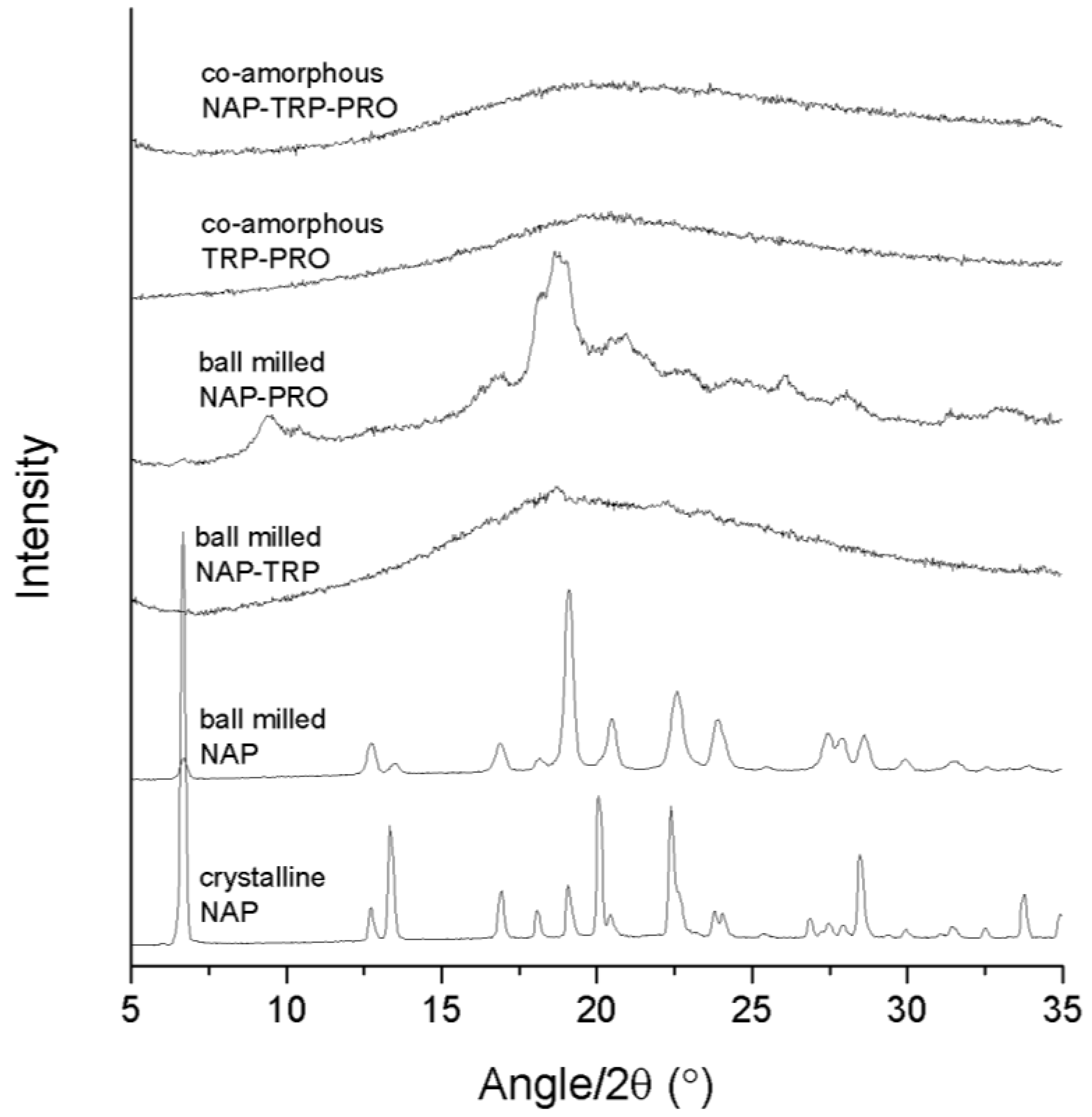
3.1. Determination of Successful Amorphization by XRPD
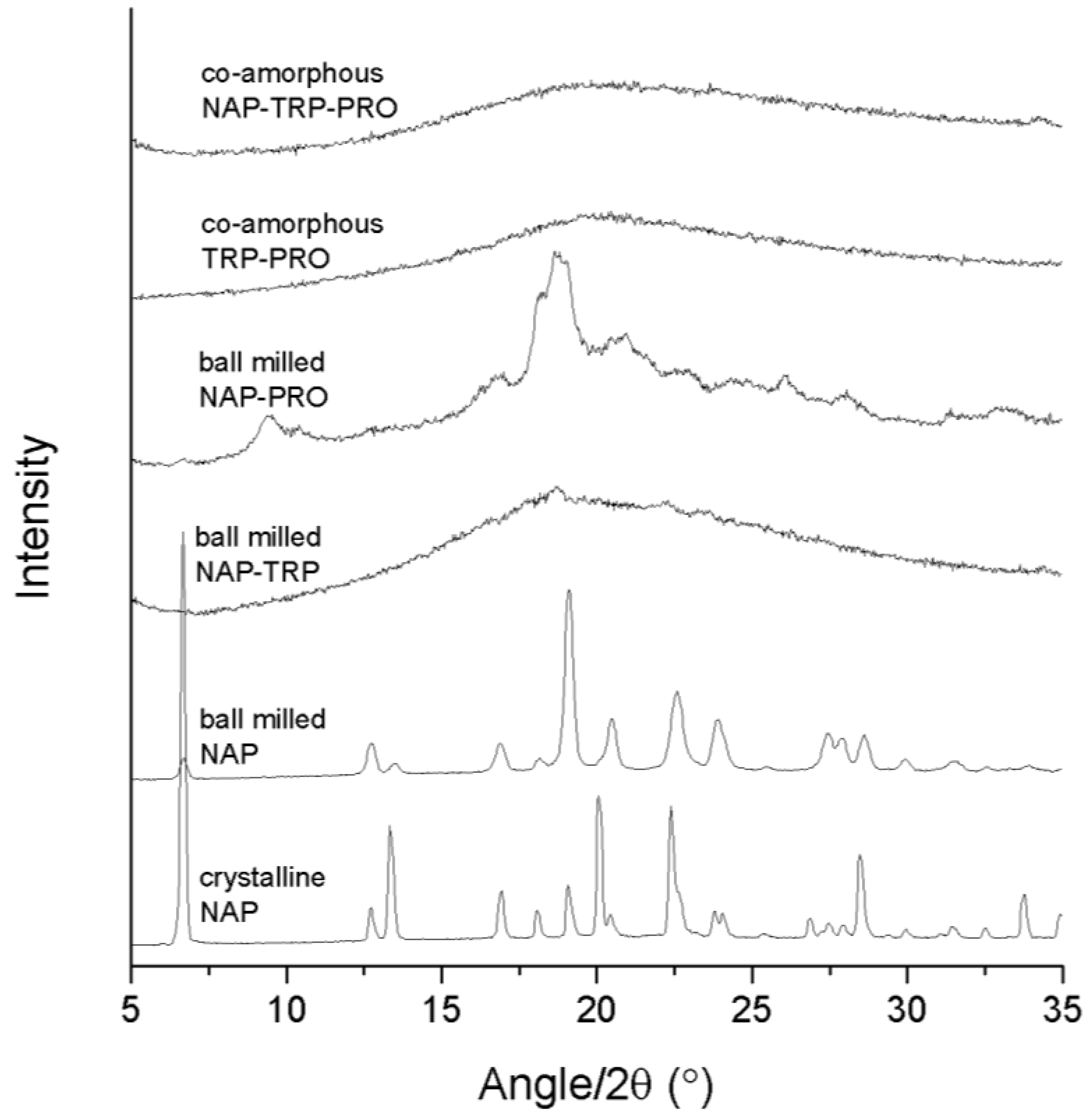
Success in amorphization was ascertained by the detection of a halo as the only feature in the XRPD diffractogram. The analysis of the pure compounds revealed that neither NAP (
Figure 1) nor any of the amino acids could be transformed into an amorphous solid when ball milled alone for 90 min (data not shown). The preferred orientation of the NAP molecule caused the observed changes in the relative intensity of the X-ray reflections upon milling. In the following, various amino acid and drug combinations with and without PRO were prepared to investigate the effect of adding a highly soluble amino acid to a co-amorphous system. All samples were prepared at a molar ratio of 1:1 (drug:amino acid) or 1:1:1 (drug:amino acid:PRO).
Figure 1.
X-ray powder diffraction (XRPD) diffractograms of crystalline NAP, ball-milled NAP, ball-milled NAP–TRP, ball-milled NAP–PRO, co-amorphous TRP–PRO and co-amorphous NAP–TRP–PRO.
Figure 1.
X-ray powder diffraction (XRPD) diffractograms of crystalline NAP, ball-milled NAP, ball-milled NAP–TRP, ball-milled NAP–PRO, co-amorphous TRP–PRO and co-amorphous NAP–TRP–PRO.
The analysis of the NAP–TRP mixture upon ball milling revealed small remaining reflections in the diffractogram, which can be related to the crystalline form of NAP (
Figure 1). It has been shown that process parameters can have a great influence on the reduction in the reflection intensities towards a completely amorphous mixture [
10]. However, remaining reflections could still be observed for NAP–TRP after 6 h of ball milling (data not shown). These results are in contrast to previous findings, where the use of TRP was found to be particular advantageous when forming co-amorphous mixtures with a drug [
13]. The size and flexibility of the molecule are known to be important characteristics when choosing a good amorphous stabilizer [
3,
23]. The previous results suggest that TRP tends to have the molecular size and flexibility required to be a good amorphous stabilizer. However interactions between TRP and NAP appear to be insufficient to create a completely co-amorphous mixture at the applied process conditions.
Ball milling of NAP–PRO showed remaining crystallinity in the XRPD diffractogram, which can be related to crystalline NAP and crystalline PRO (see
Figure 2). This indicated that PRO alone is not sufficient in stabilizing a co-amorphous system with NAP. Generally, rigid molecules, like PRO, are not easily transformed into their amorphous form, because of their stiff structure and high tendency to arrange in an ordered crystal lattice, in contrast to more flexible molecules [
3,
23]. Co-milling of TRP–PRO resulted in a co-amorphous mixture, suggesting interactions between the two amino acids (
Figure 1). Ball milling of the ternary mixture, NAP–TRP–PRO, resulted in an amorphous blend (
Figure 1). Thus, the addition of PRO had a positive effect on the amorphization of NAP, which could not be achieved by any of the amino acids alone.
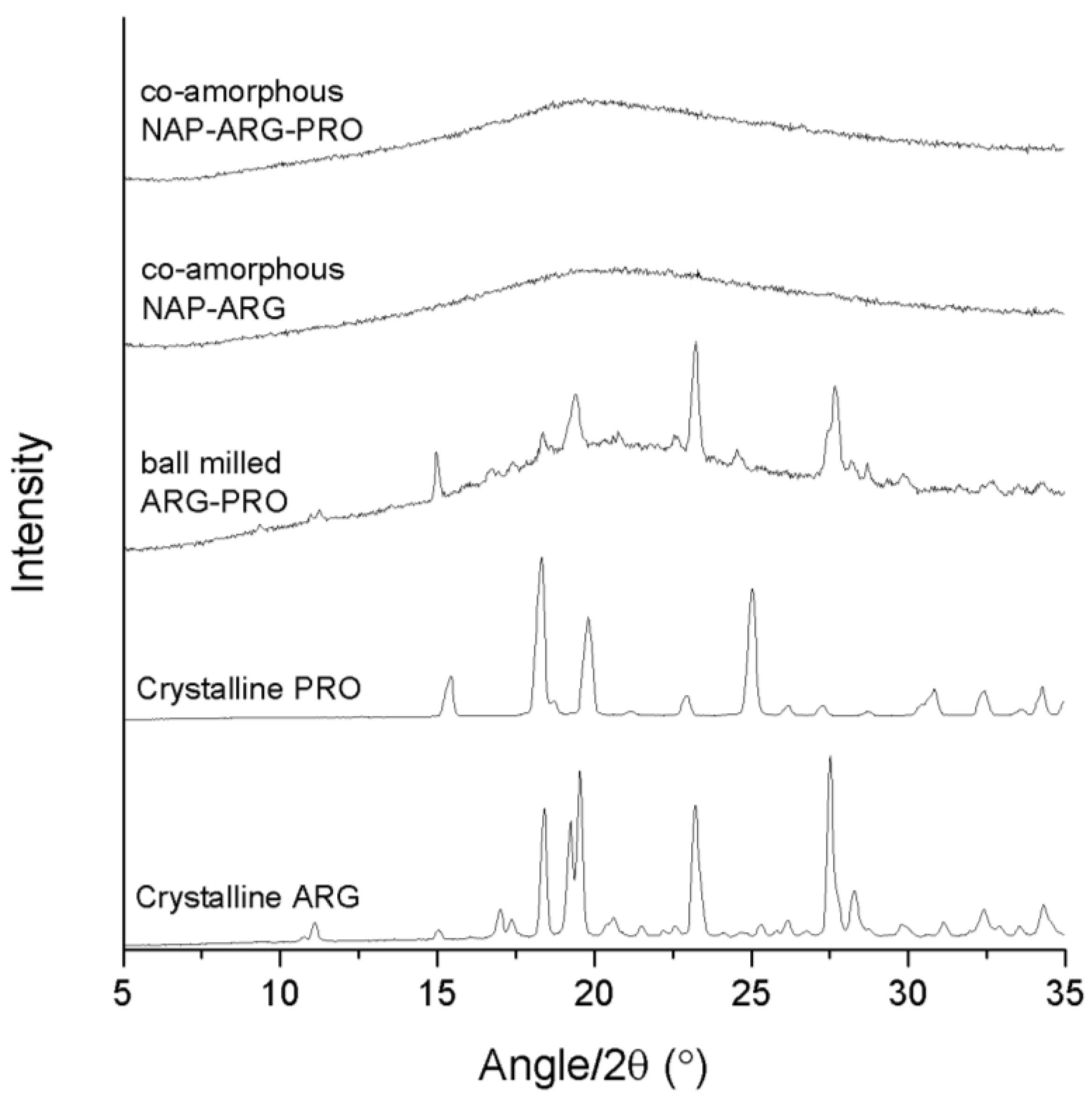
Figure 2.
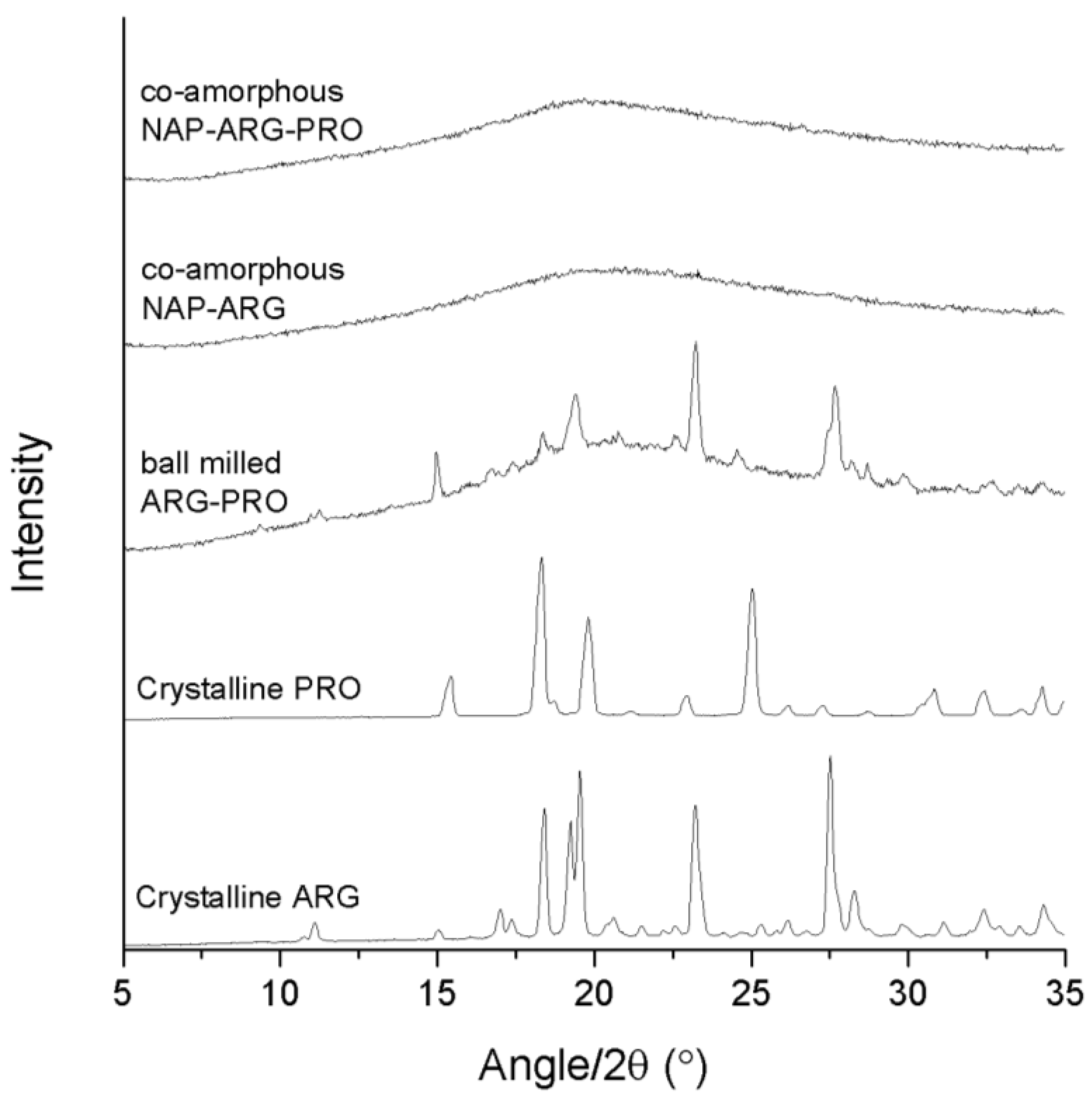
XRPD diffractograms of crystalline ARG, crystalline PRO, ball-milled ARG–PRO, co-amorphous NAP–ARG and co-amorphous NAP–ARG–PRO.
Figure 2.
XRPD diffractograms of crystalline ARG, crystalline PRO, ball-milled ARG–PRO, co-amorphous NAP–ARG and co-amorphous NAP–ARG–PRO.
The binary mixture, NAP–ARG, was found to be amorphous after ball milling, in contrast to an earlier publication, where small crystalline reflections were seen in the X-ray diffractogram upon ball milling of NAP–ARG (
Figure 2) [
18]. Variation within these results probably is due to differences in milling time and milling frequency. Ball-milled ARG–PRO did not result in a co-amorphous mixture, and the remaining reflections can be correlated to a combination of crystalline ARG and PRO being present in the mixture (
Figure 2). The ternary mixture NAP–ARG–PRO was found to be amorphous, as only a halo was detected by XRPD. Summarizing, the presence of ARG stabilized co-amorphous mixtures as observed in co-amorphous NAP–ARG and NAP–ARG–PRO. In the case of NAP–TRP, a small remaining degree of crystallinity was observed, while NAP–TRP–PRO resulted in a co-amorphous mixture.
3.2. Determination of Thermal Properties by DSC
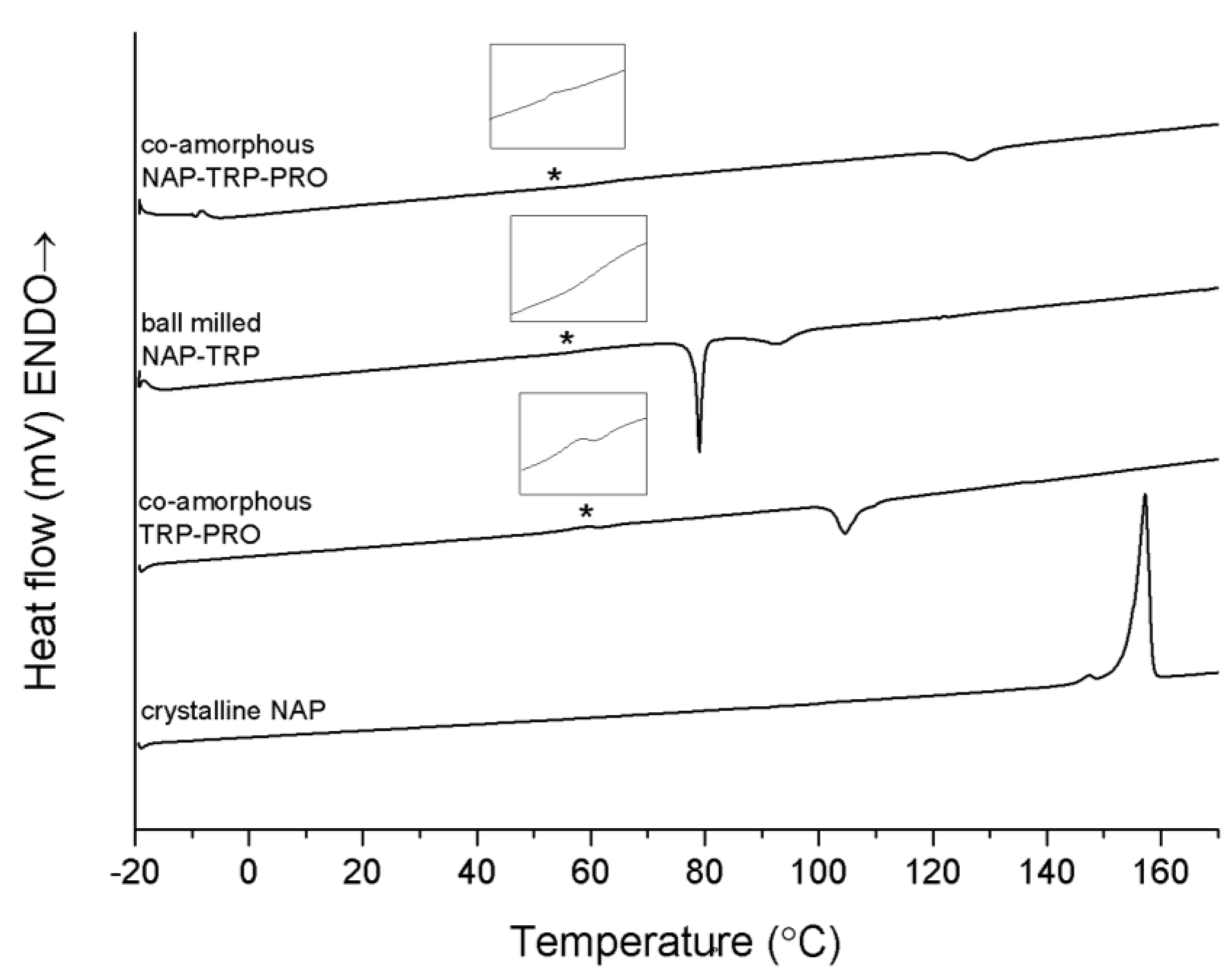
DSC studies were carried out in order to identify and characterize the thermal properties of the co-amorphous mixtures. In the DSC thermogram of pure crystalline NAP, a large endothermic event at approximately 157 °C was observed, which represented the melt of the crystalline anhydrous state (
Figure 3) [
15,
24,
25]. As NAP on its own cannot be transferred into an amorphous form by ball milling, the T
g of NAP (5.0 ± 0.4 °C) was determined via
in situ quench cooling in a DSC using a heating rate of 10 K/min, as previously published [
26].
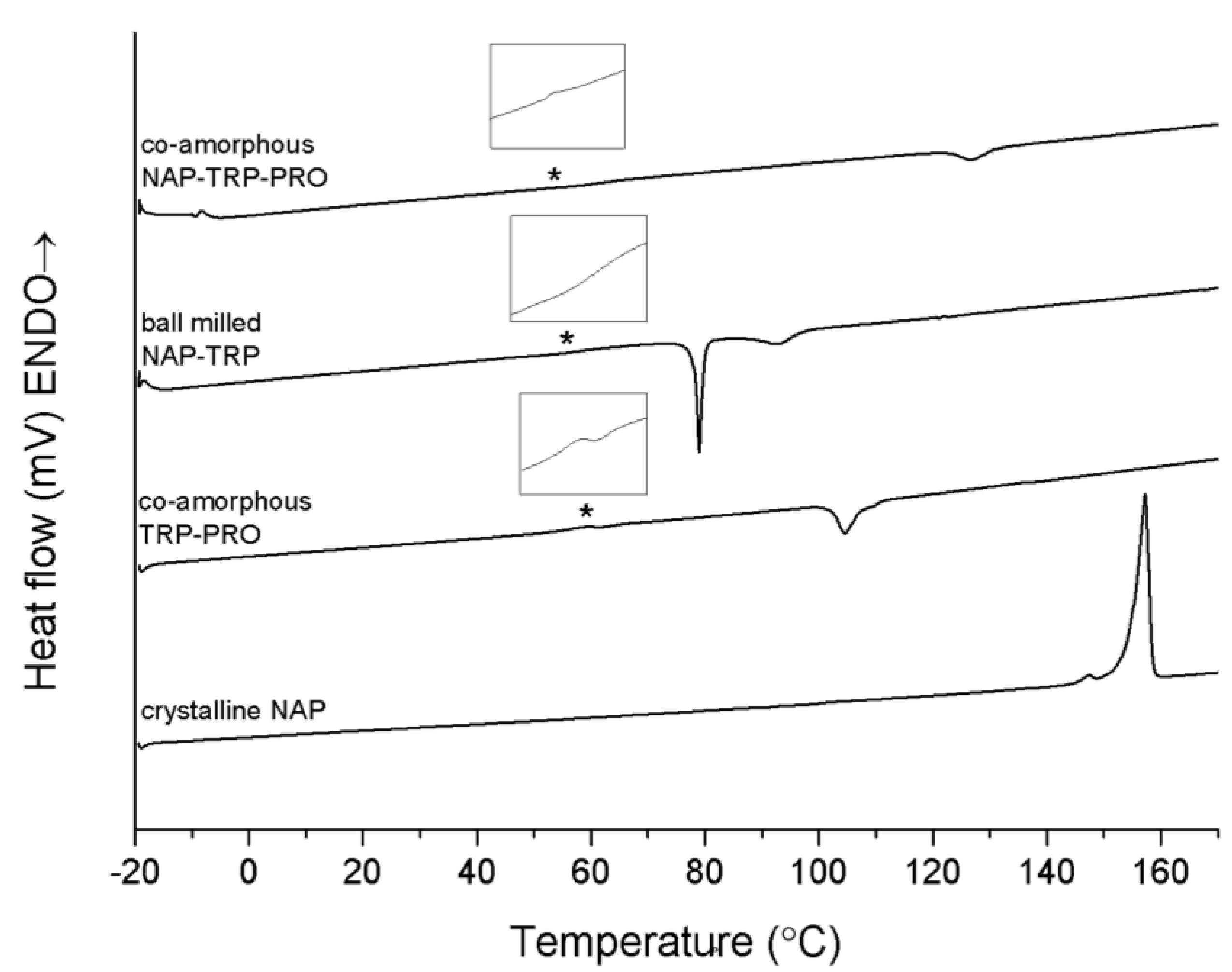
Figure 3 further shows the thermograms of the binary and ternary co-amorphous systems containing TRP. The sample containing NAP–TRP had a T
g around 58 °C, followed by a re-crystallization of TRP. No melting of crystalline NAP was observed in the thermogram, although the XRPD diffractogram showed small remaining crystalline NAP reflections in this sample (see
Figure 1). A possible explanation could be that NAP and TRP form a homogeneous phase with one T
g, spiked with small amounts of remaining NAP crystals that can be identified with XRPD after the milling. Upon heating above T
g, the remaining NAP crystals dissolve within the NAP–TRP blend in its supercooled liquid state. However, this phase is also supersaturated with respect to TRP in the amorphous NAP, leading to TRP partly recrystallizing above the T
g. This leaves an amorphous NAP phase saturated with TRP, which is stable enough to prevent crystallization of NAP. Thus, no NAP melting endotherm was detected in the NAP–TRP thermogram.
The co-amorphous NAP–TRP–PRO mixture consisted of a homogeneous phase reflected as a single T
g in the thermogram. A small exothermic peak could be observed at approximately 120 °C, which is due to the recrystallization of TRP, confirmed by XRPD measurements of the powder after pre-heating above 95 °C (data not shown). Recrystallization of TRP can further be observed in co-amorphous TRP–PRO (
Figure 3). The re-crystallization event occurs at a higher temperature and with a lower enthalpy in the ternary mixture compared to NAP–TRP and TRP–PRO, suggesting a more stable and homogeneous mixture, even though the T
g of the NAP–TRP–PRO (55.1 ± 3.1 °C) is lower than that of NAP–TRP (58.2 ± 0.5 °C) and TRP–PRO (67.2 ± 6.8 °C). Thus, the presence of both TRP and PRO improves the ability to form a co-amorphous mixture. This is further confirmed by the XRPD measurements presented in
Figure 1; showing that complete amorphization of the formulation after 90 min of ball milling requires the presence of both TRP and PRO. However, the DSC results discussed above suggest additional interactions when NAP is added to TRP–PRO.
Figure 3.
DSC measurements of crystalline NAP, co-amorphous TRP–PRO, ball-milled NAP–TRP and co-amorphous NAP–TRP–PRO. The Tg is marked by asterisk (*) and enlarged in the insert above the graph. Endothermal events up.
Figure 3.
DSC measurements of crystalline NAP, co-amorphous TRP–PRO, ball-milled NAP–TRP and co-amorphous NAP–TRP–PRO. The Tg is marked by asterisk (*) and enlarged in the insert above the graph. Endothermal events up.
A benefit of applying TRP–PRO as an amorphous stabilizer in co-amorphous formulations is the high T
g (67.2 ± 6.8 °C) of this system compared to the T
g for amorphous NAP (5.0 ± 0.4 °C) (
Table 2) [
13]. According to the Gordon Tayler equation, the glass transition temperature of a co-amorphous system is expected to have a value between the corresponding T
gs of the components in the blend [
22]. Thus, there is a high probability to obtain an increased T
g of the co-amorphous mixture consisting of a drug together with TRP–PRO, compared to the amorphous drug alone. A theoretical T
g for co-amorphous NAP–TRP–PRO of 37.2 °C is calculated by the Gordon Taylor equation (
Table 2), which is considerably higher than for amorphous NAP alone. However, when comparing to the experimental T
g, an even further increase in T
g (55.1 ± 3.1 °C) was observed, suggesting strong molecular interactions between the molecules in the co-amorphous NAP–TRP–PRO mixture, as the Gordon Taylor equation assumes an amorphous blend without interactions [
22]. These results indicate TRP–PRO as being beneficial to combine with NAP in order to obtain a thermally-stable co-amorphous system with a high T
g. However, the T
g of amorphous TRP (140.2 ± 1.0 °C) is even higher, which explains why this amino acid previously has contributed to stable co-amorphous systems.
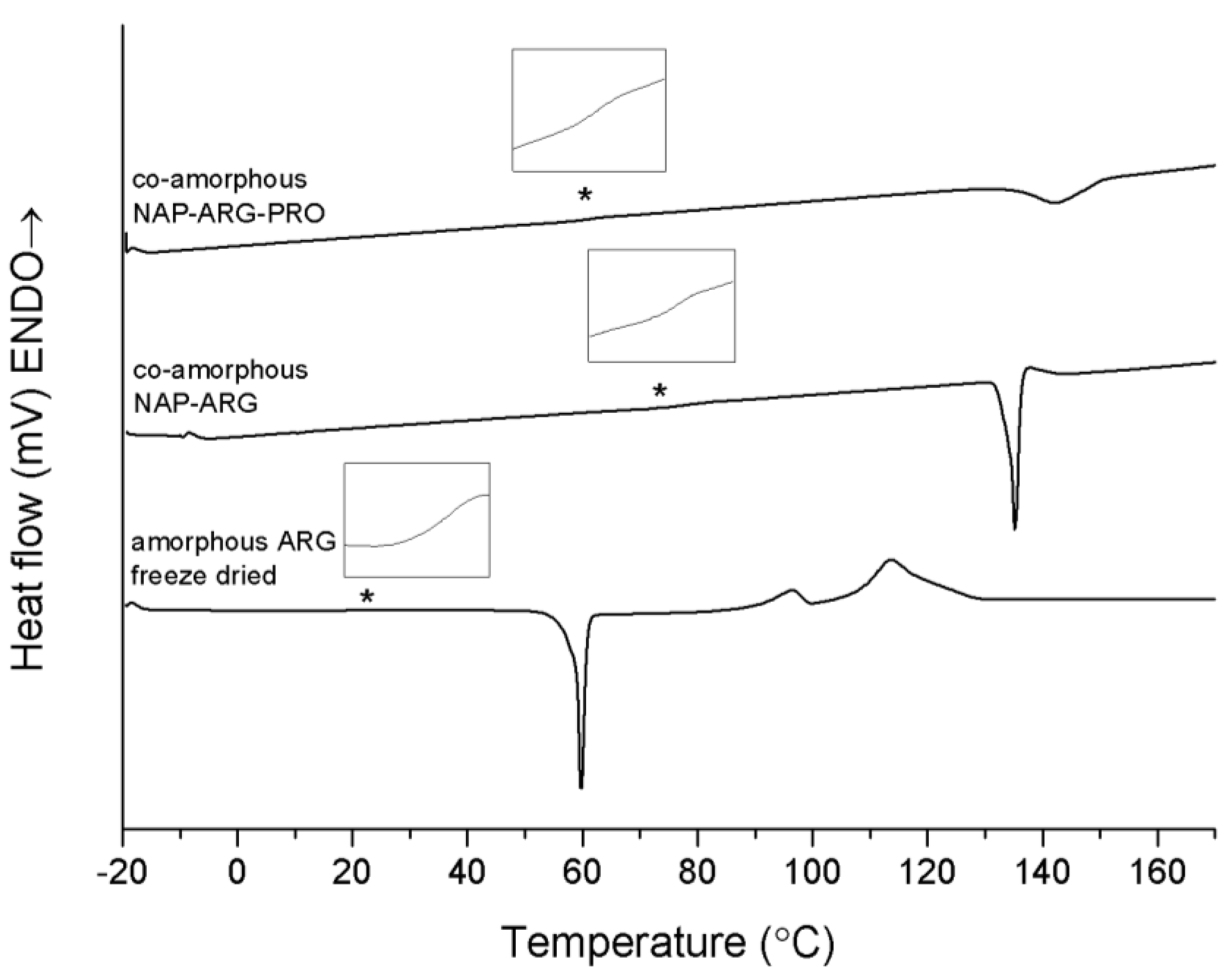
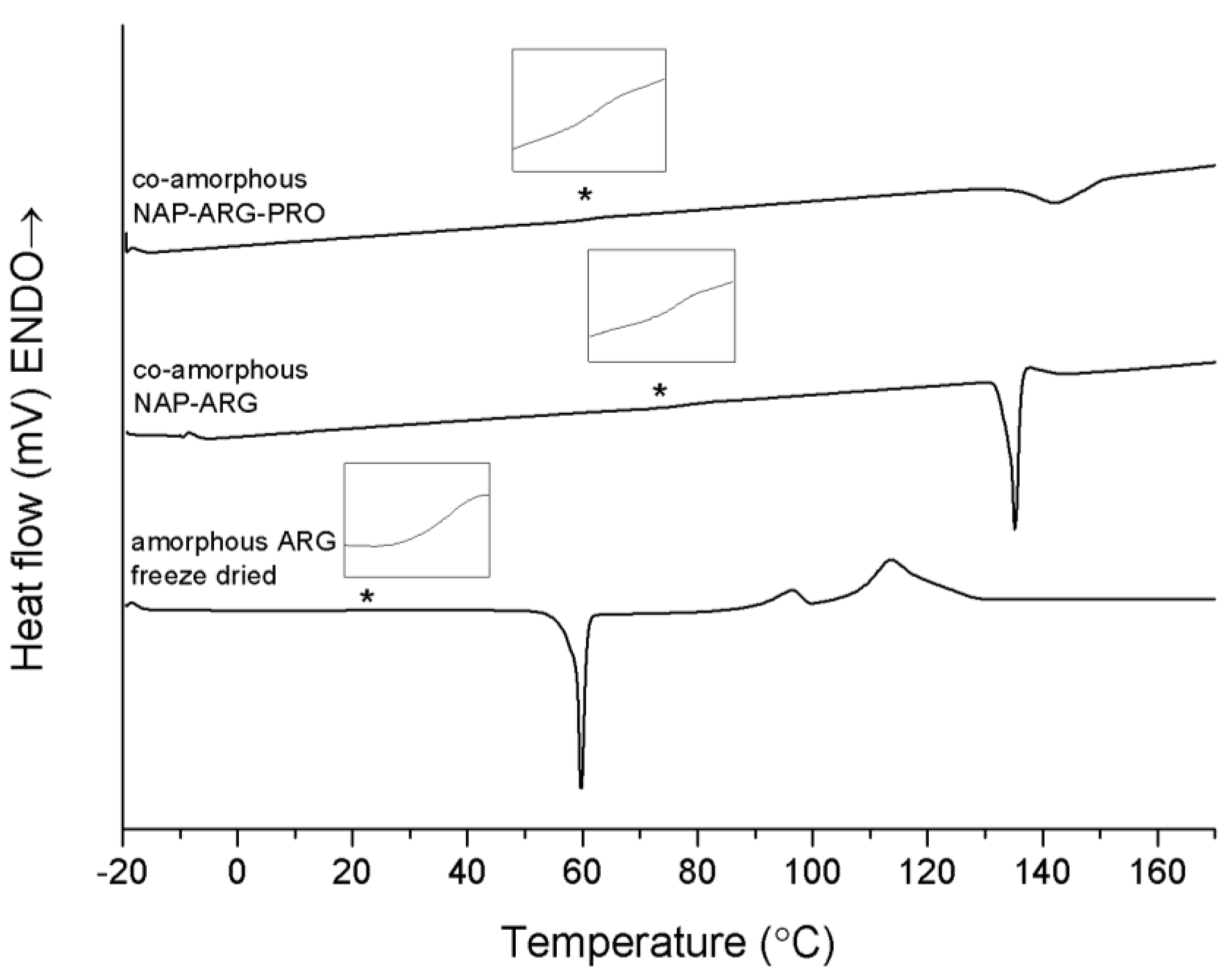
Co-amorphous NAP–ARG and NAP–ARG–PRO both showed a single T
g at 72.1 ± 2.9 °C and 63.5 ± 1.9 °C, respectively, followed by a re-crystallization exotherm (
Figure 4). XRPD measurements of the mixtures pre-heated to 160 °C suggest that NAP and ARG crystallize in both mixtures. However, no NAP endotherm can be observed in the thermogram, which indicates that cooling before the XRPD measurements could have caused NAP to crystallize. The crystallization peak observed in the thermogram is expected to be ARG, which appears at a higher temperature and with a lower enthalpy when PRO is added. These results suggest again that PRO contributed to a stabilization of the co-amorphous system, similar to NAP–TRP–PRO.
Table 2.
Experimental T
gs of the pure amorphous quench-cooled NAP [
26], amorphous freeze-dried ARG, ball-milled TRP and ball-milled co-amorphous blends (
n = 3, mean ± standard deviation).
Table 2.
Experimental Tgs of the pure amorphous quench-cooled NAP [26], amorphous freeze-dried ARG, ball-milled TRP and ball-milled co-amorphous blends (n = 3, mean ± standard deviation).
| Sample Content | Tg (°C) | Theoretical Tg (°C) |
|---|
| NAP | 5.0 (±0.4) | – |
| ARG | 18.4 (±2.7) | – |
| TRP 1 | 140.2 (±1.0) | – |
| NAP–ARG | 72.1 (±2.9) | 10.5 |
| NAP–ARG–PRO | 63.5 (±1.9) | 8.2 |
| NAP–TRP–PRO | 55.1 (±3.1) | 37.2 |
| NAP–TRP 1 | 58.2 (±0.5) | 54.6 |
| TRP–PRO | 67.2 (±6.8) | – |
| ARG–PRO 2 | 11.0 (±4.1) | – |
It is obvious from
Table 2 that mixtures containing ARG resulted in T
g values considerably higher than the theoretical calculations. This can be explained by ionic interactions, due to salt formation, which is not considered in the Gordon Taylor calculation. One of the reasons for the low theoretical calculated T
g of the co-amorphous systems is the low T
g of amorphous ARG, measured as 18.4 ± 2.7 °C of the freeze-dried substance (
Table 2). This T
g is lower than previously reported, where T
gs of 42 °C [
27] and 55 °C [
28] were found. The low T
g of amorphous ARG indicates that salt formation or other strong interactions between the amino acid and other components in the co-amorphous system are required to obtain a co-amorphous system with ARG.
Figure 4.
DSC thermograms of amorphous freeze-dried ARG, co-amorphous NAP–ARG and NAP–ARG–PRO. The Tg is marked by asterisk (*) and enlarged in the insert above the graph. Endothermal events up.
Figure 4.
DSC thermograms of amorphous freeze-dried ARG, co-amorphous NAP–ARG and NAP–ARG–PRO. The Tg is marked by asterisk (*) and enlarged in the insert above the graph. Endothermal events up.
The Tg of both ternary systems is lower than those of the corresponding binary mixtures without PRO. This is likely to be a consequence of the low Tg of PRO, decreasing the Tg of the co-amorphous blend. Unfortunately, this could not be confirmed experimentally, as it was not possible to prepare amorphous PRO.
3.3. Investigation of Molecular Interactions by FTIR
A qualitative assessment of molecular interaction in the co-amorphous mixtures, as well as the molecular arrangement was performed, based on vibrational spectroscopy. Ideally, the IR spectra of the co-amorphous samples are compared to the IR spectra of each of the components in the amorphous form, as the spectrum of the amorphous form has a tendency to exhibit reduced signals, peak shifts and slightly broader peaks compared to the corresponding crystalline spectrum [
29,
30]. As neither NAP nor any of the amino acids were amorphous after 90 min of ball milling, reference spectra were obtained for the amorphous quench-cooled NAP, amorphous freeze-dried ARG and amorphous TRP prepared by 6 h of ball milling. These spectra, as well as the spectra of co-amorphous TRP–PRO and crystalline amino acids were used as the reference. The spectral region between 1000 and 1800 cm
−1 was chosen for analysis, as it contains information regarding changes in the aromatic ring systems (1100–1500 cm
−1), as well as hydrogen bonded carboxylic acids (1700 cm
−1) and amides (1600 cm
−1) [
30].
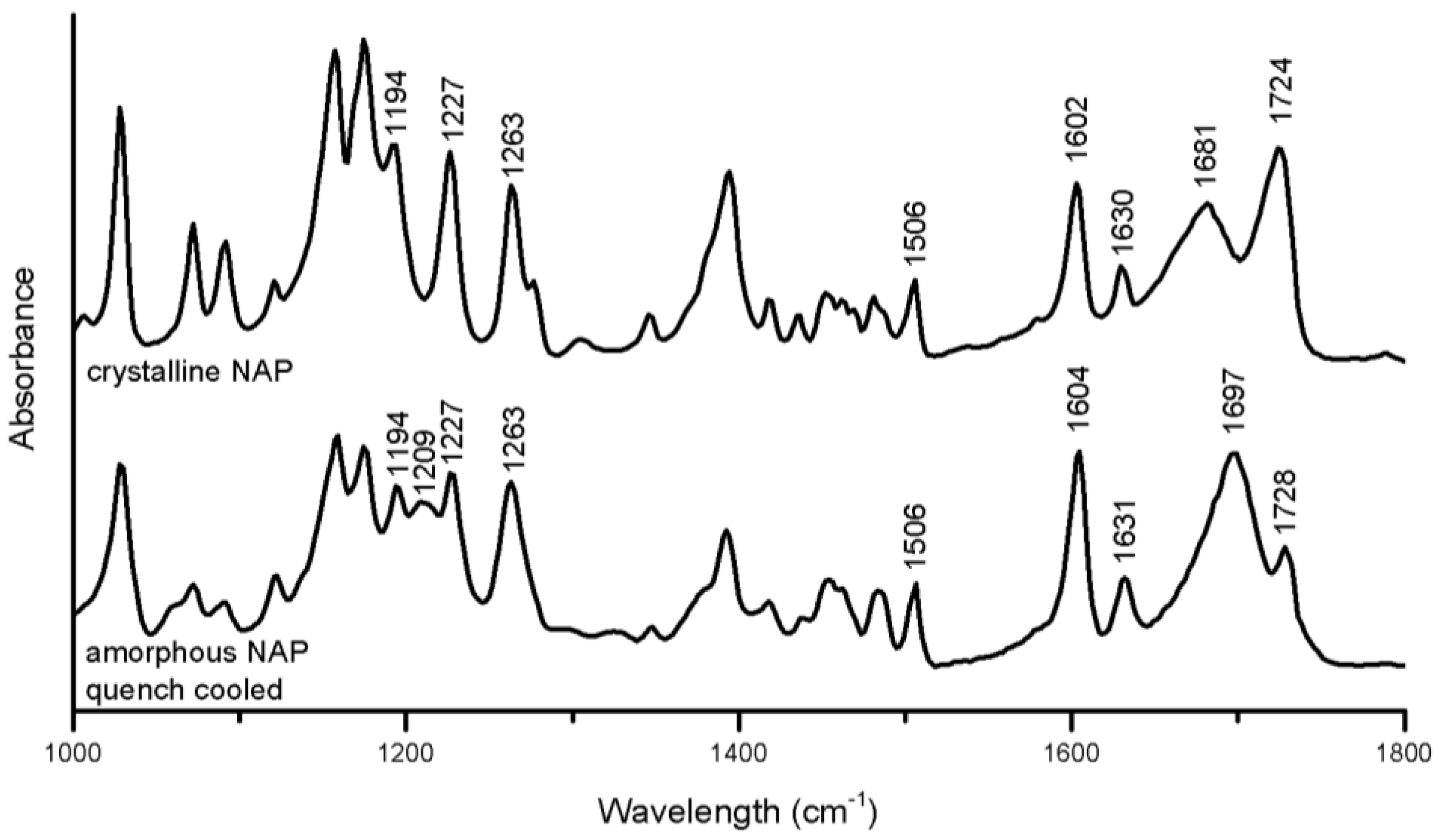
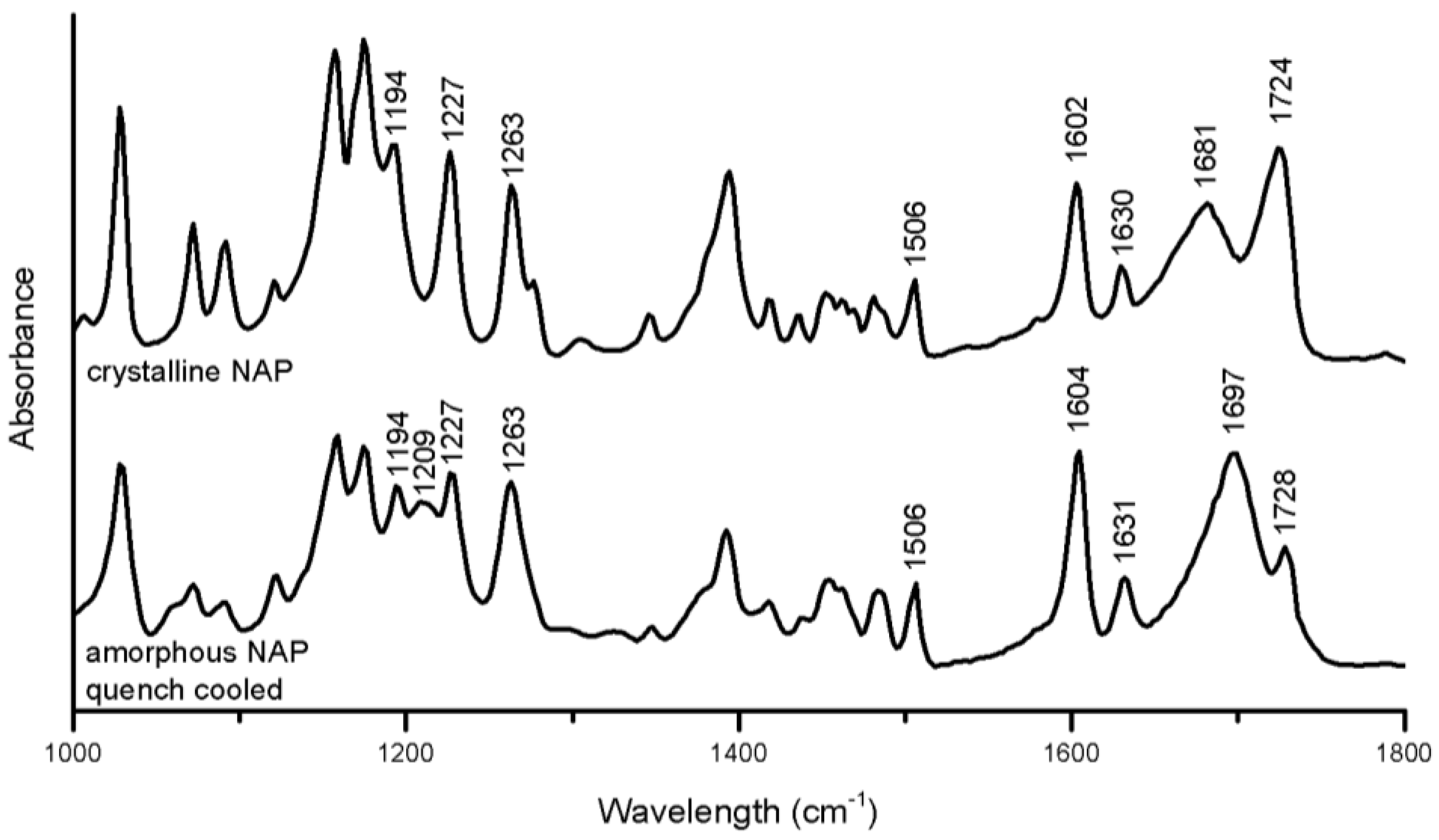
The crystalline to amorphous transformation of NAP induced the following changes in the spectrum (
Figure 5): peak alterations were observed in the aromatic region related to changes in the molecular environment of the naphthalene moiety (1194–1263 cm
−1) [
30] the peak at 1681 cm
−1 referring to the hydrogen-bonded carbonyl stretch was increased and shifted to 1697 cm
−1, and the free carboxylic acid group peak at 1724 cm
−1 was reduced and shifted to 1728 cm
−1 [
31]. Generally, shifts of functional groups upon amorphous to crystalline transformations or in amorphous mixtures indicate changes of intermolecular interactions between the molecules, e.g., a changed hydrogen bonding pattern [
14,
30]. The changes observed in amorphous NAP suggest the formation of NAP dimers interacting via hydrogen-bonds, including the carboxylic acid groups upon amorphization, as previously reported [
30]. The changes in the aromatic region further indicated a change in the π–π interactions.
Figure 5.
FTIR spectra of crystalline and amorphous NAP prepared by quench cooling.
Figure 5.
FTIR spectra of crystalline and amorphous NAP prepared by quench cooling.
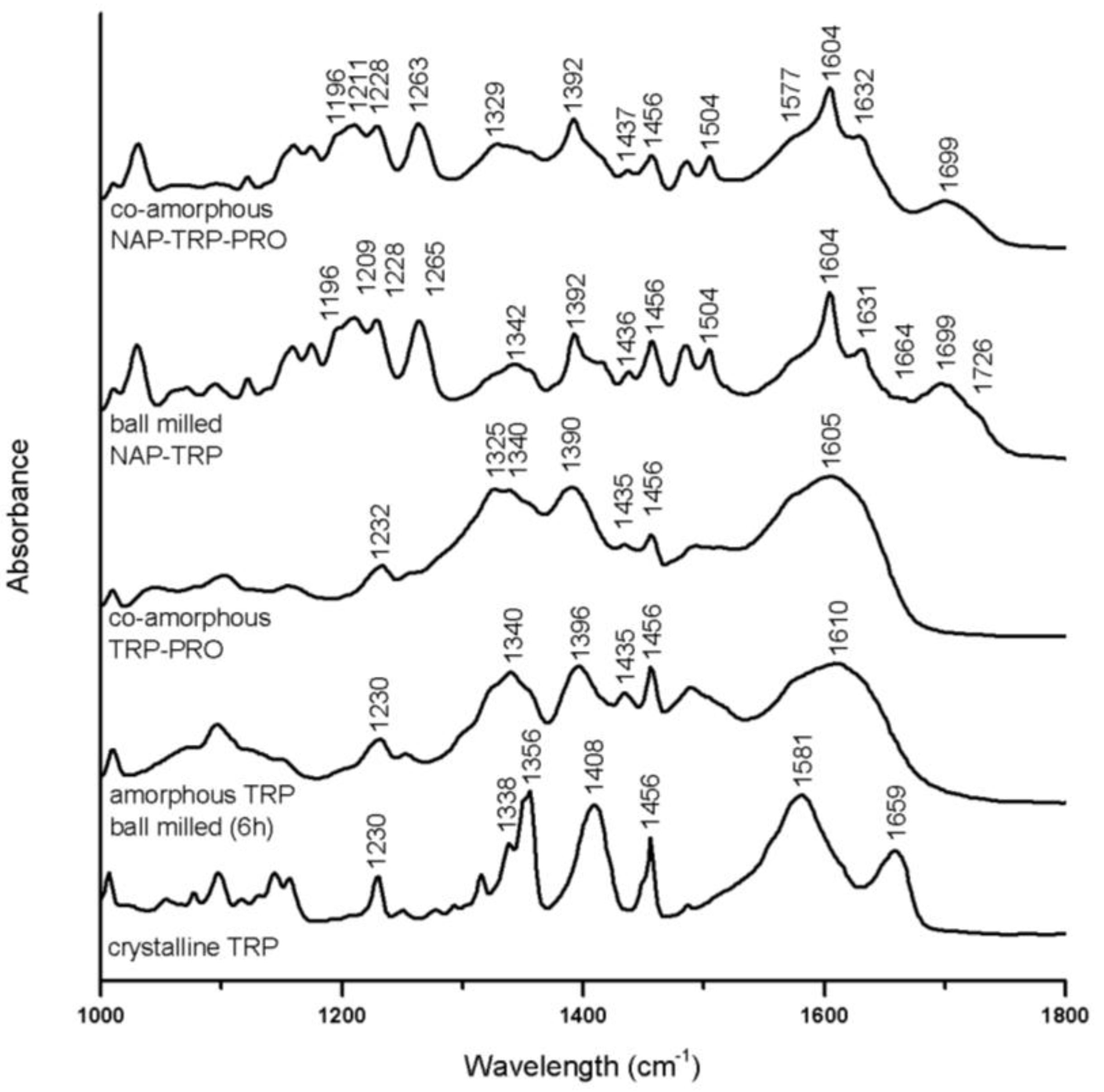
3.3.1. Interaction in Tryptophan (TRP) Containing Mixtures
Several alterations can be observed in the TRP spectra upon amorphization after 6 h of ball milling. Broader bands, as well as decreased peak intensities can be observed for the aromatic region (1200–1500 cm
−1) (
Figure 6) [
32]. Besides the general broadening of the peaks, the most pronounced change is related to the CO
2− asymmetric stretch at 1659 cm
−1 and the amide band at 1581 cm
−1 in crystalline TRP, which are combined to one broad band around 1610 cm
−1 in amorphous TRP [
33].
In co-amorphous TRP–PRO, the C=O stretches or amid bands usually found between 1500 cm
−1 and 1700 cm
−1 in the crystalline spectra fused to a broad band with a maximum around 1605 cm
−1, as also observed for amorphous TRP. These findings, as well as a further broadening of the peaks in the aromatic region compared to amorphous TRP are an indication of molecular interactions. Strong interactions between the TRP and PRO molecules comply well with the DSC data, which showed a particularly high T
g for the mixture of these amino acids (
Table 2).
This combined band together with amorphous NAP peaks is seen in the spectrum of amorphous NAP–TRP as a peak with a maximum at 1604 cm
−1. The carbonyl symmetric stretch at 1396 cm
−1 in amorphous TRP is further decreased in NAP–TRP, which indicates that this group is involved in interactions with NAP. The crystalline TRP band at 1659 cm
−1 is transformed into a peak with decreased intensity and maximum at 1664 cm
−1 in the NAP–TRP spectrum, which supports the occurrence of interactions involving CO
2− [
33]. The peaks in the aromatic region of the NAP–TRP spectrum are similar to amorphous TRP and can further be compared to the alterations previously observed upon forming co-amorphous carbamazepine–TRP, where hydrogen bonds, including the TRP carboxylic acid and amide group, as well as π–π interactions between the aromatic rings, were observed [
14]. The major alterations in the NAP–TRP spectrum related to NAP are an additional reduction and shifts in the hydrogen bonded (1699 cm
−1) and the free carboxylic acid (1726 cm
−1) peaks, compared to amorphous NAP. These findings support that NAP–TRP heterodimers are formed upon ball milling, interacting via hydrogen bonds involving the carboxylic group in NAP in contrast to the homodimers in amorphous NAP. Similar heterodimers have been observed between NAP and indomethacin upon amorphization [
26,
30]. The peak observed at 1726 cm
−1 in the spectrum for ball-milled NAP–TRP indicates free carboxylic groups, which can be related to unbound NAP molecules, as also observed for crystalline and amorphous NAP (
Figure 6). In a completely co-amorphous mixture, all NAP molecules are expected to be involved in interactions. Thus, free C=O groups confirm the presence of crystalline NAP molecules in the mixture, observed as small remaining reflections by the XRPD (see above). Changes in the aromatic region of the spectra upon amorphization of NAP together with TRP are very similar to the spectra for amorphous NAP, further suggesting π–π interactions, as observed as a consequence of molecular changes of TRP.
In the ternary co-amorphous NAP–TRP–PRO mixture, the distinct peaks referring to free acids and carbonyls (1650–1750 cm
−1) are transformed into a broad peak with a maximum at 1699 cm
−1. Additional changes are observed in this region of the NAP–TRP–PRO spectrum compared to NAP–TRP, indicating that all three components are involved in interactions, which is in line with the findings of the DSC measurements (
Figure 3). The crystalline TRP amide bond at 1581 cm
−1 appears to remain as a small shoulder at 1577 cm
−1 in co-amorphous NAP–TRP–PRO. However a similar pattern is observed in the spectra of amorphous TRP and co-amorphous TRP–PRO, suggesting that the shoulder is not an indication of remaining crystallinity (
Figure 6). Alterations related to the TRP amide, as well as the aromatic vibrations in NAP and TRP are almost identical in the ternary mixture and ball-milled NAP–TRP, which indicates π–π interactions besides hydrogen bonds in co-amorphous NAP–TRP–PRO.
Overall, the changes in the aromatic region of NAP and TRP indicate π–π interactions in ball-milled NAP–TRP and co-amorphous NAP–TRP–PRO. Additional changes in vibrations indicate hydrogen bonding between the TRP carboxylic acid or amide group and the NAP carboxylic acid group in both mixtures. However, unbound NAP molecules appear in ball-milled NAP–TRP, observed as remaining free carbonyl groups, as this mixture is not completely amorphous. PRO contributed to further hydrogen bonding in co-amorphous NAP–TRP–PRO, observed as additional changes in the bands around 1699 cm−1, which could explain the absence of unbound NAP molecules, leading to a completely amorphous mixture.
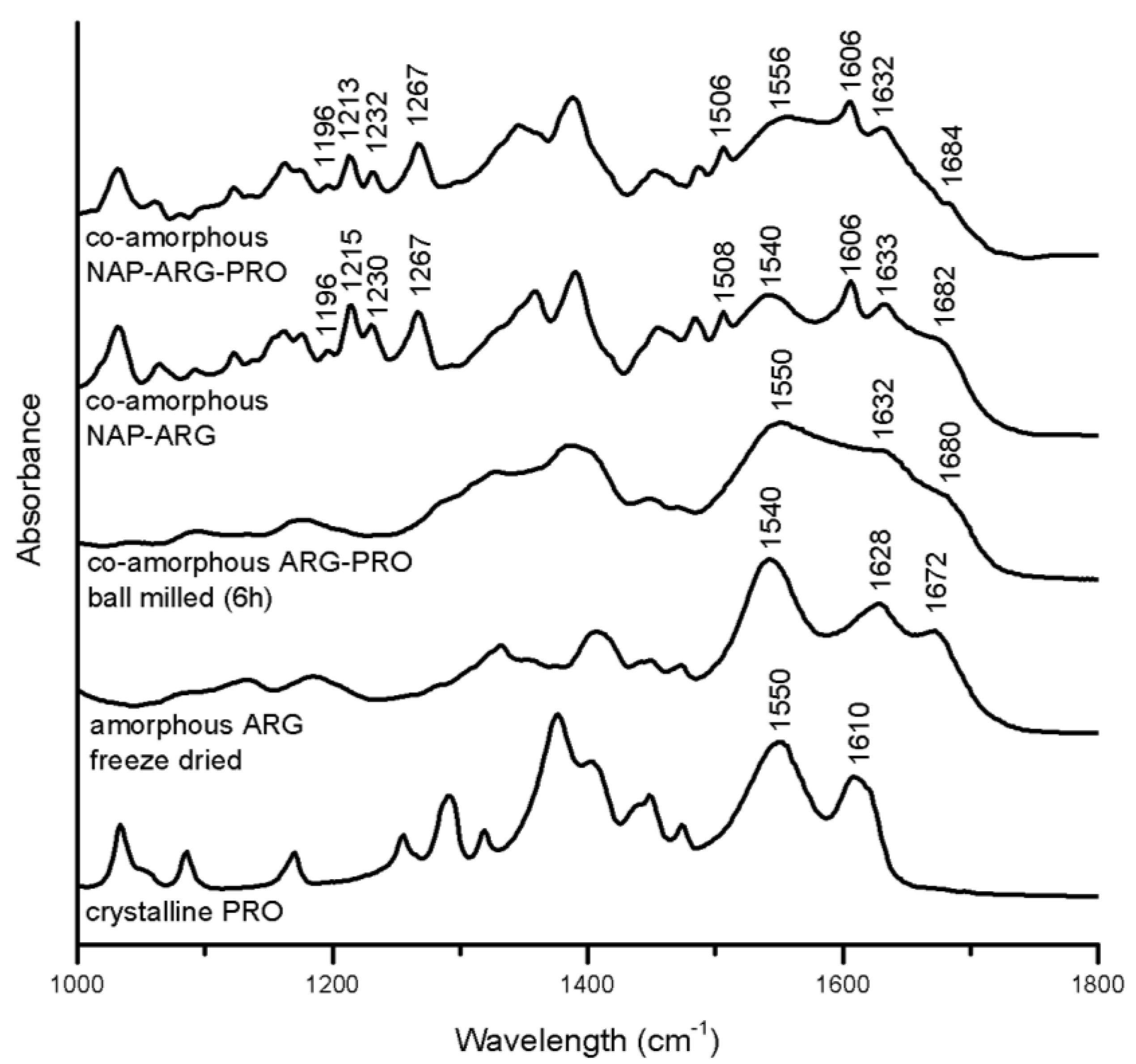
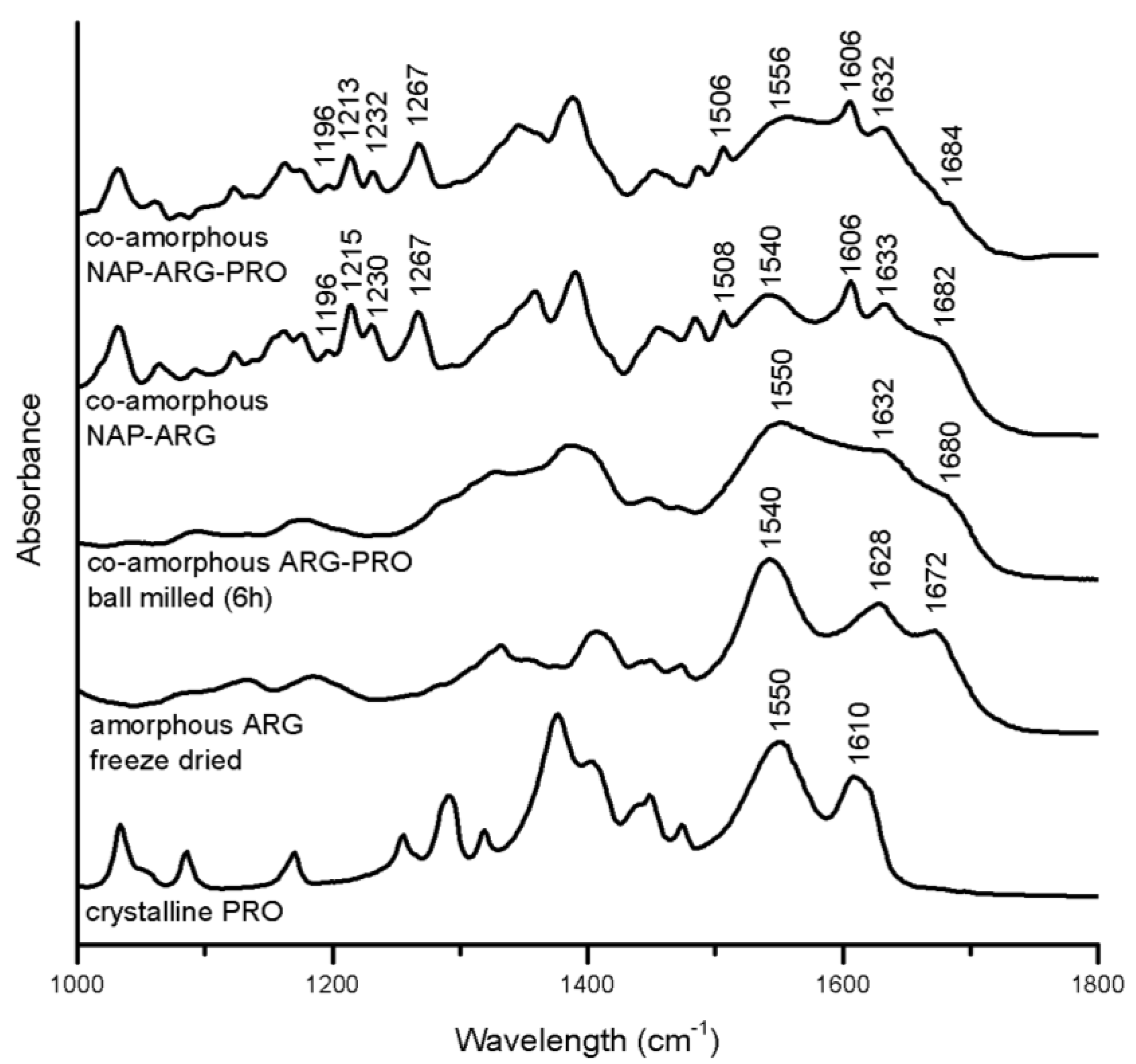
3.3.2. Salt Formation in Arginine (ARG) Containing Mixtures
Several indications for interactions in the ARG containing mixtures are seen with FTIR (
Figure 7). In the spectrum of co-amorphous NAP–ARG, the free carboxylic acid band of NAP (1697 and 1728 cm
−1) cannot be observed, indicating a salt formation between the acidic drug and the basic amino acid, as also suggested earlier [
18]. The corresponding ionized carboxyl group, which usually appears around 1505–1610 cm
−1 [
34], is difficult to identify, due to overlapping peaks, similar to previously investigated co-amorphous indomethacin–ARG mixtures [
14]. Furthermore, the amide vibration at 1540 cm
−1, as well as vibrations related to the guanidyl group (1600–1700 cm
−1) are reduced compared to amorphous ARG, supporting the likelihood of salt formation with NAP [
32]. Besides the salt formation, it is possible that the guanidinium group in ARG interacts with the aromatic rings in NAP, resulting in π–π interactions, as described between ARG and other aromatic drugs [
16].
In the spectrum of co-amorphous NAP–ARG–PRO, the amide vibration of PRO was shifted from 1550 cm
−1 in the crystalline spectrum to 1556 cm
−1, similar to the shoulder in co-amorphous ARG–PRO at 1550 cm
−1 [
35]. Furthermore, the C=O band at 1610 cm
−1 in the spectrum of PRO cannot be found in the co-amorphous blend [
32]. In co-amorphous ARG–PRO, this band has changed into a shoulder at 1632 cm
−1, which cannot be observed in the spectrum of co-amorphous NAP–ARG–PRO, as it is overlapping with NAP vibrations. These changes are indicative of molecular interactions between PRO with either one or two of the other components, NAP and ARG, in the ternary mixture. The alteration in the NAP aromatic region observed in NAP–ARG and NAP–ARG–PRO could be associated with π–π interactions, as could the changes in the aliphatic vibration in PRO (1390–1300 cm
−1) [
32]. Similar interactions between the guanidinium group in ARG and the aromatic rings of other drugs have previously been described [
16]. The most pronounced interactions in both co-amorphous mixtures, including NAP and ARG, was the salt formation; however, additional interactions involving PRO are suggested to occur in the ternary mixture.
Figure 6.
FTIR spectra of crystalline TRP, amorphous TRP ball milled for 6 h, co-amorphous TRP–PRO, the ball-milled mixture, NAP–TRP, and co-amorphous NAP–TRP–PRO.
Figure 6.
FTIR spectra of crystalline TRP, amorphous TRP ball milled for 6 h, co-amorphous TRP–PRO, the ball-milled mixture, NAP–TRP, and co-amorphous NAP–TRP–PRO.
Figure 7.
FTIR spectra of crystalline PRO, as well as the spectra of amorphous freeze-dried ARG, co-amorphous ARG–PRO ball milled for 6 h, co-amorphous NAP–ARG and co-amorphous NAP–ARG–PRO.
Figure 7.
FTIR spectra of crystalline PRO, as well as the spectra of amorphous freeze-dried ARG, co-amorphous ARG–PRO ball milled for 6 h, co-amorphous NAP–ARG and co-amorphous NAP–ARG–PRO.
3.4. Storage Stability
Co-amorphous NAP–ARG and NAP–TRP–PRO were stable throughout the period of the stability study (332 days at room temperature and at 40 °C under dry conditions). The samples where considered stable when observing a halo in the XRPD diffractogram. The co-amorphous NAP–ARG–PRO mixture was stable for approximately 84 days on storage at room temperature and 40 °C (
Table 3). Interestingly, the powder collapsed, resulting in a hard solid matrix after 84 days of storage. The XRPD diffractogram of the stored sample revealed small reflections, indicating crystalline PRO in the matrix. The glass transition temperature has often been shown to correlate with the stability of an amorphous system [
36], and the reduced stability of co-amorphous NAP–ARG–PRO compared to co-amorphous NAP–ARG can be explained by a lowered T
g. However, this tendency was not shown for the NAP–TRP–PRO blend, even though the T
g was similar to the T
g of NAP–ARG–PRO. It is known that strong molecular interactions can have a higher impact on the physical stability of a co-amorphous system than a higher T
g [
11,
26]. From the stability data, it is suggested that PRO contributed less to the overall interactions in a mixture with the NAP–ARG salt, compared to stronger interactions between the molecules in co-amorphous NAP–TRP–PRO. Thus, the suitability of PRO in a co-amorphous mixture might depend on its ability to interact with the other components. In the examples of this study, PRO appears to be a good stabilizer in the non-salt NAP–TRP–PRO blend, enhancing the interactions between all three components and, thus, influencing its amorphization behavior during ball milling, recrystallization upon heating in the DSC and stability during storage, as well as its dissolution behavior (see below).
Table 3.
Stability of co-amorphous NAP–TRP–PRO, NAP–ARG and NAP–ARG–PRO upon storage at room temperature and 40 °C under dry conditions.
Table 3.
Stability of co-amorphous NAP–TRP–PRO, NAP–ARG and NAP–ARG–PRO upon storage at room temperature and 40 °C under dry conditions.
| Sample Content | Stability at Room Temperature | Stability at 40 °C |
|---|
| NAP–TRP–PRO | >332 days | >332 days |
| NAP–ARG | >332 days | >332 days |
| NAP–ARG–PRO | 84 days | 84 days |
3.5. Intrinsic Dissolution Testing
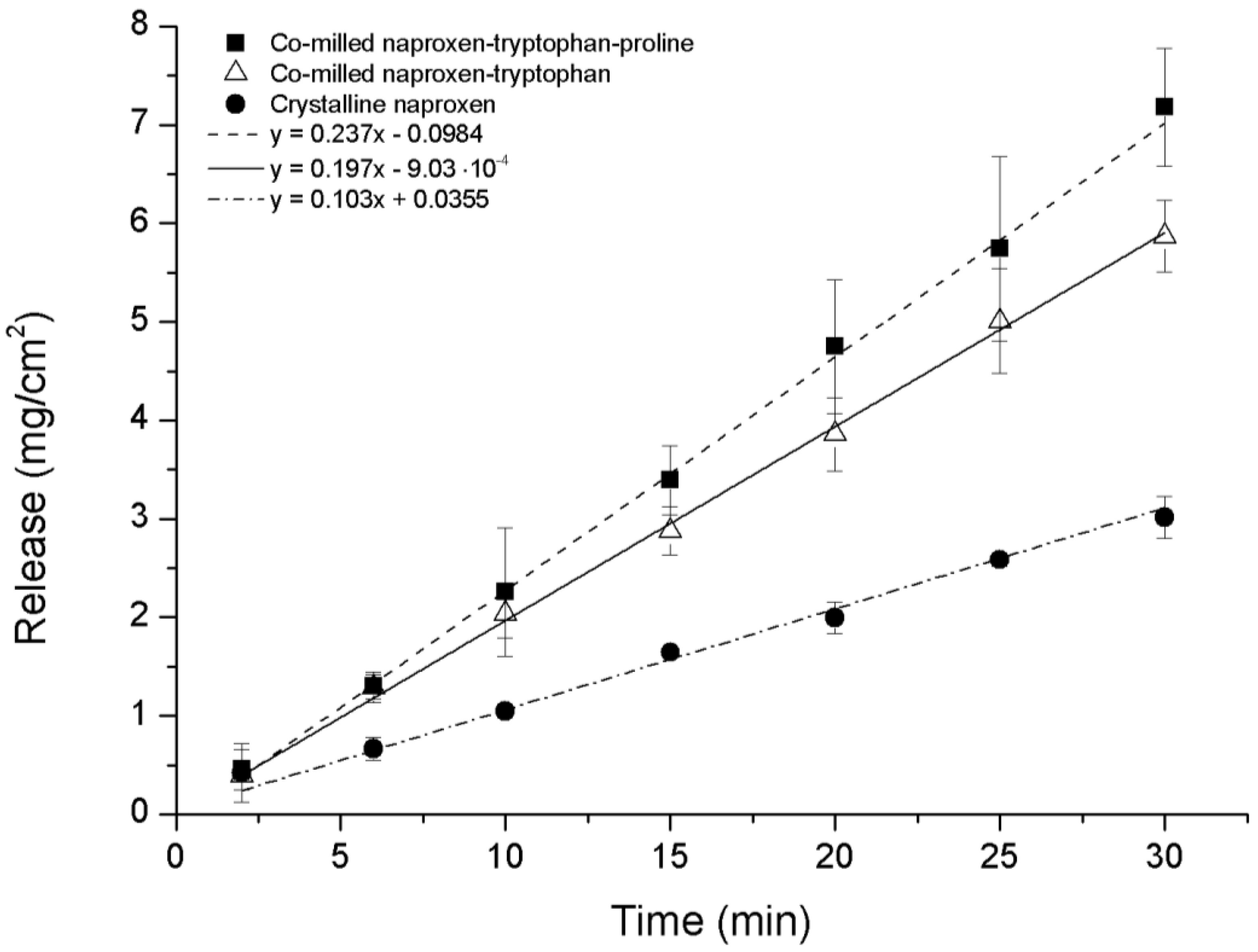
The intrinsic dissolution profile of NAP was measured for ternary co-amorphous mixtures containing PRO and binary co-amorphous mixtures and compared to crystalline NAP. Although it was not possible to transform NAP–TRP into a completely co-amorphous mixture, the intrinsic dissolution rate (IDR) of co-milled NAP–TRP was measured and resulted in a 1.9-fold increase compared to crystalline NAP. An even higher dissolution rate of NAP could be found for the ternary co-amorphous systems (
Figure 8). Linear regression showed that the curves where significantly different from one another (
p < 0.05). Further, the amount of NAP dissolved from NAP–TRP–PRO was significantly higher than for NAP–TRP after 15 min and onwards. The improved IDR of NAP can be assigned to the high solubility of PRO combined with the improved stabilization of the co-amorphous system and might be indicative of a three way interaction in the mixture between the components, where the overall gain in the dissolution rate is determined by the solubility of all three components. These findings suggest that it can beneficial to consider the solubility of the amino acid when choosing components for a co-amorphous mixture, resulting in the high dissolution rate of the drug.
Figure 8.
Intrinsic dissolution profiles of crystalline NAP, non-amorphous ball-milled NAP–TRP and co-amorphous NAP–TRP–PRO (n = 3, mean ± standard deviation).
Figure 8.
Intrinsic dissolution profiles of crystalline NAP, non-amorphous ball-milled NAP–TRP and co-amorphous NAP–TRP–PRO (n = 3, mean ± standard deviation).
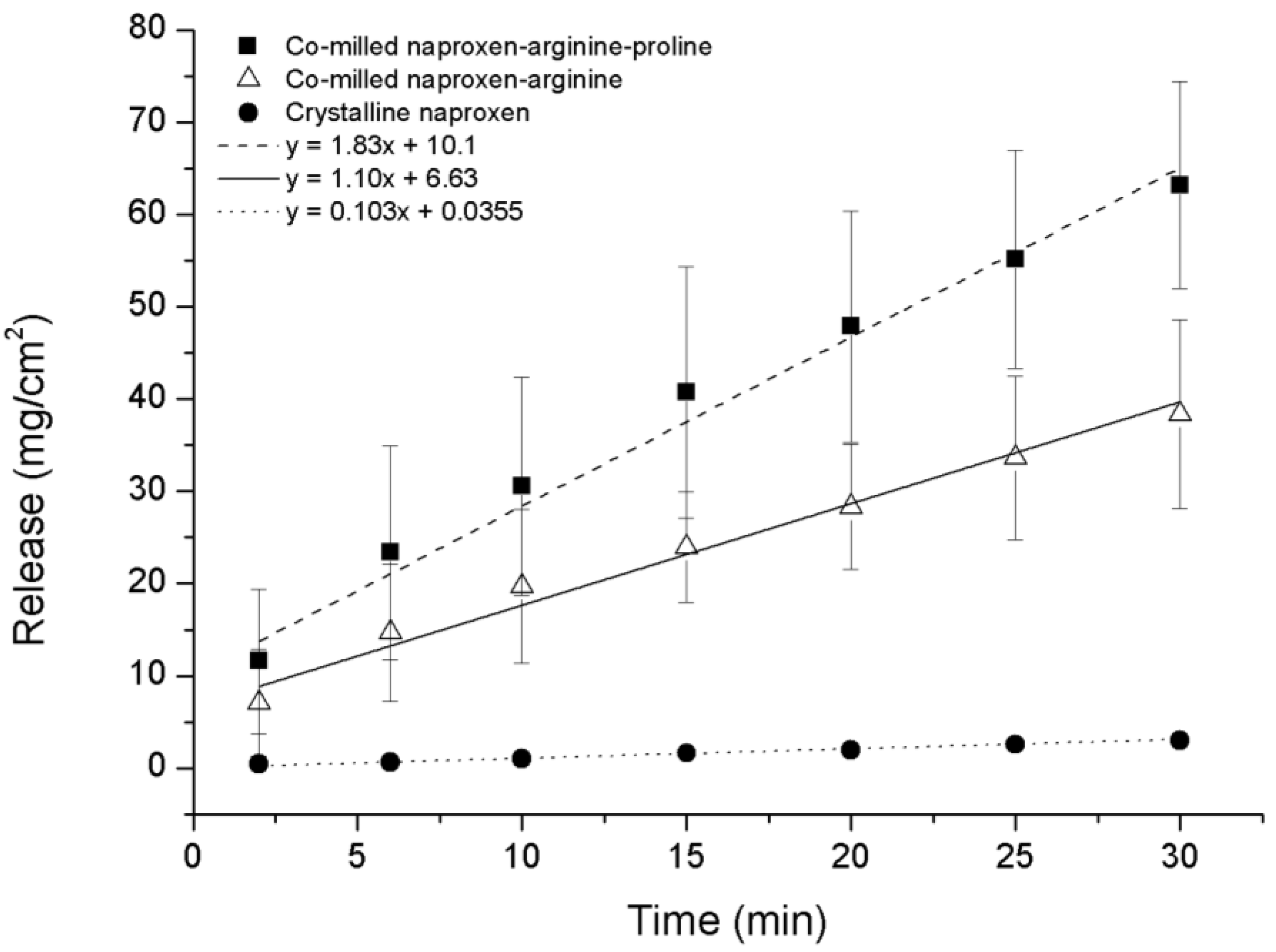
As expected, the IDR of the NAP–ARG salt in the co-amorphous mixture was faster compared to crystalline NAP (
Figure 9). A further improvement in the dissolution rate could be observed from the ternary co-amorphous mixture additionally including PRO, resulting in 1.6-fold and 18-fold increases when compared to the NAP–ARG salt and pure crystalline NAP, respectively. Linear regression and ANOVA showed significant improvement in the amount of NAP dissolved from NAP–ARG–PRO observed at all time points, as well as the slope compared to NAP–ARG and crystalline NAP.
In
Figure 9, a high
y-intercept is observed for the dissolution profile of NAP from co-amorphous NAP–ARG, as well as NAP–ARG–PRO systems, indicating a burst release of NAP. A possible explanation could be supersaturation occurring in the area surrounding the disc at the beginning of the dissolution experiment, resulting in the re-crystallization of NAP at the surface of the disc. This theory was supported by XRPD measurements of the disc surface after the dissolution experiment, where crystalline NAP reflections could be observed. Re-crystallization of the drug at the disc surface has previously been observed for other co-amorphous mixtures [
11].
The main focus of co-amorphous formulations has previously been to investigate non-salt forming drugs, as alternative methods are needed in order to improve the solubility and dissolution rate of such drugs [
13]. Combining the model drug NAP with TRP and PRO confirms the ability of a co-amorphous system to fulfil this formulation purpose by improving the dissolution rate of the drug without forming a salt. However, the high dissolution rate of NAP achieved by combining NAP, ARG and PRO suggested that a co-amorphous system can also further improve the dissolution rate of an amorphous salt. This confirms the potential of the co-amorphous formulation approach, even for salt-forming drug candidates.
Figure 9.
Intrinsic dissolution profiles of crystalline NAP, co-amorphous NAP–ARG and co-amorphous NAP–ARG–PRO (n = 3, mean ± standard deviation).
Figure 9.
Intrinsic dissolution profiles of crystalline NAP, co-amorphous NAP–ARG and co-amorphous NAP–ARG–PRO (n = 3, mean ± standard deviation).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}