2. Materials and Methods
2.1. General
All the chemicals and solvents used in the organic synthesis were purchased from Sigma-Aldrich, TCI, or Acros organics and were used without further purification. Baicalein, trifluoroacetic acid (TFA), glipizide, 7-EC, enalapril maleate, formic acid, MeOH, 1-octanol, UDPGA, NADPH, PAPS, GSH, hydroxypropyl-β-cyclodextrin (HPβCD), MgCl2, and alamethicin were purchased from Sigma-Aldrich. Tris-hydrochloride (Tris-HCl) was purchased from Roche Diagnostics Gmb. HPLC-grade water and acetonitrile were purchased from Avantor Performance Materials. Pooled mouse liver S9 fractions were purchased from BD Biosciences. Reactions were monitored by TLC on 0.25 mm Merck precoated silica gel plates (60 F254). Reaction progress was monitored by TLC analysis using a UV lamp and/or KMnO4 staining for detection purposes. Column chromatography was performed on silica gel (230–400 mesh, Merck, Darmstadt, Germany). 1H and 13C NMR spectra were recorded at room temperature (298 K) in CDCl3 (7.26/77.16 ppm) or DMSO-d6 (2.50/39.50 ppm) on Bruker Ultrashield 600 MHz Plus spectrometer and referenced to an internal solvent. Chemical shifts are reported in parts per million (ppm). Coupling constants (J) are given in Hertz. Splitting patterns are indicated as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad for 1H NMR data. High resolution mass spectra (HRMS) were recorded on an Agilent 6530 Accurate mass Q-TOF LC/MS spectrometer. Low resolution mass spectra (LRMS) analyses were obtained from an API 150EX ESI-MS spectrometer. The purity of final compounds was measured by analytical reverse-phase HPLC on an Agilent 1260 Infinity (Agilent) with a C18 column (Phenomenex, 150 mm × 4.6 mm, 3 μm, 110 Å).
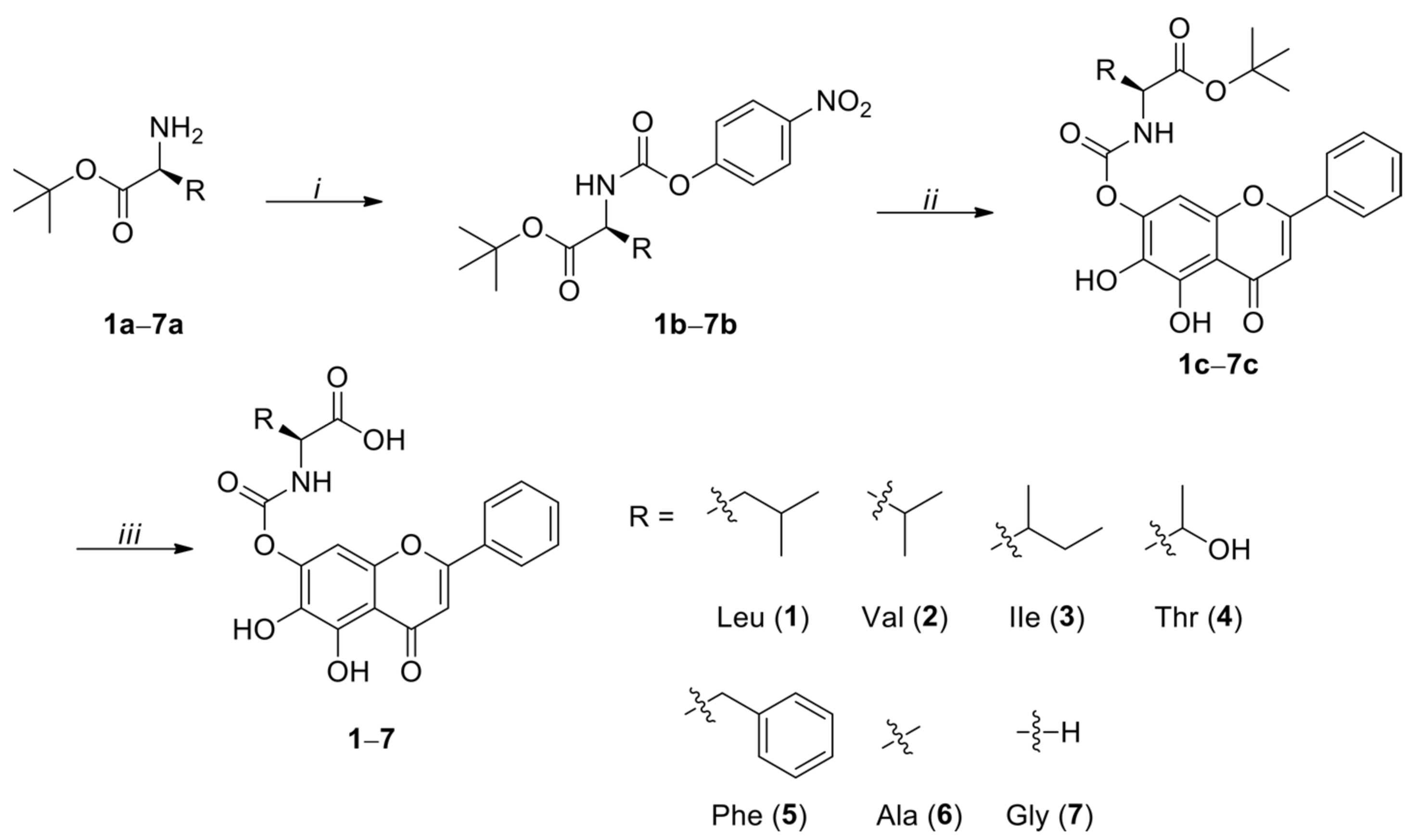
2.2. General Procedure for the Preparation of Activated 4-Nitrophenyl Urethanes (1b–7b)
N,N-Diisopropylethylamine (DIPEA, 2.86 mL, 16.4 mmol, 2.0 eq.) was added to a stirred a solution of amino acid tert-butyl ester 1a–7a (8.2 mmol, 1.0 eq.) and bis(4-nitrophenyl) carbonate (2.74 g, 9.0 mmol, 1.1 eq.) in THF (15 mL) at 0 °C, and stirring was continued for 12 h at room temperature. The reaction mixture was evaporated, and the residue was purified by silica gel column chromatography eluted with dichloromethane (DCM)/MeOH (100:1 to 50:1, v/v) to give 4-nitrophenyl urethanes 1b–7b.
(S)-tert-Butyl 4-methyl-2-(((4-nitrophenoxy)carbonyl)amino)pentanoate (1b). Pale yellow oil, 52% yield. 1H NMR (600 MHz, CDCl3) δ 8.25 (d, J = 9.0 Hz, 2H), 7.34 (d, J = 9.0 Hz, 2H), 5.68 (d, J = 9.0 Hz, 1H), 4.24 (q, J = 4.8 Hz, 1H), 1.85–1.52 (m, 2H), 1.51 (s, 9H), 1.49 (s, 1H), 1.03 (d, J = 6.6 Hz, 3H), 0.96 (d, J = 6.6 Hz, 3H). LRMS (ESI) m/z 353.2 [M+H]+.
(S)-tert-Butyl 3-methyl-2-(((4-nitrophenoxy)carbonyl)amino)butanoate (2b). Pale yellow oil, 67% yield. 1H NMR (600 MHz, CDCl3) δ 8.24 (d, J = 9.0 Hz, 2H), 7.33 (d, J = 9.0 Hz, 2H), 5.56 (d, J = 9.0 Hz, 1H), 4.24 (q, J = 4.8 Hz, 1H), 2.28–2.21 (m, 1H), 1.51 (s, 9H), 1.03 (d, J = 6.6 Hz, 3H), 0.96 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 170.7, 155.9, 153.2, 144.6, 124.9, 121.6, 82.3, 67.8, 59.6, 31.3, 27.7, 25.5, 18.8, 17.3. LRMS (ESI) m/z 339.2 [M+H]+.
(2S,3R)-tert-Butyl 3-methyl-2-(((4-nitrophenoxy)carbonyl)amino)pentanoate (3b). Pale yellow oil, 64% yield. 1H NMR (600 MHz, CDCl3) δ 8.24 (d, J = 9.6 Hz, 2H), 7.33 (d, J = 9.6 Hz, 2H), 5.70 (d, J = 8.4 Hz, 1H), 4.27 (q, J = 4.8 Hz, 1H), 2.05–1.87 (m, 1H), 1.51 (s, 9H), 1.51–1.47 (m, 1H), 1.35–1.16 (m, 1H), 0.99 (d, J = 7.2 Hz, 3H), 0.97 (d, J = 7.2 Hz, 3H). LRMS (ESI) m/z 353.2 [M+H]+.
(2S,3S)-tert-Butyl 3-(tert-butoxy)-2-(((4-nitrophenoxy)carbonyl)amino)butanoate (4b). Pale yellow oil, 56% yield. 1H NMR (600 MHz, CDCl3) δ 8.24 (d, J = 5.4 Hz, 2H), 7.33 (d, J = 5.4 Hz, 2H), 5.88 (d, J = 9.0 Hz, 1H), 4.27 (q, J = 1.8 Hz, 1H), 4.11 (d, J = 1.8 Hz, 1H), 1.50 (s, 9H), 1.27 (d, J = 6.6 Hz, 3H), 1.19 (s, 9H). LRMS (ESI) m/z 341.1 [M+H]+.
(S)-tert-Butyl 2-(((4-nitrophenoxy)carbonyl)amino)-3-phenylpropanoate (5b). Pale yellow oil, 61% yield. 1H NMR (600 MHz, CDCl3) δ 8.24 (d, J = 7.2 Hz, 2H), 7.36–7.31 (m, 2H), 7.29–7.26 (m, 3H), 7.23–7.19 (m, 2H), 5.62 (d, J = 7.8 Hz, 1H), 4.27 (q, J = 2.4 Hz, 1H), 3.31–3.08 (m, 2H), 1.45 (s, 9H). LRMS (ESI) m/z 387.2 [M+H]+.
(S)-tert-Butyl 2-(((4-nitrophenoxy)carbonyl)amino)propanoate (6b). Pale yellow oil, 63% yield. 1H NMR (600 MHz, CDCl3) δ 8.23 (d, J = 9.0 Hz, 2H), 7.32 (d, J = 9.0 Hz, 2H), 5.86 (d, J = 7.2 Hz, 1H), 4.33–4.29 (m, 1H), 1.49(s, 9H), 1.47 (d, J = 7.2 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 171.7, 155.6, 152.4, 144.9, 125.2, 122.1, 82.7, 50.5, 28.0, 18.7. LRMS (ESI) m/z 311.1 [M+H]+.
tert-Butyl 2-(((4-nitrophenoxy)carbonyl)amino)acetate (7b). Pale yellow oil, 60% yield. 1H NMR (600 MHz, CDCl3) δ 8.19 (d, J = 10.2 Hz, 2H), 7.33 (d, J = 10.2 Hz, 2H), 6.26 (t, J = 6.0 Hz, 1H), 3.95 (d, J = 5.4 Hz, 2H), 1.49 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 168.5, 155.7, 153.2, 144.5, 124.8, 121.8, 82.3, 43.2, 27.7. LRMS (ESI) m/z 297.1 [M+H]+.
2.3. General Procedure for the Preparation of 7-N-Monosubstituted-Baicalein Carbamate Esters (1c–7c)
DIPEA (0.55 mL, 3.16 mmol, 2.0 eq.) was added to a stirred a solution of baicalein (1.58 mmol, 1.0 eq.) and activated 4-nitrophenyl urethane 1a–7a (1.58 mmol, 1.0 eq.) in THF (10 mL) at 0 °C, and stirring was continued for 12 h at room temperature, when TLC (DCM/MeOH, 20/1, v/v) indicated that reaction was complete. The reaction mixture was evaporated, and the residue was purified twice by silica gel column chromatography (DCM/acetone, 100/1 to 50/1, v/v, followed by DCM/MeOH, 100/1, v/v) to provide 7-N-monosubstituted-baicalein carbamate esters 1c–7c.
(S)-tert-Butyl 2-((((5,6-dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)-4-methylpentanoate (1c). Yellow powder, 78% yield. 1H NMR (600 MHz, DMSO-d6) δ 12.93 (s, 1H), 8.13–8.07 (m, 3H), 7.66–7.56 (m, 3H), 7.01 (s, 1H), 6.66 (s, 1H), 3.97–3.91 (m, 1H), 1.79–1.70 (m, 1H), 1.64–1.56 (m, 1H), 1.54–1.46 (m, 1H), 1.40 (s, 9H), 0.93 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 182.7, 172.1, 163.9, 158.1, 154.2, 153.9, 153.5, 132.6, 131.2, 129.6, 127.0, 123.4, 105.3, 104.4, 94.5, 81.0, 53.8, 28.1, 24.7, 23.2, 21.9. HRMS (ESI) m/z calculated for C26H29NO8– [M-H]–: 482.1823; found: 428.1828.
(S)-tert-Butyl 2-((((5,6-dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)-3-methylbutanoate (2c). Yellow powder, 47% yield. 1H NMR (600 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.10–8.06 (m, 2H), 8.04 (d, J = 8.4 Hz, 1H), 7.63–7.56 (m, 3H), 6.99 (s, 1H), 6.66 (s, 1H), 3.83 (q, J = 6.6 Hz, 1H), 2.10–2.04 (m, 1H), 1.44 (s, 9H), 0.96 (d, J = 5.4 Hz, 3H), 0.94 (d, J = 5.4 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 170.5, 163.5, 157.6, 153.8, 153.7, 153.0, 132.1, 130.7, 129.2, 126.5, 122.9, 104.8, 104.0, 94.1, 80.6, 60.6, 54.9, 30.1, 27.7, 18.9, 18.1. HRMS (ESI) m/z calculated for C25H27NO8– [M-H]–: 468.1664; found: 468.1645.
(2S,3R)-tert-Butyl 2-((((5,6-dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)-3-methylpentanoate (3c). Yellow powder, 69% yield. 1H NMR (600 MHz, DMSO-d6) δ 12.95 (s, 1H), 8.09 (d, J = 7.2 Hz, 2H), 8.06 (d, J = 8.4 Hz, 1H), 7.63–7.56 (m, 3H), 7.01 (s, 1H), 6.66 (s, 1H), 3.88 (q, J = 6.6 Hz, 1H), 1.89–1.73 (m, 1H), 1.44 (s, 9H), 1.38–1.21 (m, 2H), 0.92 (d, J = 7.2 Hz, 3H), 0.89 (d, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 170.5, 163.5, 157.6, 153.8, 153.7, 153.7, 153.0, 132.1, 130.7, 129.2, 126.5, 122.9, 104.8, 104.0, 94.1, 80.6, 59.5, 36.6, 27.7, 24.9, 15.4, 11.3. HRMS (ESI) m/z calculated for C26H29NO8– [M-H]–: 482.1820; found: 482.1798.
(2S,3R)-tert-Butyl 3-(tert-butoxy)-2-((((5,6-dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)butanoate (4c). Yellow powder, 60% yield. 1H NMR (600 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.07 (d, J = 7.2 Hz, 2H), 7.64–7.53 (m, 3H), 7.44 (d, J = 9.0 Hz, 1H), 6.99 (s, 1H), 6.64 (s, 1H), 4.06–4.00 (m, 1H), 3.97 (dd, J = 4.0 and 9.3 Hz, 1H), 1.42 (s, 9H), 1.14 (s, 9H), 1.13 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 169.3, 163.5, 157.6, 153.8, 153.6, 153.0, 132.1, 130.7, 129.1, 126.5, 122.9, 104.8, 104.0, 94.1, 80.9, 73.4, 67.2, 60.5, 28.3, 27.7, 19.9. HRMS (ESI) m/z calculated for C28H33NO9– [M-H]–: 526.2082; found: 526.2099.
(S)-tert-Butyl 2-((((5,6-dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)-3-phenylpropanoate (5c). Yellow powder, 66% yield. 1H NMR (600 MHz, DMSO-d6) δ 8.21 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 7.2 Hz, 1H), 7.63–7.54 (m, 3H), 7.32–7.21 (m, 5H), 6.99 (s, 1H), 6.63 (s, 1H), 4.13 (q, J = 8.4 Hz, 1H), 3.03–2.95 (m, 2H), 1.34 (s, 9H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 170.5, 163.4, 153.8, 153.3, 153.0, 137.3, 132.1, 130.7, 129.4, 129.3, 129.2, 128.2, 126.5, 126.5, 122.9, 104.8, 103.8, 94.1, 80.7, 56.5, 36.8, 27.5. HRMS (ESI) m/z calculated for C29H27NO8– [M-H]–: 516.1659; found: 516.1673.
(S)-tert-Butyl 2-((((5,6-dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)propanoate (6c). Yellow powder, 80% yield. 1H NMR (600 MHz, DMSO-d6) δ 12.93 (s, 1H), 8.14 (d, J = 7.2 Hz, 1H), 8.09 (d, J = 7.2 Hz, 1H), 7.65–7.56 (m, 3H), 7.01 (s, 1H), 6.66 (s, 1H), 3.99–3.93 (m, 1H), 1.42 (s, 9H), 1.31 (d, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 172.0, 163.5, 157.6, 153.7, 153.1, 153.0, 132.1, 130.7, 129.2, 126.5, 122.8, 104.8, 104.0, 94.0, 80.4, 50.3, 27.6, 17.1. HRMS (ESI) m/z calculated for C23H23NO8+ [M+H]+: 442.1497; found: 442.1507.
tert-Butyl 2-((((5,6-dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)acetate (7c). Yellow powder, 74% yield. 1H NMR (600 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.13–8.05 (m, 3H), 7.66–7.55 (m, 3H), 7.01 (s, 1H), 6.66 (s, 1H), 3.72 (d, J = 6.0 Hz, 2H), 1.43 (s, 9H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 168.9, 163.4, 157.5, 153.8, 153.7, 153.0, 132.1, 130.7, 129.1, 126.5, 122.8, 104.8, 103.9, 94.0, 80.7, 43.1, 27.7. HRMS (ESI) m/z calculated for C22H21NO8+ [M+Na]+: 450.1159; found: 450.1171.
2.4. General Procedure for the Tert-Butyl Ester Deprotection in Baicalein-Amino Acid Cabamoyl Conjugate (1–7)
The tert-butyl protected baicalein-amino acid carbamoyl conjugates 1c–7c (0.20 mmol) was dissolved in DCM (3 mL) at 0 °C. Trifluoroacetic acid (TFA, 1 mL) was added and the reaction mixture was stirred for 6 h at room temperature, when TLC (DCM/MeOH, 20/1, v/v) indicated the disappearance of stating material. The solvent and residual TFA were removed under reduced pressure. The residue was suspended in DCM-hexane (24 mL, 1/5, v/v) and filtered, and the filtrate was washed with DCM-hexane (12 mL, 1/5, v/v) to yield the baicalein-amino acid carbamoyl conjugates (1–7).
(S)-2-((((5,6-Dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)-4-methylpentanoic acid (1). Yellow amorphous powder, quantitative yield. 1H NMR (600 MHz, DMSO-d6) δ 12.93 (s, 1H), 11.28 (s, 1H), 8.10 (d, J = 7.2 Hz, 1H), 8.08 (d, J = 10.8 Hz, 1H), 7.63 (t, J = 7.2 Hz, 1H), 7.59 (t, J = 7.2 Hz, 2H), 7.01 (s, 1H), 6.66 (s, 1H), 4.06–4.01 (m, 1H), 1.79–1.72 (m, 1H), 1.67–1.59 (m, 1H), 1.58–1.49 (m, 1H), 0.93 (d, J = 6.6 Hz, 3H), 0.90 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 174.0, 163.5, 157.5, 153.7, 153.5, 153.0, 132.1, 130.7, 129.2, 126.5, 123.0, 104.8, 104.0, 94.0, 52.5, 24.3, 22.9, 21.3. HRMS (ESI) m/z calculated for C22H21NO8+ [M+H]+: 428.1340; found: 428.1351.
(S)-2-((((5,6-Dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)-3-methylbutanoic acid (2). Yellow amorphous powder, quantitative yield. 1H NMR (600 MHz, DMSO-d6) δ 12.93 (s, 1H), 11.33 (s, 1H), 8.09 (d, J = 7.2 Hz, 2H), 8.02 (d, J = 8.4 Hz, 1H), 7.64–7.58 (m, 3H), 7.01 (s, 1H), 6.67 (s, 1H), 3.91 (dd, J = 6.0 and 9.0 Hz, 1H), 2.17–2.07 (m, 1H), 0.95 (d, J = 7.2 Hz, 3H), 0.95 (d, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 172.9, 163.5, 157.5, 153.8, 153.7, 153.0, 132.1, 130.7, 129.2, 126.5, 123.0, 104.8, 104.0, 94.1, 59.8, 30.0, 19.1, 18.0. HRMS (ESI) m/z calculated for C21H19NO8+ [M+H]+: 414.1184; found: 414.1194.
(2S,3R)-2-((((5,6-Dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)-3-methylpentanoic acid (3). Yellow amorphous powder, quantitative yield. 1H NMR (600 MHz, DMSO-d6) δ 12.93 (s, 1H), 8.08 (d, J = 7.2 Hz, 2H), 8.01 (d, J = 8.4 Hz, 1H), 7.62 (t, J = 7.2 Hz, 1H), 7.58 (t, J = 7.8 Hz, 2H), 7.00 (s, 1H), 6.66 (s, 1H), 3.95 (dd, J = 6.6 and 8.4 Hz, 1H), 1.89–1.81 (m, 1H), 1.53–1.42 (m, 1H), 1.31–1.21 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 7.8 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 172.9, 163.5, 157.7, 153.7, 153.7, 153.0, 132.1, 130.7, 129.2, 126.5, 123.0, 104.8, 104.0, 94.1, 59.0, 36.5, 24.6, 15.5, 11.3. HRMS (ESI) m/z calculated for C22H21NO8+ [M+H]+: 428.1340; found: 428.1349.
(2S,3R)-2-((((5,6-Dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)-3-hydroxybutanoic acid (4). Yellow amorphous powder, quantitative yield. 1H NMR (600 MHz, DMSO-d6) δ 12.95 (s, 1H), 8.09 (d, J = 7.8 Hz, 2H), 7.67–7.53 (m, 3H), 7.48 (d, J = 9.0 Hz, 1H), 7.01 (s, 1H), 6.66 (s, 1H), 4.19–4.11 (m, 1H), 4.09–3.99 (m, 1H), 1.16 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 171.9, 163.5, 157.5, 153.8, 153.7, 153.0, 132.1, 130.7, 129.2, 126.5, 122.9, 104.9, 104.0, 94.1, 66.8, 60.2, 20.2. HRMS (ESI) m/z calculated for C20H17NO9+ [M+H]+: 416.0976; found: 416.0984.
(S)-2-((((5,6-Dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)-3-phenylpropanoic acid (5). Yellow amorphous powder, quantitative yield. 1H NMR (600 MHz, DMSO-d6) δ 12.89 (s, 1H), 11.25 (s, 1H), 8.17 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 7.2 Hz, 1H), 7.67–7.66 (m, 3H), 7.36–7.29 (m, 4H), 7.27–7.22 (m, 1H), 7.01 (s, 1H), 6.64 (s, 1H), 4.23–4.16 (m, 1H), 3.09 (dd, J = 5.4 and 13.5 Hz, 1H), 2.96 (dd, J = 9.6 and 13.8 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 172.9, 163.5, 157.5, 153.7, 153.4, 153.0, 137.7, 132.1, 130.7, 129.2, 129.2, 128.2, 126.5, 126.5, 122.8, 104.8, 104.0, 94.0, 55.8, 36.6. HRMS (ESI) m/z calculated for C25H19NO8+ [M+H]+: 462.1184; found: 462.1190.
(S)-2-((((5,6-Dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)propanoic acid (6). Yellow amorphous powder, quantitative yield. 1H NMR (600 MHz, DMSO-d6) δ 12.93 (s, 1H), 11.27 (s, 1H), 8.11 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 7.2 Hz, 2H), 7.62 (t, J = 7.2 Hz, 1H), 7.58 (t, J = 7.2 Hz, 1H), 7.00 (s, 1H), 6.66 (s, 1H), 4.09–4.02 (m, 1H), 1.33 (d, J = 7.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 174.0, 163.5, 157.5, 153.7, 153.2, 153.0, 132.1, 130.7, 129.2, 126.5, 122.9, 104.9, 104.0, 94.1, 49.5, 17.3. HRMS (ESI) m/z calculated for 19H15NO8+ [M+H]+: 386.0871; found: 386.0877.
2-((((5,6-Dihydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)carbonyl)amino)acetic acid (7). Yellow amorphous powder, quantitative yield. 1H NMR (600 MHz, DMSO-d6) δ 12.94 (s, 1H), 11.29 (s, 1H), 8.09 (d, J = 7.2 Hz, 2H), 8.05 (t, J = 6.0 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.58 (t, J = 7.8 Hz, 2H), 7.01 (s, 1H), 6.66 (s, 1H), 3.76 (d, J = 6.0 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 182.2, 171.2, 163.5, 157.5, 153.9, 153.7, 153.0, 132.1, 130.7, 129.2, 126.5, 122.9, 104.9, 104.0, 94.1, 42.4. HRMS (ESI) m/z calculated for C18H13NO8+ [M+H]+: 372.0714; found: 372.0722.
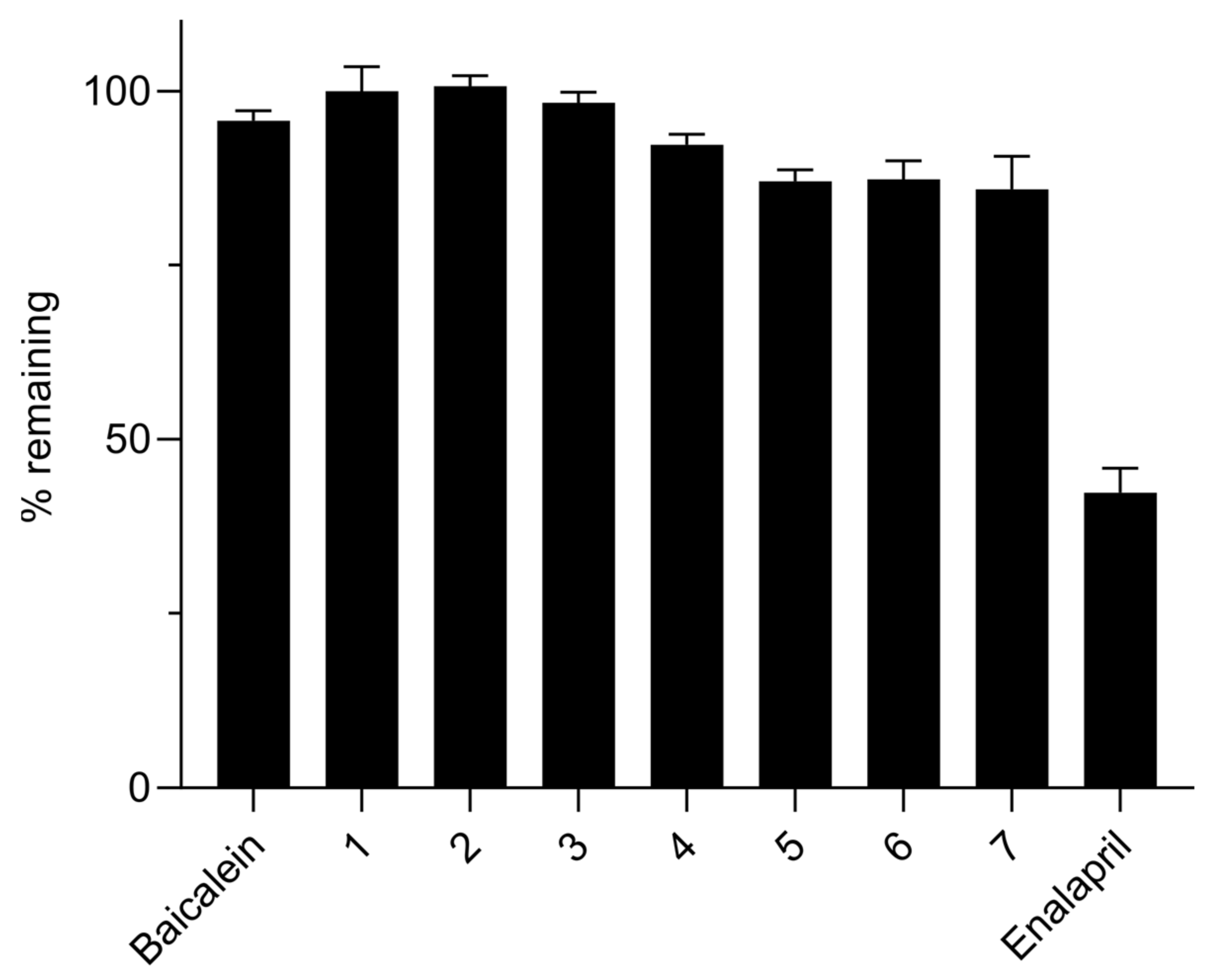
2.5. Chemical Stability
Stock solutions of the test compounds (baicalein, compounds 1–7) prepared in MeOH at 10 mM were diluted in 0.1 N HCl and 0.1 M potassium phosphate buffer (pH 6.5 and 7.4) in triplicate to a final concentration of 10 μM. The mixtures were then incubated at 37 °C in a shaking water bath. Samples taken at 0, 1, 2, 3 and 4 h were analyzed by Thermo UltiMate 3000 series HPLC system. Chromatographic separation was performed on an Agilent Eclipse Plus C18 column (100 × 4.6 mm, 3.5 µm) kept at 40 °C with isocratic elution at a flow rate of 0.6 mL/min using a mixture of 0.1 vol% TFA in deionized water and acetonitrile as the mobile phase. The detection wavelengths were 277 and 270 nm for baicalein and compounds 1–7, respectively. Peak areas of the analytes on the UV chromatograms were used to calculate % remaining values at each time points.
2.6. Solubility
Stock solutions of the test compounds (baicalein and compounds 1–7) prepared at 10 mM in DMSO were diluted with 0.1 M potassium phosphate buffer (pH 7.4) in Mini-UniPrep filter vial (PTFE membrane, 0.45 μm) to a final concentration of 500 µM. The mixture was then shaken at 800 rpm for 90 min at rt and filtered by slowly pressing the upper tube of the Mini-Uniprep vial. The filtrate (300 µL) was then transferred to an HPLC vial containing same volume of acetonitrile. Sample analysis was conducted as described above in Chemical stability assay.
2.7. logD7.4
The assay was conducted as reported previously [
14]. Briefly, 100 µM test solutions were prepared by diluting 10 mM stock solutions of the test compounds (baicalein and compounds
1–7) in 0.1 M potassium phosphate buffer (pH 7.4) saturated with 1-octanol. Standard solutions were prepared by diluting each test solution with the same volume of DMSO in HPLC vials. Partition solutions were prepared by mixing each test solution and 1-octanol saturated with 0.1 M potassium phosphate buffer (pH 7.4) at three different volume ratios, namely 1:0.02, 1:0.2 and 1:2, in HPLC vials. The partition solutions were then vortexed for 1 h at 1500 rpm at rt. The standard solutions and the aqueous phase (bottom layer) of each partition solution were analyzed directly from the HPLC vials as described above in Chemical stability assay. The
logD7.4 values were calculated by the following equation:
where
As and
Ap are, respectively, the peak areas of the standard and the aqueous phase of the partition solutions and
Vt and
Vo the volumes of test solution and 1-octanol of the partition solutions.
2.8. PAMPA
The assay was conducted as reported previously [
15]. The samples were analyzed as described below in mouse plasma stability assay. Low permeability references (atenolol, hydrochlorothiazide, nadolol and ranitidine) had
Papp values <1 nm/sec, while high permeability references (chloramphenicol, ketoprofen, metoprolol, propranolol and verapamil) showed
Papp values >10 nm/sec.
2.9. Metabolic Stability
2.9.1. Mouse Plasma
Working solutions of the test compounds (baicalein, enalapril, and compounds 1–7) prepared in 50 vol% aqueous MeOH at 40 μM were diluted in mouse plasma in triplicate to a final concentration of 1 μM. The mixtures were then incubated at 37 °C in a shaking water bath. A 50 μL aliquot was removed at 0 and 60 min from each incubation mixture followed by immediate mixing with 150 μL ice-cold MeOH containing 1 μg/mL glipizide as the internal standard (IS). The resulting mixtures, following a 5-min vortexing and sonication, were then centrifuged at 3000 rpm for 30 min at 4 °C. The supernatant was analyzed by Agilent 6460 QQQ LC−MS/MS system in a positive multiple reaction monitoring (MRM) mode. Gradient elution was conducted on an Agilent Eclipse Plus C18 column (100 × 2.1 mm, 3.5 µm) at 40 °C with 0.1 vol% formic acid in deionized water and 0.1 vol% formic acid in acetonitrile running at 0.45 mL/min. MRM transitions of the analytes were set as follows: baicalein: m/z 271→123, enalapril: m/z 377→234, 1: m/z 428→271, 2: m/z 414→271, 3: m/z 428→271, 4: m/z 416→271, 5: m/z 462→271, 6: m/z 386→271, 7: m/z 372→271, and glipizide: m/z 446→321. The analytical data were analyzed using Agilent MassHunter Quantitative Analysis Software (version B.05.00). Peak area ratios of the analytes vs. IS on the MRM chromatograms were used to calculate % remaining at 60 min.
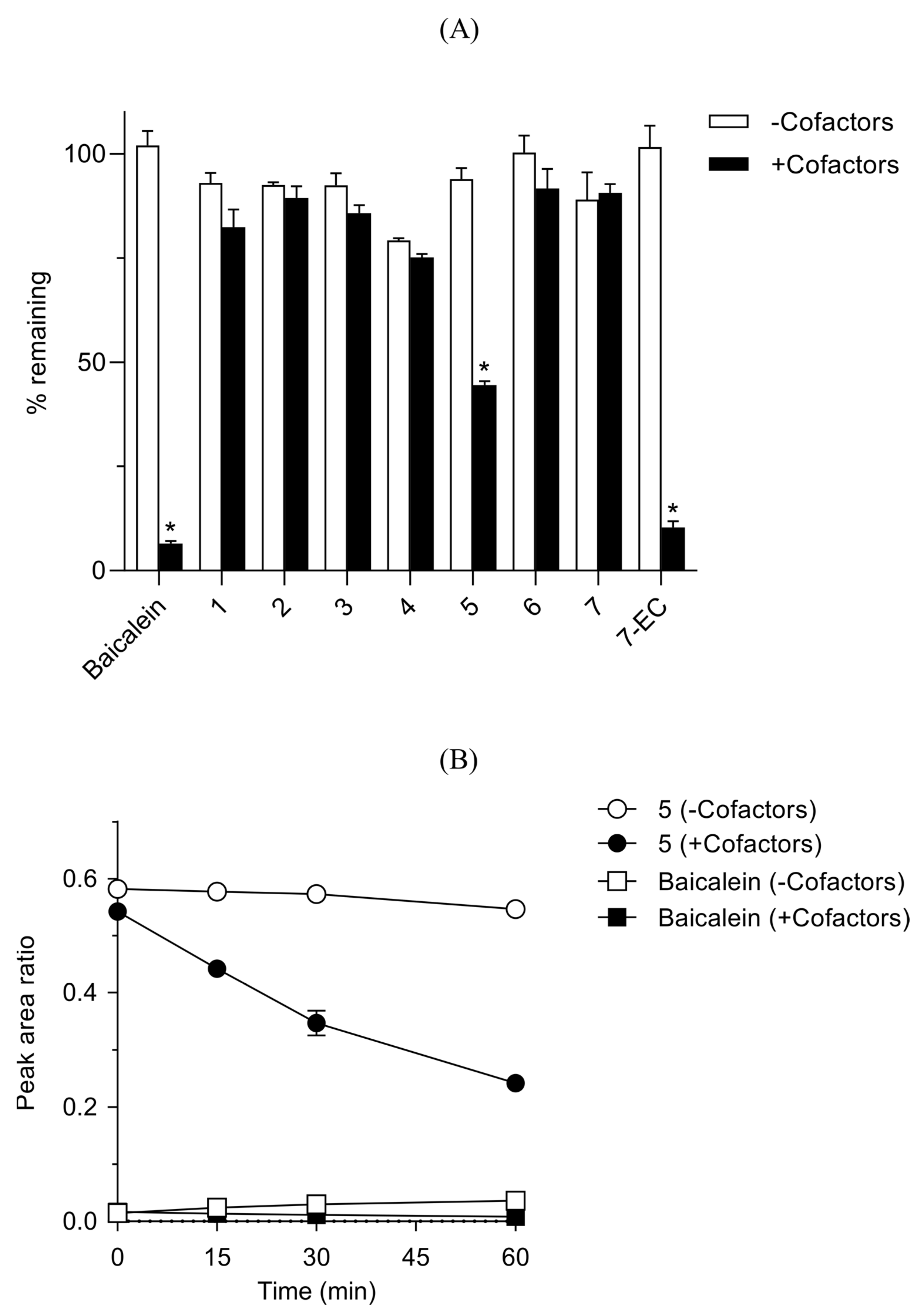
2.9.2. Mouse Liver S9 Fractions
Working solutions of the test compounds (baicalein, 7-EC, and compounds 1–7) prepared in 50 vol% aqueous MeOH at 120 μM were added in triplicate to a 200 mM tris buffer (pH 7.4) containing mouse liver S9 fractions (1 mg protein/mL), alamethicin (25 μg/mL) and MgCl2 (2 mM). The final concentration of test compound was 3 μM. This mixture was pre-incubated at 37 °C in a shaking water bath for 5 min followed by addition of a cofactor mixture to following final concentrations: 1 mM NADPH, 0.5 mM UDPGA, 0.1 mM PAPS and 2.5 mM GSH. A 50 μL aliquot was taken at 0, 15, 30 and 60 min from each incubation mixture. These samples were then treated in the same way as described above in Mouse plasma stability assay. The supernatant was analyzed by Agilent 6530 Q−TOF LC−MS/MS system in a positive auto MS/MS scan mode. The liquid chromatography was conducted in the same condition as described above in mouse plasma stability assay. The analytical data were analyzed using Agilent MassHunter Quantitative Analysis Software (version B.05.00). MS chromatograms were extracted using calculated exact masses ([M+H]+) for the analytes. Peak area ratios of the analytes vs. IS on the extracted MS chromatograms were used to calculate % remaining values at each time points.
2.9.3. Mouse Pharmacokinetics
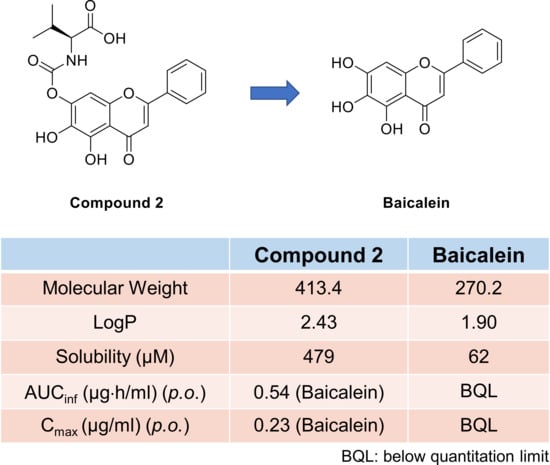
Pharmacokinetic studies were conducted with male ICR mice (8 weeks old; 30–35 g body wt.) purchased from Koatech Company (Pyeongtaek, Korea), as reported previously [
16]. Intravenous dosing solutions were prepared in EtOH/20% aqueous HPβCD (10/90 vol%) at 2 and 3 mg/mL for baicalein and compound
2, respectively. Oral dosing solutions were prepared in EtOH/tween 80/20% aqueous HPβCD (10/10/80 vol%) at 1 and 1.5 mg/mL for baicalein and compound
2, respectively. Plasma sample analysis was performed as described above in mouse plasma stability assay.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}