease. Nature 1992, 360, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Mann, D.M.; Ruoslahti, E. Negative regulation of transforming growth factor-β by the proteoglycan decorin. Nature 1990, 346, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Border, W.A.; Ruoslahti, E. Transforming growth factor-β in disease: The dark side of tissue repair. J. Clin. Investig. 1992, 90, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vial, C.; Gutierrez, J.; Santander, C.; Cabrera, D.; Brandan, E. Decorin interacts with connective tissue growth factor (CTGF)/CCN2 by LRR12 inhibiting its biological activity. J. Biol. Chem. 2011, 286, 24242–24252. [Google Scholar] [CrossRef] [Green Version]
- Buraschi, S.; Neill, T.; Iozzo, R.V. Decorin is a devouring proteoglycan: Remodeling of intracellular catabolism via autophagy and mitophagy. Matrix Biol. 2019, 75–76, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Goldoni, S.; Humphries, A.; Nystrom, A.; Sattar, S.; Owens, R.T.; McQuillan, D.J.; Ireton, K.; Iozzo, R.V. Decorin is a novel antagonistic ligand of the Met receptor. J. Cell Biol. 2009, 185, 743–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishioka, Y.; Thomas, M.; Wakamatsu, J.; Hattori, A.; Sharma, M.; Kambadur, R.; Nishimura, T. Decorin enhances the proliferation and differentiation of myogenic cells through suppressing myostatin activity. J. Cell Physiol. 2008, 215, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Järvinen, T.A.H.; Rashid, J.; May, U.; Valmari, T.; Ahsan, F. Systemic targeted delivery of multi-functional recombinant proteins and nanoparticles in regenerative medicine. ACS Biomater. Sci. Eng. 2017, 3, 1273–1282. [Google Scholar] [CrossRef] [Green Version]
- Järvinen, T.A. Design of target-seeking antifibrotic compounds. Methods Enzymol. 2012, 509, 243–261. [Google Scholar] [PubMed]
- Cochran, S.; Li, C.P.; Ferro, V. A surface plasmon resonance-based solution affinity assay for heparan sulfate-binding proteins. Glycoconj. J. 2009, 26, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Heydemann, A.; Huber, J.M.; Demonbreun, A.; Hadhazy, M.; McNally, E.M. Genetic background influences muscular dystrophy. Neuromuscul. Disord. 2005, 15, 601–609. [Google Scholar] [CrossRef]
- Seidler, D.G.; Goldoni, S.; Agnew, C.; Cardi, C.; Thakur, M.L.; Owens, R.T.; McQuillan, D.J.; Iozzo, R.V. Decorin protein core inhibits in vivo cancer growth and metabolism by hindering epidermal growth factor receptor function and triggering apoptosis via caspase-3 activation. J. Biol. Chem. 2006, 281, 26408–26418. [Google Scholar] [CrossRef] [Green Version]
- Goldoni, S.; Iozzo, R.V. Tumor microenvironment: Modulation by decorin and related molecules harboring leucine-rich tandem motifs. Int. J. Cancer 2008, 123, 2473–2479. [Google Scholar] [CrossRef]
- Ma, W.; Cai, S.; Du, J.; Tan, Y.; Chen, H.; Guo, Z.; Hu, H.; Fang, R.; Cai, S. SDF-1/54-DCN: A novel recombinant chimera with dual inhibitory effects on proliferation and chemotaxis of tumor cells. Biol. Pharm. Bull. 2008, 31, 1086–1091. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, L.R.; Schonherr, E.; Waddington, R.; Niland, S.; Seidler, D.G.; Aeschlimann, D.; Eble, J.A. Decorin regulates endothelial cell motility on collagen I through activation of insulin-like growth factor I receptor and modulation of α2β1 integrin activity. J. Biol. Chem. 2008, 283, 17406–17415. [Google Scholar] [CrossRef] [Green Version]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93. [Google Scholar] [CrossRef]
- Farini, A.; Gowran, A.; Bella, P.; Sitzia, C.; Scopece, A.; Castiglioni, E.; Rovina, D.; Nigro, P.; Villa, C.; Fortunato, F.; et al. Fibrosis Rescue Improves Cardiac Function in Dystrophin-Deficient Mice and Duchenne Patient-Specific Cardiomyocytes by Immunoproteasome Modulation. Am. J. Pathol. 2019, 189, 339–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quattrocelli, M.; Spencer, M.J.; McNally, E.M. Outside in: The matrix as a modifier of muscular dystrophy. Biochem. Biophys. Acta Mol. Cell Res. 2017, 1864, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Howard, Z.M.; Lowe, J.; Blatnik, A.J.; Roberts, D.; Burghes, A.H.M.; Bansal, S.S.; Rafael-Fortney, J.A. Early Inflammation in Muscular Dystrophy Differs between Limb and Respiratory Muscles and Increases with Dystrophic Severity. Am. J. Pathol. 2021, 191, 730–747. [Google Scholar] [CrossRef] [PubMed]
- Järvinen, T.A.H.; Ruoslahti, E. Generation of a multi-functional, target organ-specific, anti-fibrotic molecule by molecular engineering of the extracellular matrix protein, decorin. Br. J. Pharmacol. 2019, 176, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Demonbreun, A.R.; Allen, M.V.; Warner, J.L.; Barefield, D.Y.; Krishnan, S.; Swanson, K.E.; Earley, J.U.; McNally, E.M. Enhanced Muscular Dystrophy from Loss of Dysferlin Is Accompanied by Impaired Annexin A6 Translocation after Sarcolemmal Disruption. Am. J. Pathol. 2016, 186, 1610–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Putten, M.; Putker, K.; Overzier, M.; Adamzek, W.A.; Pasteuning-Vuhman, S.; Plomp, J.J.; Aartsma-Rus, A. Natural disease history of the D2-mdx mouse model for Duchenne muscular dystrophy. FASEB J. 2019, 33, 8110–8124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coley, W.D.; Bogdanik, L.; Vila, M.C.; Yu, Q.; Van Der Meulen, J.H.; Rayavarapu, S.; Novak, J.S.; Nearing, M.; Quinn, J.L.; Saunders, A.; et al. Effect of genetic background on the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 2016, 25, 130–145. [Google Scholar] [CrossRef] [Green Version]
- Vohra, R.; Batra, A.; Forbes, S.C.; Vandenborne, K.; Walter, G.A. Magnetic Resonance Monitoring of Disease Progression in mdx Mice on Different Genetic Backgrounds. Am. J. Pathol. 2017, 187, 2060–2070. [Google Scholar] [CrossRef] [Green Version]
- Heydemann, A.; Ceco, E.; Lim, J.E.; Hadhazy, M.; Ryder, P.; Moran, J.L.; Beier, D.R.; Palmer, A.A.; McNally, E.M. Latent TGF-β-binding protein 4 modifies muscular dystrophy in mice. J. Clin. Investig. 2009, 119, 3703–3712. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, L.E.; Williams, J.E.; Deering, M.; Brazeau, D.; Koury, S.; Martinez, D.A. Localization and early time course of TGF-β1 mRNA expression in dystrophic muscle. Muscle Nerve. 2004, 30, 645–653. [Google Scholar] [CrossRef]
- Bernasconi, P.; Di Blasi, C.; Mora, M.; Morandi, L.; Galbiati, S.; Confalonieri, P.; Cornelio, F.; Mantegazza, R. Transforming growth factor-β1 and fibrosis in congenital muscular dystrophies. Neuromuscul. Disord. 1999, 9, 28–33. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Zhu, J.; Sun, B.; Branca, M.; Tang, Y.; Foster, W.; Xiao, X.; Huard, J. Decorin gene transfer promotes muscle cell differentiation and muscle regeneration. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1616–1622. [Google Scholar] [CrossRef]
- Wagner, K.R.; Liu, X.; Chang, X.; Allen, R.E. Muscle regeneration in the prolonged absence of myostatin. Proc. Natl. Acad. Sci. USA 2005, 102, 2519–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.L.; Walton, K.L.; Hagg, A.; Colgan, T.D.; Johnson, K.; Qian, H.; Gregorevic, P.; Harrison, C.A. Specific targeting of TGF-β family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E5266–E5275. [Google Scholar] [CrossRef] [Green Version]
- Estrellas, K.M.; Chung, L.; Cheu, L.A.; Sadtler, K.; Majumdar, S.; Mula, J.; Wolf, M.T.; Elisseeff, J.H.; Wagner, K.R. Biological scaffold-mediated delivery of myostatin inhibitor promotes a regenerative immune response in an animal model of Duchenne muscular dystrophy. J. Biol. Chem. 2018, 293, 15594–15605. [Google Scholar] [CrossRef] [Green Version]
- Crone, M.; Mah, J.K. Current and Emerging Therapies for Duchenne Muscular Dystrophy. Curr. Treat. Options Neurol. 2018, 20, 31. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Haginoya, K.; Wu, Y.; Chiba, Y.; Nakanishi, T.; Onuma, A.; Sato, Y.; Takigawa, M.; Iinuma, K.; Tsuchiya, S. Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J. Neurol. Sci. 2008, 267, 48–56. [Google Scholar] [CrossRef]
- Song, Y.; Yao, S.; Liu, Y.; Long, L.; Yang, H.; Li, Q.; Liang, J.; Li, X.; Lu, Y.; Zhu, H.; et al. Expression levels of TGF-β1 and CTGF are associated with the severity of Duchenne muscular dystrophy. Exp. Ther. Med. 2017, 13, 1209–1214. [Google Scholar] [CrossRef] [Green Version]
- Morales, M.G.; Cabello-Verrugio, C.; Santander, C.; Cabrera, D.; Goldschmeding, R.; Brandan, E. CTGF/CCN-2 over-expression can directly induce features of skeletal muscle dystrophy. J. Pathol. 2011, 225, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Karstoft, K.; Pedersen, B.K. Skeletal muscle as a gene regulatory endocrine organ. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, A.P.; Cooper, R.G. The role of myokines in muscle health and disease. Curr. Opin. Rheumatol. 2016, 28, 661–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemmari, T.; Ivanova, L.; May, U.; Lingasamy, P.; Pasternack, A.; Prince, S.; Ritvos, O.; Makkapati, S.; Teesalu, T.; Cairo, M.S.; et al. Exposed CendR domain in homing peptide yields skin-targeted therapeutic in epidermolysis bullosa. Mol. Ther. 2020, 28, 1833–1845. [Google Scholar] [CrossRef]
- Rider, C.C. Heparin/heparan sulphate binding in the TGF-β cytokine superfamily. Biochem. Soc. Trans. 2006, 34 Pt 3, 458–460. [Google Scholar] [CrossRef] [Green Version]
- Rider, C.C.; Mulloy, B. Heparin, Heparan Sulphate and the TGF-β Cytokine Superfamily. Molecules 2017, 22, 713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, M.M.; Briquez, P.S.; Guc, E.; Tortelli, F.; Kilarski, W.W.; Metzger, S.; Rice, J.J.; Kuhn, G.A.; Muller, R.; Swartz, M.A.; et al. Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science 2014, 343, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Ehara, Y.; Li, J.; Inada, K.; Ohno, K. Protein-Anchoring Therapy of Biglycan for Mdx Mouse Model of Duchenne Muscular Dystrophy. Hum. Gene Ther. 2017, 28, 428–436. [Google Scholar] [CrossRef]
- Amenta, A.R.; Yilmaz, A.; Bogdanovich, S.; McKechnie, B.A.; Abedi, M.; Khurana, T.S.; Fallon, J.R. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc. Natl. Acad. Sci. USA 2011, 108, 762–767. [Google Scholar] [CrossRef] [Green Version]
- Fallon, J.R.; McNally, E.M. Non-Glycanated Biglycan and LTBP4: Leveraging the extracellular matrix for Duchenne Muscular Dystrophy therapeutics. Matrix Biol. 2018, 68–69, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Krusius, T.; Ruoslahti, E. Primary structure of an extracellular matrix proteoglycan core protein deduced from cloned cDNA. Proc. Natl. Acad. Sci. USA 1986, 83, 7683–7687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, D.M.; Yamaguchi, Y.; Bourdon, M.A.; Ruoslahti, E. Analysis of glycosaminoglycan substitution in decorin by site-directed mutagenesis. J. Biol. Chem. 1990, 265, 5317–5323. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Peptides & Recombinant Proteins |
|---|---|
| CAR | CARSKNKDC |
| mCAR | CAQSNNKDC |
| DCN | 5′-hDCN—HHHHHH-3′ |
| CAR-DCN | 5′-hDCN—CARSKNKDC—HHHHHH-3′ |
| mCAR-DCN | 5′-hDCN—CAQSNNKDC—HHHHHH-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, A.; May, U.; Prince, S.N.; Järvinen, T.A.H.; Heydemann, A. Systemically Administered Homing Peptide Targets Dystrophic Lesions and Delivers Transforming Growth Factor-β (TGFβ) Inhibitor to Attenuate Murine Muscular Dystrophy Pathology. Pharmaceutics 2021, 13, 1506. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13091506
Iqbal A, May U, Prince SN, Järvinen TAH, Heydemann A. Systemically Administered Homing Peptide Targets Dystrophic Lesions and Delivers Transforming Growth Factor-β (TGFβ) Inhibitor to Attenuate Murine Muscular Dystrophy Pathology. Pharmaceutics. 2021; 13(9):1506. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13091506
Chicago/Turabian StyleIqbal, Aqsa, Ulrike May, Stuart N. Prince, Tero A.H. Järvinen, and Ahlke Heydemann. 2021. "Systemically Administered Homing Peptide Targets Dystrophic Lesions and Delivers Transforming Growth Factor-β (TGFβ) Inhibitor to Attenuate Murine Muscular Dystrophy Pathology" Pharmaceutics 13, no. 9: 1506. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13091506