
40 Years of Helicobacter pylori: A Revolution in Biomedical Thought

1
Interdepartmental Research Center for Pre-Latin, Latin and Oriental Rights and Culture Studies (CEDICLO), University of Bari, 70121 Bari, Italy
2
Department of Clinical Disciplines, School of Technical Medical Sciences, University of Elbasan “A. Xhuvani”, 3001 Elbasan, Albania
*
Authors to whom correspondence should be addressed.
Gastroenterol. Insights 2021, 12(2), 111-135; https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12020011
Submission received: 26 February 2021
/
Revised: 16 March 2021
/
Accepted: 22 March 2021
/
Published: 24 March 2021
(This article belongs to the Collection Advances in Gastrointestinal Cancer)
Abstract
:Background: Various microorganisms such as bacteria, virus, and fungi can infect humans and cause not just a simple infection but septic conditions, organ dysfunction, and precancerous conditions or cancer involving various organ systems. After the discovery of the microscope, it was easier to discover and study such microorganisms, as in the case of Helicobacter pylori, a pathogen that was seen in the distant era of the nineteenth century but without being recognized as such. It took 100 years to later discover the pathogenesis and the cancer that this bacterium can cause. Since it was discovered, until today, there has been a continuous search for the understanding of its pathogenetic mechanisms, and the therapeutic approach is continuously updated. Methods: We investigated how diagnosis and therapy were dealt with in the past and how researchers sought to understand, exactly, the pathogenetic biomolecular mechanisms of H. pylori, from the genesis of the infection to the current knowledge, with an analysis of carcinogenic mechanisms in the stomach. We have examined the scientific evolution of the knowledge of the disease over these 40 years in the gastroenterological and pharmacological fields. This was possible through a search in the databases of Medline, the WHO website, the Centers for Disease Control and Prevention (CDC) website, PubMed, and Web of Science to analyze the earlier and the latest data regarding H. pylori. Results: With the scientific discoveries over time, thanks to an increasing number of progressions in scientific research in the analysis of the gastric mucosa, the role of Helicobacter pylori in peptic ulcer, carcinogenesis, and in some forms of gastric lymphoma was revealed. Furthermore, over the years, the biomolecular mechanism involvement in some diseases has also been noted (such as cardiovascular ones), which could affect patients positive for H. pylori. Conclusions: Thanks to scientific and technological advances, the role of the bacterium H. pylori in carcinogenesis has been discovered and demonstrated, and new prospective research is currently attempting to investigate the role of other factors in the stomach and other organs. Cancer from H. pylori infection had a high incidence rate compared to various types of cancer, but in recent years, it is improving thanks to the techniques developed in the detection of the bacterium and the evolution of therapies. Thus, although it has become an increasingly treatable disease, there is still continuous ongoing research in the field of treatment for resistance and pharma compliance. Furthermore, in this field, probiotic therapy is considered a valid adjuvant.
1. Introduction
Helicobacter pylori is a Gram-negative bacterium, microaerophilic (requires O2 at a lower concentration than in the atmosphere: 5% O2, 15% CO2, and 80% N), has a helical shape, and is categorized as a curved rod not spirochaete bacterium (3 μm long, diameter about 0.5 μm) that usually colonizes the gastric mucosa [1]. It has the ability to transform from bacillary to coccoid form (a survival form) [2] and can form organized bacterial surface colonies (bio-membranes). In its spiral form, it can survive for at least a week in water, and in the coccoid form, probably for much longer periods. The outer layer of the membrane is made up of phospholipids and lipopolysaccharides (LPS) but also cholesterol glycosides (which are present in a few other Gram-negative bacteria). Is highly motile thanks to the presence of flagella (four to six flagella at the same location) as with all the Helicobacter spp. (gastric and enterohepatic ones), composed of two copolymerized flagellins, FlaA and FlaB [3,4]. It contains a hydrogenase that uses H2 from intestinal bacteria by oxidizing it for energy and also produces oxidase, catalase, and urease. H. pylori consists of a wide diversity of strains and genomes that have been completely sequenced. H. pylori strain 26,695 has a circular genome of approximatively 1.7 million base pairs and 1590 genes. Studies on the genome have been necessary to understand the pathogenetic mechanisms, or its ability to cause diseases [5,6,7].
H. pylori infection is extremely common and is thought to affect more than 50% of the world population. The annual incidence rate of H. pylori differs significantly between developed and developing countries, with this difference mainly affecting childhood. Later in adulthood, the annual rate of infection increases for developing countries and decreases in developed countries due to socioeconomic conditions. Transmission is via fecal–oral, oral–oral (dental), gastro–oral, and sexual behavior routes [8]. H. pylori can be detected in dental plaque (oral biofilms) and from the feces of infected adults both by culture and biomolecular techniques (PCR). The infection appears to be acquired throughout life, mainly starting in infancy [9]. Interfamilial transmission is responsible for the higher incidence of infections in parents and siblings of H. pylori-positive children and children of H. pylori-positive parents [10,11]. The isolation of H. pylori strains of the same genotype in mothers and their offspring—sometimes different genotypes from that of fathers—highlights the role of close contact [12,13]. This study seeks to analyze all the knowledge of the biomolecular mechanisms, diagnosis, and treatment of infection to date through a historical scientific path. Of particular interest is the association of H. pylori infection with extra-digestive diseases.
2. Materials and Methods
A search of manuscripts was carried out in large databases from 1981 until present. From the results, the most innovative ones of each period concerning the history of the discovery, the epidemiology, and the physiopathology of carcinogenesis of H. pylori infection were chosen.
2.1. Historical Helicobacter pylori Traces
Since the time of ancient Egypt (Ebers Papyrus, 1600 BC), and later in the classical world with Hippocrates, Galen, Dioscurides, and others, gastrointestinal symptoms were described and various remedies, e.g., extracts from specific herbs, were used to treat various gastric symptoms (such as Elettaria cardamomum for indigestion and flatulence) or antidotes such as Theriaca for gastrointestinal or other poisoning [14,15,16].
Later, at the end of the sixteenth century, a separate invention was the beginning to forever change the study of organisms. In 1595, the Dutch pair Hans and Zacharias Janssen (father and son) built the first microscope to enlarge an image 20 times. Thereafter, in 1674, Anthony van Leeuwenhoek (1632–1723), a naturalist and optician, created his microscope and magnified objects 300 times (Figure 1) [17,18]. Furthermore, he made important biological discoveries such as red blood cells, numerous bacteria and protozoa (describing, with a good precision, Volvox, Vorticella, Opaline, Nyctotherus, Trichomonas vaginalis, Bacterium coli, Azotobacter, Amylobacter saccarobutyricum, and others). He was the first person to see microorganisms, which he called small “insects” (animalcules). In fact, he noticed a sample of his teeth and this was the first time that bacteria were observed. Van Leeuwenhoek noted: “There are smaller bugs in a man’s toothpick than there are in an entire kingdom“. It soon became clear that the creatures discovered by Leeuwenhoek not only live in the world around them, but also have a significant impact on it [18].
At the same time, the English scientist Robert Hooke (1635–1703) built his microscope that could enhance 500 times and observed thin sections of cork containing many “cells”, a term we still use today for the life unit. They discovered that they are also responsible for human and animal diseases, and the most contagious historical epidemic infections such as plague of cholera, syphilis, and others. In the following years, microscopes continuously evolved, and today, we have optical microscopes and also powerful electronic microscopes that allow us to see the structures of microorganisms, including viruses, because the magnification is more than 300,000 times [18,19].
The study of bacteria of the gastric mucosa by microscope began from 1875 onwards, when G. Bottcher and M. Letulle demonstrated the presence of bacteria in the margins of ulcers (Table 1) [20,21]. Subsequently, in 1892, the Italian scientist G. Bizzozero was one of the pioneers of histology and of the use of the microscope in medical research. He identified spirochetes (spiral bacteria) in the gastric mucosa of dogs, thus demonstrating that some bacteria can survive in the acidic environment of the stomach (Figure 2) [20,22,23,24,25]. Later, the term peptic ulcer or peptic ulcer disease (PUD) was used for the first time in 1882 by Heinrich Irenaeus Quincke, emphasizing that gastric juice is responsible for gastric ulcers compared to the previous hypothesis that pepsin is responsible (digestion theory). In the first 10 years after its discovery in 1981, it was proven that H. pylori accounts for 80% of peptic ulcers, even though scientists for years ignored the possibility that it was due to a germ [20,21,23].
In 1906, the first medical report on the presence of spirochetes in the gastric contents of patients with human target spiral gastric carcinoma bacteria was published by W. Krienitz. Later, in the 1920s, the urease enzyme was discovered in the gastric mucosa of primates. In the 1930s, the first necropsy study on gastric tissue samples in patients that died with gastric carcinoma was performed in the United States, which had a high incidence rate at that time. In the analyzed samples, the presence of "spirochetes" bacteria was observed in 43% of the samples [20,21,23]. Subsequently, the evolution of endoscopic techniques allowed to obtain a more accurate observation of gastric sites where colonies of Gram-negative bacteria were identified, especially in patients with gastric pathologies [21,23]. In 1981 began the first trial by Robin Warren of patients selected by Barry Marshall. Subsequently, in 1983, the culture of spiral microbes from gastric mucosal biopsies from patients with chronic active gastritis was announced for the first time. Indeed, the researchers Robin Warren and Barry Marshall performed the culture and isolation of spiral bacteria in 58 gastric biopsies, which were named Campylobacter pyloridis, because the bacterium had the morphological characteristics of the Campylobacter genus. This is due to the similar biological characteristics and the similar guanine/cytosine ratio (G+C ratio: 30–48% molar) in Campylobacter spp. [24,25,26]. Experimental evidence of the role of H. pylori in causing gastric damage was subsequently confirmed by the fulfillment of Koch’s criteria for pathogenic microorganisms by B. Marshall [27]. Through the ingestion of H. pylori from a patient with gastritis, as a result, he became sick himself. The two Australian researchers, Marshall and Warren, were the first to carry out the cultivation. Subsequently, in 1987, it was decided to rename the bacterium Campylobacter pylori. Additionally, it was the first demonstration of a relationship between H. pylori and gastric damage, and in the same year, the foundation of the European H. pylori Study Group (EHSG) was created. However, in 1989, after the morphological and genetic identification of this microorganism showed that it could not be considered a member of the Campylobacter genus, the name Helicobacter pylori prevailed, a name that is still used internationally [8,28,29,30,31,32]. The need to identify the new genus Helicobacter was indicated by the sequences of Campylobacter jejuni and H. pylori, the results of an extensive analysis of DNA–DNA hybridization experiments, the sequence of rRNA genes 16S and 23S, the fatty acid profile of membranous phospholipids and respiratory mitochondrial chain components [33,34,35], Meanwhile, since 1990 and until 2000, many other Helicobacter spp. have been discovered such as those of H. felis, H. bilis, and others [36]. Subsequently, the etiological association of H. pylori infection and its correlation with gastric pathologies such as gastritis, gastroduodenal ulcer, gastric cancer, and, finally, mucosa-associated lymphoid tissue (MALT) lymphoma (MALToma) radically changed the treatment of gastric pathologies. The discovery was so important that it was recognized by the International Medical Community, which awarded Marshall and Warren with the Nobel Prizes for Medicine in 2005 [21,36,37,38].
The time period during which H. pylori colonized the human stomach remains unknown, but the phylogenetic characteristics of the different geographic populations of H. pylori reflect significant events in human prehistory. Studies have shown that thanks to the migration of populations, the bacterium migrated from the African continent to the rest of the world about 58,000 years ago and has been estimated to have colonized humans before 115,000 years (Figure 3) [39,40]. Phylogenetic comparisons showed a similar evolution in humans and H. pylori and that the bacterium’s natural host is humans. Therefore, zoonotic transmission is not possible, despite sporadic isolation of gastric strains of H. pylori in some mammals such as felines, monkeys, and others [41].
2.2. The Biomolecular Knowledge Developments
H. pylori, in order to finally exert its pathogenic action, must firstly survive in the acidic gastric environment and penetrate the protective barrier of the gastric mucosa to eventually colonize the gastric mucosa (Table 2). Subsequently, to multiply inside the mucosa, it must escape the host’s immune defense mechanisms [42].
H. pylori possesses five major families of outer membrane proteins (OMPs) [24] such as adhesins, porins, iron transporters, flagellar proteins, and other proteins with unknown function [43]. Thus, the virulence factors to achieve this are divided into two categories. The first includes the factors necessary for the initial colonization of the gastric mucosa, common to all H. pylori strains (except for some mutant strains). The second includes flagella, adhesins (BabA/B, SabA, AlpA/B, OipA, and HopZ), and urease. The flagellum has a spiral shape which allows it to move from the gastric lumen (where the pH is acidic) deep into the mucus, where the pH is almost neutral, and allows it to grow and multiply (Figure 4) [44,45].
Adhesins are proteins which are considered essential for the stable adhesion of H. pylori to gastric epithelial cells (Figure 5) Finally, the production of urease was initially thought to be a necessary factor for the infection, needed in large amounts [48,49]. In fact it was demonstrated through a complete transcription analysis using differential RNA-seq, which confirmed the main virulence loci such as the urease operongene cluster ureABIEFGH and the pathogenicity island of the cag. In addition, H. pylori arginase has a role in allowing the bacterium to not be recognized from the immune system by competing with nitric oxide (NO) synthase for L-arginine, thereby reducing NO production. However, NO is not only an antimicrobial agent but also a useful component of innate immunity [48,50].
The second group of virulence factors is not considered necessary for initial colonization and consists of polysaccharidases, adhesion protein bab A2, cip A proteins (encoded by the HPO 638 gene), Heat shock HspA and HspB proteins (hspA and hspB heat-shock gene cluster), exotoxins such as vacuolating cytotoxin A (VacA), and lytic enzymes (mucinases, proteases, and lipases) that cause damage to the mucosa [53]. The infection, therefore, promotes a greater production of cytokines by itself (interleukins and tumor necrosis factor-α (TNF-α)), which has a negative effect on the cells and the whole organism. The toxigenic gene vacuolating cytotoxin autotransporter (vacA Q48245) and its subtypes s1/m1 and s1/m2 promote disruption of the calcium channel TRPML1, thus increasing the risk for the development of gastric carcinoma. [54] However, among the virulence agents of H. pylori is the antigenic protein cytotoxin-associated protein A (CagA, molecular weight 125–140 kDa). CagA is an immunomodulatory extracellular protein of some strains of H. pylori (Figure 6) [55,56].
After 1997, the cag region of microbial DNA was identified, which was designated as cag pathogenicity island—cag-PAI (dimension ~40 kbp, composed by ~30 genes). It is considered the main virulence factor of H. pylori [57]. Some researchers compared this region of pathogenicity to those found in uropathogenic strains of Escherichia coli. Considering this and the presence of a low GC content in cag-PAI, we can hypothesize that cag-PAI was probably acquired by horizontal transfer from another bacterial species such as E. coli [58,59,60]. The cag-PAI region is responsible for encoding the CagA protein as well as for the transfer of this protein and possibly other Helicobacter proteins from the bacterium to the cytoplasm of the epithelial cells of the gastric mucosa (Figure 7) [60,61,62]. Upon entering the epithelial cell, the CagA protein is phosphorylated by modifying the action of many intracellular enzyme systems such as that of the SHP-2(protein tyrosine phosphatase) system [63]. This action will lead to the activation of apoptotic mechanisms, interruption of the normal cell proliferation of the gastric epithelium and promoting a more intense inflammatory response from the host [64,65]. In fact, the strains of CagA(+) H. pylori have a high infectious power, determined by both the high anti-inflammatory response and the dysregulation of the proliferative system of gastric epithelial cells by the CagA protein. In fact, high production of pro-inflammatory cytokines (L8, IL6, IL1 α, IL1 β) in the stomach has been found in patients infected with CagA(+) H. pylori strains. The serine protease HtrA also plays a main role in the pathogenesis because enables the translocation of CagA and aids H. pylori to transmigrate across the epithelium [66,67].
2.3. The Associated Diseases
Colonization of the stomach by H. pylori in most patients can cause acute gastritis or chronic non-atrophic gastritis (diffuse antral predominant gastritis or superficial gastritis) from the first moment of infection at the site of colonization (Table 3). Therefore, we can have acute gastritis and also a severe neutrophilic gastritis with transient hypochlorhydria. Infection causes no symptoms in about 80% of those infected. Histologically, an inflammatory infiltration of the stomach wall is observed in which H. pylori bacteria are found [71,72,73]. Inflammatory infiltration is limited in the pyloric area, while inflammatory lesions are limited or not observed in the stomach body. This is due to the production of ammonia which is cytotoxic (to counteract and regulate the pH in its favor) together with proteases, some phospholipases, vacuolating cytotoxin A (VacA), and the CagA gene (potentially carcinogenic). Additionally, cysteine-rich Hcp proteins (particularly HcpA), in turn, cause inflammation by triggering host immune responses. Finally, the bacterium also increases cyclooxygenase-2 (COX-2) levels in chronic gastritis, which can probably be the basis for extragastric diseases [71,74,75,76]. H. pylori is the main agent of peptic ulcer, and not all patients can develop this condition (the reason still remains unknown). On the other hand, 20% of patients with H. pylori infection will develop peptic ulcer during their lifetime. Ulcers in the stomach and duodenum occur when the consequences of inflammation destroy the mechanisms that protect the mucous membranes from stomach acid and pepsin [73,74]—hence, the site of H. pylori colonization, and the one that will be affected by the two factors. H. pylori colonizes the pyloric antrum in subjects with large acid production, whereas in those producing normal or small amounts, it can colonize various locations in the stomach. If it is then located near the pyloric antrum, the inflammatory response induces the G cells of the antrum to secrete gastrin, which stimulates the parietal cells that increase the secretion of acid which damages the duodenum, causing ulcers with an increase in these cells over time [75]. When colonizing other sites in the stomach, the inflammatory response can cause gastric atrophy and, ultimately, ulcers, increasing the risk of cancer [73,76]. Finally, the growth hormone-releasing peptides (GHRPs) that inhibit normal food intake and reduce gastrointestinal motility, which increases after eradication therapy of H. pylori, decrease [77]. Thus, another disease associated with H. pylori is gastric cancer (>50% of adenocarcinomas), and it was suggested to be a high risk factor for stomach cancer in 1991; in 1994, the World Health Organization classified H. pylori as a type I carcinogen and the National Institutes of Health (NIH), USA, recommends eradication of the bacterium in peptic ulcer as a definite curing treatment. Later, in 2001, the first observational trial confirmed that H. pylori eradication prevents gastric cancer [24]. According to data in the literature, approximately 1–3% of infected patients in the world develop gastric cancer compared to 0.13% of people with no infection. Regarding stomach cancer, the WHO, in 2020, reported that incidence cases before the age of 75 are in fifth place for males (7.2%) and seventh for women (4.1%) [78,79]. It is certain that intestinal type cancer, and, to a lesser extent, cancer of the diffuse type, is more common in patients with H. pylori. Among the patients at risk, those with atrophic multifocal gastritis and concomitant intestinal metaplasia are those who have the highest risk of gastric carcinogenesis. Instead, the specific form of gastric cardias cancer is not associated with H. pylori. Over time, two probable theories about cancerogenesis have been developed. The first hypothesis takes into account the increased production of free radicals near the H. pylori infection site and an increase in the mutation rate of host cells from double-stranded DNA breaks [80]. Gastric inflammation is characterized by infiltration of neutrophils and macrophages into the epithelium, leading to an increase in pro-inflammatory cytokines, and they are reactive to oxygen (reactive oxygen species, ROS) and to nitrogen (reactive nitrogen species, RNS). This would cause damage to DNA, including the 8-oxo-2’-deoxyguanosine (8-OHdG) [81]. The presence of the cytotoxic gene cagA can increase 8-OHdG more in the gastric cells than in non-presence of this gene in certain H. pylori strains. The second hypothesis assumes that H. pylori induces inflammation and locally elevated levels of TNF-α and/or interleukin 6 (IL-6). According to the perigenetic mechanism, the increase in inflammation of Tumor necrosis factor alpha (TNF-α) and/or interleukin 6 (IL-6) leads to an alteration of the cells of the gastric epithelium. Therefore, it will lead to an alteration of their union, resulting in dispersion and migration without occurring in additional mutations of the tumor suppressor genes. This proposed mechanism has been called the perigenetic pathway [82,83,84]. Finally, the gastric stump can also be infected by H. pylori or Epstein–Barr virus (more frequent). It is considered a long-term precancerous condition that can progress to cancer. In fact, gastrectomy (distal or proximal) is a carcinogenic risk factor for several reasons. The etiopathogenesis could be related to duodenogastric reflux, hypergastrinemia, atrophic gastritis, and achlorhydria, and some polymorphisms of cyclooxygenase-2 (COX-2) and interleukin-1β (IL-1 beta) could also probably be involved. Furthermore, it has been noted that H. pylori eradication therapy may reduce the risk of carcinogenesis in patients undergoing previous gastrectomy for early gastric cancer. It has been noticed that bile reflux interferes with the prevalence of H. pylori infection because it also has bactericidal action against this bacterium. However, this carcinogenic correlation with H. pylori appears to be rare. To improve the prognosis of gastric stump cancer, surveillance endoscopies of the gastric remnant and anastomosis site with multiple gastroenterostomy biopsies should be performed [85,86]. Finally, H. pylori is associated with a lymphoma derived from mucosa-associated lymphoid tissue (MALT) [87]. From the various research data, three main considerations emerge. The first is that after infection of the gastric mucosa, a percentage of patients develop a clonal hyperplasia of B lymphocytes, lymphatic tissue of the mucosa, and, therefore, a lymphatic tissue (which does not normally exist). The second is that this tissue (MALT) is probably malignant in some patients, giving rise to lymphoma in 72–98% of patients with MALT lymphoma that carry H. pylori. Finally, the third highlights that the eradication of the bacterium is not sufficient for the complete disappearance of low-grade MALT lymphoma. Other mucosa-associated lymphomas in H. pylori carriers may arise from lymphoid tissue in the esophagus, colon, rectum, and around the periorbital area of the eye (extranodal marginal zone B cell lymphoma). Therefore, it remains unknown why MALToma does not occur in all patients [24,86,88]. In a first study in 1997, other subsequent researchers noted the remission of the pathology for 62–95% of patients after the eradication of the bacterium as a cure in low-grade MALT lymphoma [89,90,91,92].
Extra-digestive diseases associated with H. pylori, for the most part, derived from epidemiological studies via case–control studies. In 2000, in the Maastricht II Consensus Report, for the first time, a relationship between H. pylori and other extra-digestive diseases was revealed. Hence, a high antibody positivity for H. pylori was detected in patients with coronary heart disease, rosacea dandruff, and active bronchiectasis. The association between peptic ulcer and atherosclerotic ischemic lesions was already known based on necropsy or surgical patients’ studies. It was considered that a diet rich in dairy products for the treatment of ulcers was sometimes considered to cause atherosclerotic lesions over time [24,93]. The prevailing pathogenetic opinion is that the chronic activation of inflammatory mediators induced by H. pylori contributes to the occurrence of non-specific inflammatory reactions such as those observed in the diseases mentioned above [94].
In 1999, for the first time, it was noted that in coronary patients, there is an increase in the prevalence of CagA(+) H. pylori strains. Subsequent prospective studies involving many patients confirmed this result [24,95,96]. In most cases, the difference in CagA(+) H. pylori strains between patients and controls was statistically significant at the level of p < 0.001. Indeed, in some studies, a difference was only observed in CagA(+) and not in all H. pylori strains. In conclusion, a review of the literature leads to the conclusion that there is, to some extent, an association between H. pylori infection and ischemic myocardial disease, and therefore, various hypotheses are proposed (Figure 8) [97]. According to one theory, infection/inflammation in the stomach is due to the release of a number of pro-inflammatory cytokines, mainly interleukins (IL-8, IL-6, IL-1a, and IL-1b) and anti-TNF, that can increase the risk of coronary events [98]. In addition, it is noted that the intracellular activity of the CagA protein provokes an inflammatory response and higher concentrations of pro-inflammatory cytokines that are finally re-contracted in patients with CagA (+) infections in the stomach. The second hypothesis is that the lower B12 absorption in these patients results in a consequent increased homocysteinemia, which is a factor predisposing patients to coronary artery disease. Finally, the infection can lead to a procoagulant state which will lead to atherogenesis [99]. Among the proteins of H. pylori is hsp60, which is also a vascular endothelial antigen, and a cross-reactive autoimmunity reaction may develop. This can cause endothelial damage by producing autoantibodies that destroy the vascular endothelium. However, this concept is purely theoretical since, at least up until now, no such antibodies have been detected in patients. Patients with the bacterium have a reduced plasma HDL (High-Density Lipoproteins), which can also lead to atherosclerosis. Finally, a hypothesis would be that the increase in free radicals due to the decrease in antioxidants could induce an increased risk of coronary heart disease, but it has not been proven [100].
It has long been known that patients with peptic ulcer have a higher prevalence of chronic obstructive pulmonary disease (COPD) and active pulmonary tuberculosis than the general population. It is known that chronic inflammation and autoimmune activation reactions have been implicated in the pathogenesis of many respiratory diseases such as chronic bronchitis and bronchiectasis [101]. Hence, several studies have investigated a possible association between H. pylori infection and respiratory diseases such as chronic bronchitis, active pulmonary tuberculosis, bronchiectasis, and lung cancer. Chronic bronchitis was associated with peptic ulcer after a long time of identification of H. pylori. In 1998, in a prospective pilot study on a sample of 60 patients with chronic bronchitis, it was found that they had a higher prevalence of H. pylori infection than healthy subjects. Subsequently, a larger epidemiological study in 2000 showed that IgG (Immunoglobulin G) positive for H. pylori had a particularly high prevalence of chronic bronchitis in women. It has been shown that pro-inflammatory cytokines (interleukins and tumor necrosis factor-α) are also involved in the pathogenesis of chronic bronchitis by promoting non-specific inflammation of the bronchial tree [102]. Thus, it can be assumed that H. pylori infection can cause bronchitis through the action of the produced cytokines and in combination with environmental, genetic, and other unknown factors. A possible translocation of H. pylori into the respiratory tree leading to chronic inflammation has been hypothesized (the bacterium was also cultured in the tracheobronchial secretions but was not found in histological preparations from bronchial tissue, nor was it isolated from bronchial alveolar lavage (BAL)) [103]. The reaction leading to bronchiectasis may be caused by the activation of systemic inflammatory mediators from chronic H. pylori infection. Pro-inflammatory cytokines (interleukins and tumor necrosis factor-α) are involved in the pathogenesis of COPD. Therefore, it can be deduced that H. pylori infection could, through pro-inflammatory action and in combination with the social environment and idiosyncrasy of the patient, have a role in the pathogenesis of COPD. Several studies have shown that chronic bronchitis is more common in anti-H. pylori IgG positive cases and has a higher prevalence of infection compared to healthy subjects. Hence, patients with peptic ulcer show a higher prevalence for COPD [101,102].
The association between H. pylori and skin diseases would be due to endogenous or exogenous vasodilatory factors and was noted many years before. Chronic idiopathic urticaria in many studies has an epidemiological link, but for others, there are opposite reports [103,104]. Rosacea is a relatively common inflammatory skin condition. Initially, a particularly high prevalence of H. pylori infection was reported in patients with rosacea. The disease remission observed after H. pylori eradication has not been assessed, as antibiotics given as an eradication treatment are, at the same time, effective drugs against rosacea. Other diseases have occasionally been associated with H. pylori, mostly supported by case reports or small studies such as alopecia areata, nodular acne, psoriasis, atopic dermatitis, and lichen planus [105].
The hypothesis that H. pylori infection may be a cause for some autoimmune diseases has been known for 20 years ago. Sjogren’s syndrome was one of the first extra-gastric diseases to be associated with infection, and a recurrence associated with successful eradication therapy has been described. These studies involved small numbers of patients, possibly due to the rarity of the syndrome. Henoch–Schönlein purpura is a rare disease and there are two case reports in the literature in which complete reversal of the symptoms was found after eradication of the bacterium. Other implicated diseases have been considered, e.g., autoimmune thrombocytopenia, autoimmune thyroiditis, and acute autoimmune polyneuropathy. Hypotheses have been advanced that the high concentration of inflammation mediators or the antigens of the bacterium itself cause the production of autoantibodies through a cross-reactive mechanism that can be correlated with H. pylori infection [106].
In patients with chronic cholecystitis, primary biliary cirrhosis, and primary sclerosing cholangitis, genetic material of H. pylori was detected by PCR. Therefore, regarding hepatic cirrhosis and H. pylori, they are still controversial. However, it seems to link the infection to a complication of cirrhosis, which is encephalopathy. It appears that in most studies in cirrhotic patients with encephalopathy, they have a significantly higher rate of H. pylori infection than in cirrhotic patients without hepatic encephalopathy. However, references to the contrary are not absent from the literature. This relationship breaks down the justification that H. pylori has a high ammonia production due to the increased activity of urease and cannot be easily catabolized in the diseased liver [107,108,109]. However, research data from clinical trials on the role of H. pylori in biliary diseases are conflicting, as only a few strains of H. pylori can survive in the gallbladder (H. hepaticus, H. bilis, H. rappini, and H. pullorum). Therefore, although some of these strains have been isolated from histological preparations of patients with chronic cholecystitis, the clinical significance of this finding remains obscure. Other diseases associated with H. pylori are type I diabetes mellitus, chronic pancreatitis, glaucoma, idiopathic thrombocytopenic purpura, iron deficiency anemia, and angioneurotic edema but also some neurological and ophthalmological ones [109,110].
2.4. Microbiota and H. pylori Interactions
The human microbiota is the set of microorganisms (approximately 10,000,000 different species), mostly bacteria but also archaea, fungi, and viruses, located in the various parts of the human body (such as oral, gut, and others). In particular, one of the most important parts is that of the intestinal microbiota because there is cross-talk with the gut/lung, gut/brain, and gut/skin which influences the host immune homeostasis. This ecosystem includes numerous microorganisms which belong to approximately 500 different genera [111]. The intestinal microbiota overcomes other functions such as aiding the digestion and absorption of food, the synthesis of vitamins, the catabolism of biomolecules, the metabolism of bile salts, and the regulation of inflammatory reactions. The disruption of this symbiotic balance (eubiosis) can create local and general homeostasis problems of the immune system as it would lead to dysbiosis, which, in turn, can lead to an increase in pathogens such as Candida albicans [112,113]. Common causes of disruption to the intestinal microbiota are alcohol abuse, reckless use of antibiotics, or infectious agents (such as H. pylori), which can lead to significant metabolic (such as obesity), inflammatory, and allergic disorders and even to precancerous conditions [114,115,116,117,118,119,120,121,122,123]. Therefore, if the population of one genus of microbes decreases or increases, it can affect populations of other genera by disrupting the overall balance of the intestinal microbiota and both innate and acquired immunity. An immune system acts in the gut mucosa (gut-associated lymphoid tissue, GALT), whose function is induced by the intestinal microbiota through antigenic stimuli, influencing both cellular and chemical stimulation of innate immunity (80% of B lymphocytes are detected in the gut) [123,124]. It has been noted that during H. pylori infection, there is an increase in the facultative pathogen Haemophilus spp., but there is also a decrease in the genera Parasutterella and Pseudoflavonifractor (important producers of SCFAs (Short-chain fatty acids) that modulate the immune response through regulatory T cells). In addition, an abundance in Bacteroides and Prevotella was noted. This condition can lead to an imbalance of the three cross-talking axes, causing global imbalance of the body’s homeostasis [124,125,126,127,128].
2.5. The Diagnosis Over Time
The various methods of diagnosing H. pylori infection can be divided into non-invasive and invasive. The rapid urease test (Campylobacter-like organisms test, CLO-test) is based on the property of H. pylori to produce urease, as we have described. For the test, material from the stomach is taken and then immersed in a urea substrate. If there is a decomposition of the substrate and production of ammonia and carbon dioxide which varies the pH, such a variation (detected by an indicator, it can change color) is positive for the presence of H. pylori [129]. In addition, gastric biopsy samples can be used to determine the type of eventual gene mutations to demonstrate resistant clarithromycin strains in patients being treated or to be treated. Such a biomolecular assessment of resistance to clarithromycin and the search for H. pylori strains can be performed by fluorescent in situ hybridization (FISH). This resistance was found to be associated with point mutations in domain V of 23S rRNA in the 50S ribosomal subunit. This has a blocking effect on the link between the antibiotic and the ribosome, and consequently, protein synthesis is not inhibited by the drug. Furthermore, mutations have also been found in A2143G, A2144G, A2143C, and T2182C, which cause resistance to clarithromycin in H. pylori strains [130,131].
An accurate method is histological diagnosis after endoscopy and biopsy. To confirm the diagnosis, usually, four biopsies (two from the curves and two from the stomach body) are required. Subsequently, H. pylori in the histological specimen is detected by staining with hematoxylin–eosin. Biopsy culture has high sensitivity and is applied to determine the susceptibility of antimicrobial H. pylori strains [132]. In 1990, for the first time, the non-invasive 13C urea breath test (UBT) was introduced [24]. The breath test is a non-invasive testing procedure which is based on the property of H. pylori to produce urease, which is also used to test the successful eradication of the infection. The sensitivity of the method is low, as small concentrations of H. pylori are not detected. The subject is fed 13C-labeled urea, and the concentration of 13C in the exhaled dioxide is then measured [24,133]. Finally, PCR and detection of H. pylori antigen in stool, and fecal culture, are less used in clinical practice and mainly at the experimental level. In the mid-1980s, the first serological tests appeared to diagnose H. pylori infection. These became particularly useful due to the rapidity of the results and the low cost. Serological tests are divided into qualitative and quantitative. Whole blood is used in quality tests, and the results are available within minutes and are characterized by low sensitivity and, therefore, should not be used in either clinical practice or research protocols. Quantitative tests are based on the enzyme-linked immunosorbent assay (ELISA). This method detects the concentration of antibodies against H. pylori in blood serum, not whole blood, and the titer that is evaluated as indicative of the presence or absence of infection is that of IgG antibodies. In addition, the ELISA method offers the possibility of detecting CagA(+) [24,134].
The value of serological tests is important when considering the difficulty of finding H. pylori in biopsies from areas with atrophic gastritis. Recent studies have demonstrated the superiority of serological tests over biopsy examination for patients with atrophic gastritis. The main factor affecting their reliability is previous treatment for H. pylori eradication. However, relatively recently, laboratory methods for determining the concentration of IgG antibodies from other biological fluids (urine, saliva, smear, and mouth) have been developed. Recent studies have shown that these methods have particularly low sensitivity [24,134,135].
2.6. New Therapeutic Issues
The steps of treatment against helicobacter started long ago [24]. We must mention that in 1968, John Lykoudis (Ιωάννης Λυκούδης, 1910–1980, Greece) was the first to attempt a treatment regarding gastritis and peptic ulcer with three types of antibiotics (2 quinolines, 5,7-diiode-8-oxyquinolein 0.125 g; 5-iodo-7-chlorine-8–oxyquinoline 0.125 g, and streptomycin sulfate 0.075 g) together with vitamin A. He called this therapy "Elgaco", with a therapeutic scheme of 6–8 times a day for 10 days. He was convinced that the true cause was infectious. He treated himself and about 50,000 of his patients, but his observations “Ulcer of Stomach and Duodenum” in the Journal of the American Medical Association were, unfortunately, rejected. Finally, Lykoudis presented his therapy to the Greek National Medical Association but was rejected by the medical establishment [14]. However, after the time of treatment assessments, in 1987, the relapse of duodenal ulcer was shown to be reduced by bismuth, and in 1989, proton pump inhibitor (PPI)–amoxicillin dual therapy was proposed. However, in 1990, compliance and resistance were recognized as difficulties in treating infection. As we mentioned, the resistance to clarithromycin of certain H. pylori strains depends on point mutations in the 50S ribosomal subunit, so the antibiotic can no longer block bacterial protein synthesis. The first study in 1990 confirmed that the cure for duodenal ulcer will be healing from the H. pylori infection, and in 1992, trials for proposed vaccines started. In 1994, the first trials with PPI triple therapies for ulcer treatment began; in 1996, the Maastricht I Consensus Report (and later, the other report), screen-and-treat strategy was proposed over time, and in 2010, the bismuth quadruple therapies were revisited [23,24,130]. All patients with gastric or duodenal ulcers who are positive for H. pylori should receive primary treatment for the eradication of the bacterium, which is based on a proton pump inhibitor and is given in combination with two antibiotics for 7 days. The treatment has a 90% success rate, although there are factors that can influence its success, e.g., patient compliance, side effects, and microbial resistance to clarithromycin or metronidazole. In case of failure of the first treatment, second-line treatment should be performed, while in the case of failure again, a third attempt with a quadruple regimen or long-term maintenance treatment with acid suppression is recommended. H. pylori and NSAIDs (non-steroidal anti-inflammatory drugs) are independent risk factors for gastric ulcer. It is not known whether people requiring long-term NSAID treatment should receive eradication treatment to reduce the risk of developing ulcers [136,137,138]. An indicative regimen of current H. pylori eradication therapy consists mainly of triple therapy, based on the association of a proton pump inhibitor (PPI) and two antibiotics: amoxicillin 2000 mg/day and clarithromycin 1000 mg/day; metronidazole 1000 mg/day and clarithromycin 1000 mg/day, or levofloxacin 250–500 mg and amoxicillin 1 g (or metronidazole 500 mg or clarithromycin 500 mg) twice/day. However, for multidrug-resistant H. pylori strains, the use of rifabutin in triple therapy also seems to be effective. The triple therapy must be given for 10–14 days and only in world areas with low clarithromycin-resistant strains (<15%) or where local H. pylori eradication rates reach or exceed 85%. Thus, the modality of treatment, according to the first therapy effects, can sometimes be not only concomitant (drug administration occurs simultaneously) but also sequential (administration of drugs in two periods)—e.g., 5–7 days of PPI associated with amoxicillin and, subsequently, another 5–7 days of different antibiotics such as clarithromycin and metronidazole while maintaining the administration of the PPI. In other cases, the initial therapy should be fourfold with PPI (lansoprazole 30 mg twice/day, omeprazole 20 mg twice/day, pantoprazole 40 mg twice/day, rabeprazole 20 mg twice/day, or esomeprazole 40 mg once/day), bismuth sub-citrate (524 mg four times/day), and two antibiotics (tetracycline 500 mg four times/day and 250 mg metronidazole four times/day). People who do not have a history of peptic ulcer or indigestion and are young do not need treatment, unlike older patients with a major comorbidity or a history of old peptic ulcer. Quadruple therapy is the best initial therapy in world areas where the clarithromycin resistance rate is > 15% and the treatment must be given for 14 days. The treatment is repeated if the H. pylori is not eradicated. If two cycles are ineffective, some specialists recommend performing an endoscopic examination to obtain cultures for susceptibility testing. Eradication of H. pylori can be confirmed at or after four weeks from the end of therapy by urea breath test or fecal antigen test, or by upper digestive tract endoscopy [137,138,139].
Finally, probiotics have been shown to act in a beneficial way against various diseases, including infectious ones, e.g., from bacterial, viral (such as in the current viral pandemic due to SARS-CoV-2), fungal, and other causes [139,140,141,142,143,144,145,146,147]. The ability of probiotics to be a valid adjuvant in the eradication therapy for H. pylori infection (therefore, against growth) is in its role in bringing the gut into eubiosis, including secretion of antimicrobial substances, competition at the adhesion site of epithelial cells, and stabilization of the mucosal barrier [26,27,28,29], and there may be some hypotheses regarding the beneficial effects of probiotics. Several studies have confirmed that probiotic supplementation is able to facilitate eradication and reduce the incidence of side effects of antibiotic therapy. In particular, the use of probiotics with various selected species is more advantageous than monospecies preparations (such as Lactobacillus spp. or Bifidobacterium spp.). The combination of strains L. reuteri DSM 17938 and L. reuteri ATCC PTA 6475, or supplementation with S. boulardii, has demonstrated positive effects [148,149].
3. Discussion
As we mentioned, successful colonization of the gastric epithelium by H. pylori depends on the action of bacterial-dependent mechanisms such as pH control through urease, inactivation of oxygen and nitrogen free radicals produced by neutrophils, and activation of bacterial DNA repair/recombination signaling pathways along with the ability to integrate extracellular DNA into the bacterial chromosome that promotes genetic differentiation. Furthermore, the production of various inflammatory mediators such as IL-1, IL-8, and TNF-α due to the recognition of bacterial derivatives (VacA, LPS, neutrophil activation factor (NapA), porine) and the elimination of recognition of H. pylori by Toll-like receptor (TLR) sensors, and the consequent modulation of anti-inflammatory agents, establish chronic infection [150]. Furthermore, immune responses of types Th1 and Th17 are observed, regulated by the cytokines IL-23 and IL-12. The inability of the mechanisms of natural immunity to recognize and eliminate H. pylori leads to the development of acute inflammation and to the attraction of macrophages and T and B lymphocytes and selective differentiation with concomitant Treg (Regulatory T cell) differentiation [151]. This condition further allows the bacterium to escape from the host’s immune processes. Indeed, numerous studies indicate that the possible presence of polymorphisms in the genes of inflammatory cytokine cells IL-1β, TNF-α, and of the anti-inflammatory cytokine IL-10 determines the intensity of the inflammatory response. All this constitutes a background that probably predisposes one to the oncogenesis induced by H. pylori [152]. Antimicrobial peptides (AMPs) are key components of natural gastric tissue immunity. The two main categories of AMPs are the two Ds (α- and β-defensins) and cathelicidin LL-37/hCAP-18. The secretion of human β-defensins (hβDs 1, 2, 3, and 4) in the gastric epithelium adapts it according to the presence of pathogenic microorganisms. H. pylori can colonize the gastric mucosa, escaping or possibly suppressing the mechanisms of natural immunity including AMPs. Furthermore, the ability to produce bio-membranes is of particular importance for a more complete understanding of the survival mechanisms of H. pylori in adverse gastric conditions and the pathogenesis of chronic infection. Finally, the role of HspA and -B, which are highly antigenic, in the pathogenesis of infection has not been noticeably clear [153,154,155,156]. The interactions of H. pylori Hsp-60 and TLR in host cells contribute to the development of gastric inflammation by the bacterium. Furthermore, reactions induced by immune cross-responses between H. pylori HspB and human Hsp-60 of the gastric epithelium may be involved in the development of MALT lymphoma [157,158]. In this regard, recent studies also show that CagA and HspB increase gastric cancer risk. Instead, Hsp-60, by inducing the secretion of inflammatory cytokines and promoting angiogenesis, triggers the onset of oncogenesis and, therefore, acts as a carcinogen [159]. Chemokines have chemotactic activity in leukocytes and are, thus, directly or indirectly involved in inflammatory reactions. They are classified into two main families (according to the position of the cysteine residues): C-X-C and C-C. The first has chemotactic properties in neutrophils (no monocytes) and the second has chemotactic properties in monocytes and lymphocytes with little effect on neutrophils. Infection then increases the production of C-X-C chemokines such as IL-8 and GROα (Growth-regulated oncogene α) and is associated with an inflammatory reaction without increased production of C-C chemokines. Furthermore, B cell-attracting (BCA)-1 induces chronic gastritis and is involved in the formation of MALT and gastric lymphomas in H. pylori infections [160,161,162,163]. Hence, BCA-1 assessment is a useful indicator of the development of MALT aggregates. Finally, the IL-8-251 T/A genotype is associated with an increased risk of peptic ulcer and duodenal ulcers in H. pylori infections. Instead, the IL-8-251 AA genotype represents an important indicator of the presence of gastric cancer. Furthermore, the bacterium reduces the production of the peptides trefoil factor (TFF)-1 and trefoil factor (TFF)-2, which play a role in the repair of the gastric mucosa (such as gastritis and ulcers) and suppress the processes of carcinogenesis (Figure 9) [164,165,166,167,168,169,170].
Finally, there are two main pathogenetic mechanisms that report pathogenic correlations between H. pylori and extra-digestive diseases. The first is the induction of the release of several pro-inflammatory agents such as cytokines, eicosanoids, and acute phase proteins by H. pylori. The second concerns the possibility of microbial antigens mimicking those of the host, resulting in activation of autoimmunity [91].
4. Conclusions
H. pylori was recognized as a pathogenic infectious agent for humans only in 1985. The recognition of the pathogenic role of H. pylori has revolutionized medicine and gastric and duodenal pathology. This has led not only to the new description of the etiopathogenesis of some diseases of the digestive system, but also to re-thinking the prevention and therapy. Epidemiological studies conducted in recent years have revealed that H. pylori infection represents an endemic disease all over the world. It is well established that H. pylori infection is acquired, in most cases, in childhood. We still must discover the relationships of individual and environmental factors by incidence and pathogenesis and correlations with other systems. Infection appears to be a representative indicator of socio-economic level, and at present, there is a significant reduction in the incidence of the disease worldwide and a steady decrease in the incidence of infection in childhood, probably due to better conditions of hygiene and improvement in treatment. Treatment must address compliance and antibiotic resistance during the disease. A compromising observation is the use of probiotics as adjuvants in the therapy because the microbiota can be altered by immune-mediated reactions of the organism against the bacterium.
Author Contributions
Conceptualization, I.A.C. and L.B.; methodology, S.T.; validation, D.D. and S.T.; formal analysis, D.D.; investigation, I.A.C. and L.B.; resources, D.D. and S.T.; data curation, I.A.C. and L.B.; writing—original draft preparation, I.A.C.; writing—review and editing, L.B. and D.D.; visualization, S.T.; project administration, L.B.; funding acquisition, D.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors express their gratitude to Luigi Santacroce, Clinical Microbiologist of the Medical School at the University of Bari, for his generous and valuable contribution in preparing and reviewing this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Salama, N.R.; Hartung, M.L.; Müller, A. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 2013, 11, 385–399. [Google Scholar] [CrossRef]
- Blaser, M. Antibiotic overuse: Stop the killing of beneficial bacteria. Nature 2011, 476, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Josenhans, C.; Eaton, K.A.; Thevenot, T.; Suerbaum, S. Switching of flagellar motility in Helicobacter pylori by reversible length variation of a short homopolymeric sequence repeat in fliP, a gene encoding a basal body protein. Infect. Immun. 2000, 68, 4598–4603. [Google Scholar] [CrossRef] [Green Version]
- Lertsethtakarn, P.; Ottemann, K.M.; Hendrixson, D.R. Motility and chemotaxis in Campylobacter and Helicobacter. Annu. Rev. Microbiol. 2011, 65, 389–410. [Google Scholar] [CrossRef]
- Tomb, J.F.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann, R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997, 388, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.D.; Kling-Bäckhed, H.; Giannakis, M.; Xu, J.; Fulton, R.S.; Fulton, L.A.; Cordum, H.S.; Wang, C.; Elliott, G.; Edwards, J.; et al. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: Evolution during disease progression. Proc. Natl. Acad. Sci. USA 2006, 103, 9999–10004. [Google Scholar] [CrossRef] [Green Version]
- van Vliet, A.H. Use of pan-genome analysis for the identification of lineage-specific genes of Helicobacter pylori. FEMS Microbiol. Lett. 2017, 364, fnw296. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.S.; Ahlawat, R. Helicobacter pylori. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021; Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK534233/ (accessed on 25 February 2021).
- Logan, R.P. Adherence of Helicobacter pylori. Aliment. Pharmacol. Ther. 1996, 10 (Suppl. 1), 3–15. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J.; Atherton, J.C. Helicobacter pylori persistence: Biology and disease. J. Clin. Investig. 2004, 113, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Yamaoka, Y.; Kikuchi, S.; el-Zimaity, H.M.; Gutierrez, O.; Osato, M.S.; Graham, D.Y. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology 2002, 123, 414–424. [Google Scholar] [CrossRef]
- Mitchell, J.D.; Mitchell, H.M.; Tobias, V. Acute Helicobacter pylori infection in an infant, associated with gastric ulceration and serological evidence of intra-familial transmission. Am. J. Gastroenterol. 1992, 87, 382–386. [Google Scholar]
- Raymond, J.; Thiberg, J.M.; Chevalier, C.; Kalach, N.; Bergeret, M.; Labigne, A.; Dauga, C. Genetic and transmission analysis of Helicobacter pylori strains within a family. Emerg. Infect. Dis. 2004, 10, 1816–1821. [Google Scholar] [CrossRef]
- Topi, S.; Santacroce, L.; Bottalico, L.; Ballini, A.; Inchingolo, A.D.; Dipalma, G.; Charitos, I.A.; Inchingolo, F. Gastric Cancer in History: A Perspective Interdisciplinary Study. Cancers 2020, 12, 264. [Google Scholar] [CrossRef] [Green Version]
- Santacroce, L.; Topi, S.; Haxhirexha, K.; Hidri, S.; Charitos, I.A.; Bottalico, L. Medicine and healing in the pre-Socratic thought. A brief analysis of magics and rationalism in ancient herbal therapy. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 282–287. [Google Scholar] [CrossRef]
- Charitos, I.A.; Gagliano-Candela, R.; Santacroce, L.; Bottalico, L. Venoms and poisonings during the centuries. A narrative review. Endocr. Metab. Immune Disord. Drug Targets 2020. ahead of print. [Google Scholar] [CrossRef]
- Amos, B. Lessons from the history of light microscopy. Nat. Cell. Biol. 2000, 2, E151–E152. [Google Scholar] [CrossRef]
- Gest, H. The Discovery of Microorganisms by Robert Hooke and Antoni van Leeuwenhoek, Fellows of the Royal Societ. Notes Rec. R. Soc. Lond. 2004, 58, 187–201. [Google Scholar] [CrossRef]
- Santacroce, L.; Bottalico, L.; Topi, S.; Castellaneta, F.; Charitos, I.A. The “Scourge of the Renaissance”. A Short Review About Treponema pallidum infection. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 335–343. [Google Scholar] [CrossRef]
- Kidd, M.; Modlin, I.M. A century of Helicobacter pylori: Paradigms lost-paradigms regained. Digestion 1998, 59, 1–15. [Google Scholar] [CrossRef]
- Suzuki, H.; Warren, R.; Marshall, B. Helicobacter Pylori; Springer: Tokyo, Japan, 2016; p. 8. [Google Scholar]
- Mazzarello, P.; Calligaro, A.L.; Calligaro, A. Giulio Bizzozero: A pioneer of cell biology. Nat. Rev. Mol. Cell. Biol. 2001, 2, 776–781. [Google Scholar] [CrossRef]
- Goodwin, S. Helicobacter pylori: The story continues. Lancet 2001, 357, 2056. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Link, A.; Selgrad, M. Helicobacter pylori: Perspectives and time trends. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Suriani, R.; Mazzucco, D.; Venturini, I.; Mazzarello, M.G.; Zanella, D.; Orso Giacone, G. Helicobacter Pylori: Stato, dell’arte. Caleidosc. Riv. Mens. Med. 2020, 159, 9. [Google Scholar]
- Marshall, B. Helicobacter connections. ChemMedChem 2006, 1, 783–802. [Google Scholar] [CrossRef]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 16, 1311–1315. [Google Scholar] [CrossRef]
- Megraud, F.; Bonnet, F.; Gamier, M.; Lamouliatte, H. Characterization of “Campylobacter pyloridis” by culture, enzymatic profile, and protein content. J. Clin. Microbiol. 1985, 22, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Romaniuk, P.J.; Zoltowska, B.; Trust, T.J.; Lane, D.J.; Olsen, G.J.; Pace, N.R.; Stahl, D.A. Campylobacter pylori, the spiral bacterium associated with human gastritis, is not a true Campylobacter sp. J. Bacteriol. 1987, 169, 2137–2141. [Google Scholar] [CrossRef] [Green Version]
- Marshall, B.J.; Armstrong, J.A.; McGechie, D.B.; Glancy, R.J. Attempt to fulfill Koch’s postulates for pyloric Campylobacter. Med. J. Aust. 1985, 142, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.; Nicholson, G. Ingestion of Campylobacter pyloridis causes gastritis and raised fasting gastric pH. Am. J. Gastroenterol. 1987, 82, 192–199. [Google Scholar]
- Ferguson, D.A.; Li, C.; Patel, N.R.; Mayberry, W.R.; Chi, D.S.; Thomas, E. Isolation of Helicobacter pylori from saliva. J. Clin. Microbiol. 1993, 31, 2802–2804. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.M.; Engstrand, L.; Genta, R.M.; Graham, D.Y.; el-Zaatari, F.A. Detection of Helicobacter pylori in dental plaque by reverse transcription-polymerase chain reaction. J. Clin. Microbiol. 1993, 31, 783–787. [Google Scholar] [CrossRef] [Green Version]
- Snaith, A.; El-Omar, E.M. Helicobacter pylori: Host genetics and disease outcomes. Expert Rev. Gastroenterol. Hepatol. 2008, 2, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Collado, L.; Figueras, M.J. Taxonomy, epidemiology, and clinical relevance of the genus Arcobacter. Clin. Microbiol. Rev. 2011, 24, 174–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.G. The non-H pylori helicobacters: Their expanding role in gastrointestinal and systemic diseases. Gut 2002, 50, 273–283. [Google Scholar] [CrossRef]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santacroce, L.; Cagiano, R.; Del Prete, R.; Bottalico, L.; Sabatini, R.; Carlaio, R.G.; Prejbeanu, R.; Vermesan, H.; Dragulescu, S.I.; Vermesan, D.; et al. Helicobacter pylori infection and gastric MALTomas: An up-to-date and therapy highlight. Clin. Ter. 2008, 159, 457–462. [Google Scholar] [PubMed]
- Moodley, Y.; Linz, B. Helicobacter pylori Sequences Reflect Past HumanMigrations. Genome Dyn. 2009, 6, 62–74. [Google Scholar]
- Goodman, K.J.; Correa, P. The transmission of Helicobacter pylori. A critical review of the evidence. Int. J. Epidemiol. 1995, 24, 875–887, . [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y.; Yamaoka, Y.; Malaty, H.M. Thoughts about populations with unexpected low prevalences of Helicobacter pylori infection. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 849–851. [Google Scholar] [CrossRef] [Green Version]
- Alm, R.A.; Bina, J.; Andrews, B.M.; Doig, P.; Hancock, R.E.; Trust, T.J. Comparative genomics of Helicobacter pylori: Analysis of the outer membrane protein families. Infect. Immun. 2000, 68, 4155–4168. [Google Scholar] [CrossRef] [Green Version]
- Ansari, S.; Yamaoka, Y. Survival of Helicobacter pylori in gastric acidic territory. Helicobacter 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Odenbreit, S.; Swoboda, K.; Barwig, I.; Ruhl, S.; Borén, T.; Koletzko, S.; Haas, R. Outer membrane protein expression profile in Helicobacter pylori clinical isolates. Infect. Immun. 2009, 77, 3782–3790. [Google Scholar] [CrossRef] [Green Version]
- Croxen, M.A.; Sisson, G.; Melano, R.; Hoffman, P.S. The Helicobacter pylori chemotaxis receptor TlpB (HP0103) is required for pH taxis and for colonization of the gastric mucosa. J. Bacteriol. 2006, 188, 2656–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 46. Williams, S.M.; Chen, Y.T.; Andermann, T.M.; Carter, J.E.; McGee, D.J.; Ottemann, K.M. Helicobacter pylori chemotaxis modulates inflammation and bacterium-gastric epithelium interactions in infected mice. Infect. Immun. 2007, 75, 3747–3757. [Google Scholar] [CrossRef] [Green Version]
- George, G.; Kombrabail, M.; Raninga, N.; Sau, A.K. Arginase of Helicobacter Gastric Pathogens Uses a Unique Set of Non-catalytic Residues for Catalysis. Biophys. J. 2017, 112, 1120–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, C.Y.; Sheu, B.S.; Wu, J.J. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed. J. 2016, 39, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Haley, K.P.; Gaddy, J.A. Helicobacter pylori: Genomic Insight into the Host-Pathogen Interaction. Int. J. Genom. 2015, 2015, 386905. [Google Scholar] [CrossRef] [Green Version]
- Ilver, D.; Arnqvist, A.; Ogren, J.; Frick, I.M.; Kersulyte, D.; Incecik, E.T.; Berg, D.E.; Covacci, A.; Engstrand, L.; Borén, T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998, 279, 373–377. [Google Scholar] [CrossRef]
- Xu, C.; Soyfoo, D.M.; Wu, Y.; Xu, S. Virulence of Helicobacter pylori outer membrane proteins: An updated review. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1821–1830. [Google Scholar] [CrossRef]
- Benktander, J.; Barone, A.; Johansson, M.M.; Teneberg, S. Helicobacter pylori SabA binding gangliosides of human stomach. Virulence 2018, 9, 738–751. [Google Scholar] [CrossRef] [Green Version]
- Suerbaum, S.; Thiberge, J.M.; Kansau, I.; Ferrero, R.L.; Labigne, A. Helicobacter pylori hspA-hspB heat-shock gene cluster: Nucleotide sequence, expression, putative function and immunogenicity. Mol. Microbiol. 1994, 14, 959–974. [Google Scholar] [CrossRef]
- Miehlke, S.; Yu, J.; Schuppler, M.; Frings, C.; Kirsch, C.; Negraszus, N.; Morgner, A.; Stolte, M.; Ehninger, G.; Bayerdörffer, E. Helicobacter pylori vacA, iceA, and cagA status and pattern of gastritis in patients with malignant and benign gastroduodenal disease. Am. J. Gastroenterol. 2001, 96, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Gantuya, B.; El Serag, H.B.; Saruuljavkhlan, B.; Azzaya, D.; Matsumoto, T.; Uchida, T.; Oyuntsetseg, K.; Oyunbileg, N.; Davaadorj, D.; Yamaoka, Y. Advantage of 16S rRNA amplicon sequencing in Helicobacter pylori diagnosis. Helicobacter 2021, 17, e12790. [Google Scholar] [CrossRef]
- Tohidpour, A. CagA-mediated pathogenesis of Helicobacter pylori. Microb. Pathog. 2016, 93, 44–55. [Google Scholar] [CrossRef]
- Waskito, L.A.; Yih-Wu, J.; Yamaoka, Y. The role of integrating conjugative elements in Helicobacter pylori: A review. J. Biomed. Sci. 2018, 25, 86. [Google Scholar] [CrossRef]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef] [Green Version]
- Ritter, A.; Blum, G.; Emody, L.; Kerenyi, M.; Bock, A.; Neuhierl, B.; Rabsch, W.; Scheutz, F.; Hacker, J. tRNA genes and pathogenicity islands: Influence on virulence and metabolic properties of uropathogenic Escherichia coli. Mol. Microbiol. 1995, 17, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Blum, G.; Falbo, V.; Caprioli, A.; Hacker, J. Gene clusters encoding the cytotoxic necrotizing factor type 1, Prs-fimbriae and alpha-hemolysin form the pathogenicity island II of the uropathogenic Escherichia coli strain J96. FEMS Microbiol. Lett. 1995, 126, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Odenbreit, S.; Puls, J.; Sedlmaier, B.; Gerland, E.; Fischer, W.; Haas, R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000, 287, 1497–1500. [Google Scholar] [CrossRef]
- Stein, M.; Rappuoli, R.; Covacci, A. The cag Pathogenicity Island. In Helicobacter pylori: Physiology and Genetics; Mobley, H.L.T., Mendz, G.L., Hazell, S.L., Eds.; ASM Press: Washington, DC, USA, 2001; Chapter 31. [Google Scholar]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaoka, Y.; Kita, M.; Kodama, T.; Sawai, N.; Imanishi, J. Helicobacter pylori cagA gene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology 1996, 110, 1744–1752. [Google Scholar] [CrossRef]
- Robinson, K.; Argent, R.H.; Atherton, J.C. The inflammatory and immune response to Helicobacter pylori infection. Best Pract. Res. Clin. Gastroenterol. 2007, 21, 237–259. [Google Scholar] [CrossRef] [PubMed]
- Nedrud, J.G.; Blanchard, S.S.; Czinn, S.J. Helicobacter pylori inflammation and immunity. Helicobacter 2002, 7 (Suppl. 1), 24–29. [Google Scholar] [CrossRef] [PubMed]
- Noto, J.M.; Peek, R.M., Jr. The Helicobacter pylori cag Pathogenicity Island. Methods Mol. Biol. 2012, 921, 41–50. [Google Scholar]
- Pacchiani, N.; Censini, S.; Buti, L.; Covacci, A. Echoes of a distant past: The cag pathogenicity island of Helicobacter pylori. Cold Spring Harb. Perspect. Med. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Watari, J.; Chen, N.; Amenta, P.S.; Fukui, H.; Oshima, T.; Tomita, T.; Miwa, H.; Lim, K.J.; Das, K.M. Helicobacter pylori associated chronic gastritis, clinical syndromes, precancerous lesions, and pathogenesis of gastric cancer development. World J. Gastroenterol. 2014, 20, 5461–5473. [Google Scholar] [CrossRef]
- O’Connor, A.; Furuta, T.; Gisbert, J.P.; O’Morain, C. Review—Treatment of Helicobacter pylori infection 2020. Helicobacter 2020, 25 (Suppl. 1), e12743. [Google Scholar] [CrossRef]
- Hatakeyama, M.; Higashi, H. Helicobacter pylori CagA: A new paradigm for bacterial carcinogenesis. Cancer Sci. 2005, 96, 835–843. [Google Scholar] [CrossRef]
- Dumrese, C.; Slomianka, L.; Ziegler, U.; Choi, S.S.; Kalia, A.; Fulurija, A.; Lu, W.; Berg, D.E.; Benghezal, M.; Marshall, B.; et al. The secreted Helicobacter cysteine-rich protein A causes adherence of human monocytes and differentiation into a macrophage-like phenotype. FEBS Lett. 2009, 583, 1637–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiotani, A.; Graham, D.Y. Pathogenesis and therapy of gastric and duodenal ulcer disease. Med. Clin. N. Am. 2002, 86, 1447–1466. [Google Scholar] [CrossRef]
- Dixon, M.F. Patterns of inflammation linked to ulcer disease. Bailliere’s Best Practice & Research. Clin. Gastroenterol. 2000, 14, 27–40. [Google Scholar] [CrossRef]
- Dorer, M.S.; Cohen, I.E.; Sessler, T.H.; Fero, J.; Salama, N.R. Natural competence promotes Helicobacter pylori chronic infection. Infect. Immun. 2013, 81, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Chen, C.; Hu, J.; Su, R.; Zhang, J.; Han, Z.; Chen, H.; Li, Y. Molecular mechanism of Helicobacter pylori-induced autophagy in gastric cancer. Oncol. Lett. 2019, 18, 6221–6227. [Google Scholar] [CrossRef] [Green Version]
- Francois, F.; Roper, J.; Joseph, N.; Pei, Z.; Chhada, A.; Shak, J.R.; de Perez, A.Z.; Perez-Perez, G.I.; Blaser, M.J. The effect of H. pylori eradication on meal-associated changes in plasma ghrelin and leptin. BMC Gastroenterol. 2011, 11, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuipers, E.J. Review article: Exploring the link between Helicobacter pylori and gastric cancer. Aliment. Pharmacol. Ther. 1999, 13 (Suppl. 1), 3–11. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Report on Cancer: Setting Priorities, Investing Wisely and Providing Care for All; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Koeppel, M.; Garcia-Alcalde, F.; Glowinski, F.; Schlaermann, P.; Meyer, T.F. Helicobacter pylori Infection Causes Characteristic DNA Damage Patterns in Human Cells. Cell Rep. 2015, 11, 1703–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, M.A.; Canales, J.; Corvalán, A.H.; Quest, A.F. Helicobacter pylori-induced inflammation and epigenetic changes during gastric carcinogenesis. World J. Gastroenterol. 2015, 21, 12742–12756. [Google Scholar] [CrossRef] [PubMed]
- Raza, Y.; Khan, A.; Farooqui, A.; Mubarak, M.; Facista, A.; Akhtar, S.S.; Khan, S.; Kazi, J.I.; Bernstein, C.; Kazmi, S.U. Oxidative DNA damage as a potential early biomarker of Helicobacter pylori associated carcinogenesis. Pathol. Oncol. Res. 2014, 20, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, M.; Yamaguchi, K.; Ono, Y.; Matsumoto, H.; Hayashi, T.; Ogawa, T.; Imai, K.; Kuzuhara, T.; Nishizono, A.; Fujiki, H. TNF-alpha-inducing protein, a carcinogenic factor secreted from H. pylori, enters gastric cancer cells. Int. J. Cancer 2008, 123, 117–122. [Google Scholar] [CrossRef]
- Tsuji, S.; Kawai, N.; Tsujii, M.; Kawano, S.; Hori, M. Review article: Inflammation-related promotion of gastrointestinal carcinogenesis-a perigenetic pathway. Aliment. Pharmacol. Ther. 2003, 89 (Suppl. 1), 82–89. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Miwa, K.; Inokuchi, M.; Nakagawara, H.; Tajima, H.; Takamura, H.; Ninomiya, I.; Kitagawa, H.; Fushida, S.; Fujimura, T.; et al. Spontaneous clearance of Helicobacter pylori after pylorus-preserving gastrectomy for gastric cancer. Oncol. Rep. 2013, 30, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Baas, I.O.; van Rees, B.P.; Musler, A.; Craanen, M.E.; Tytgat, G.N.; van den Berg, F.M.; Offerhaus, G.J. Helicobacter pylori and Epstein-Barr virus infection and the p53 tumour suppressor pathway in gastric stump cancer compared with carcinoma in the non-operated stomach. J. Clin. Pathol. 1998, 51, 662–666. [Google Scholar] [CrossRef] [Green Version]
- Genta, R.M.; Hamner, H.W.; Graham, D.Y. Gastric lymphoid follicles in Helicobacterpylori infection: Frequency, distribution, and response totriple therapy. Hum. Pathol. 1993, 24, 577–583. [Google Scholar] [CrossRef]
- Wotherspoon, A.C. Gastric lymphoma of mucosa-associated lymphoid tissue and Helicobacter pylori. Annu. Rev. Med. 1998, 49, 289–299. [Google Scholar] [CrossRef]
- Neubauer, A.; Thiede, C.; Morgner, A.; Alpen, B.; Ritter, M.; Neubauer, B.; Wündisch, T.; Ehninger, G.; Stolte, M.; Bayerdörffer, E. Cure of Helicobacter pylori infection and duration of remission of low-grade gastric mucosa-associated lymphoid tissue lymphoma. J. Natl. Cancer Inst. 1997, 89, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losacco, T.; Cagiano, R.; Bottalico, L.; Carlaio, R.G.; Prejbeanu, R.; Vermesan, H.; Dragulescu, S.I.; Vermesan, D.; Motoc, A.; Santacroce, L. Our experience in Helicobacter pylori infection and gastric MALToma. Clin. Ter. 2008, 159, 239–242. [Google Scholar]
- Steinbach, G.; Ford, R.; Glober, G.; Sample, D.; Hagemeister, F.B.; Lynch, P.M.; McLaughlin, P.W.; Rodriguez, M.A.; Romaguera, J.E.; Sarris, A.H.; et al. Antibiotic treatmen t of gastric lymphoma of mucosa-associated lymphoid tissue. Ann. Intern. Med. 1999, 131, 88–95. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Mégraud, F.; O’Morain, C.; Hungin, A.P.; Jones, R.; Axon, A.; Graham, D.Y.; Tytgat, G. European Helicobacter pylori Study Group (EHPSG). Current concepts in the management of Helicobacter pylori infection—The Maastricht 2-2000 Consensus Report. Aliment. Pharmacol. Ther. 2002, 16, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Gravina, A.G.; Zagari, R.M.; De Musis, C.; Romano, L.; Loguercio, C.; Romano, M. Helicobacter pylori and extragastric diseases: A review. World J. Gastroenterol. 2018, 24, 3204–3221. [Google Scholar] [CrossRef]
- Pasceri, V.; Cammarota, G.; Patti, G.; Cuoco, L.; Gasbarrini, A.; Grillo, R.L.; Fedeli, G.; Gasbarrini, G.; Maseri, A. Association of virulent Helicobacter pylori strains with ischemic heart disease. Circulation 1998, 97, 1675–1679. [Google Scholar] [CrossRef] [Green Version]
- Aceti, A.; Are, R.; Sabino, G.; Fenu, L.; Pasquazzi, C.; Quaranta, G.; Zechini, B.; Terrosu, P. Helicobacter pylori active infection in patients with acute coronary heart disease. J. Infect. 2004, 49, 8–12. [Google Scholar] [CrossRef]
- Tousoulis, D.; Davies, G.; Stefanadis, C.; Toutouzas, P.; Ambrose, J.A. Inflammatory and thrombotic mechanisms in coronary atherosclerosis. Heart 2003, 89, 993–997. [Google Scholar] [CrossRef]
- Wang, B.; Yu, M.; Zhang, R.; Chen, S.; Xi, Y.; Duan, G. A meta-analysis of the association between Helicobacter pylori infection and risk of atherosclerotic cardiovascular disease. Helicobacter 2020, 25, e12761. [Google Scholar] [CrossRef]
- Sung, J.J.; Sanderson, J.E. Hyperhomocysteinaemia, Helicobacter pylori, and coronary heart disease. Heart 1996, 76, 305–307. [Google Scholar] [CrossRef]
- Lee, M.; Baek, H.; Park, J.S.; Kim, S.; Kyung, C.; Baik, S.J.; Lee, B.K.; Kim, J.H.; Ahn, C.W.; Kim, K.R.; et al. Current Helicobacter pylori infection is significantly associated with subclinical coronary atherosclerosis in healthy subjects: A cross-sectional study. PLoS ONE 2018, 13, e0193646. [Google Scholar] [CrossRef]
- Malfertheiner, M.V.; Kandulski, A.; Schreiber, J.; Malfertheiner, P. Helicobacter pylori infection and the respiratory system: A systematic review of the literature. Digestion 2011, 84, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Gaselli, M.; Zaffoni, E.; Ruina, M.; Sartori, S.; Trevisani, L.; Ciaccia, A.; Alvisi, V.; Fabbri, L.; Papi, A. Helicobacter pylori and chronic bronchitis. Scand. J. Gastroenterol. 1999, 34, 828–830. [Google Scholar] [CrossRef]
- Rosenstock, S.J.; Jorgensen, T.; Andersen, L.P.; Bonnevie, O. Association of Helicobacter pylori infection with lifestyle, chronic disease, body indices and age at menarche in Danish adults. Scand. J. Public Health 2000, 28, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, J.; Zhang, Y.; Lei, P. Association of Helicobacter pylori infection with chronic obstructive pulmonary disease and chronic bronchitis: A meta-analysis of 16 studies. Infect. Dis. 2015, 47, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Mitz, H.S.; Farber, S.S. Demonstration of Helicobacter pylori intracheal secretions. J. Am. Osteopath. Assoc. 1993, 93, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Wedi, B.; Kapp, A. Helicobacter pylori infection in skin diseases: A critical appraisal. Am. J. Clin. Dermatol. 2002, 3, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.; Rodrigues, J.; Delgado, L.; Fonseca, J.; Vaz, M. Is Helicobacter pylori infection associated with chronic idiopathic urticaria? Allergol. Immunopathol. 2003, 31, 209–214. [Google Scholar] [CrossRef]
- Smyk, D.S.; Koutsoumpas, A.L.; Mytilinaiou, M.G.; Rigopoulou, E.I.; Sakkas, L.I.; Bogdanos, D.P. Helicobacter pylori and autoimmune disease: Cause or bystander. World J. Gastroenterol. 2014, 20, 613–629. [Google Scholar] [CrossRef]
- Rabelo-Gonçalves, E.M.; Roesler, B.M.; Zeitune, J.M. Extragastric manifestations of Helicobacter pylori infection: Possible role of bacterium in liver and pancreas diseases. World J. Hepatol. 2015, 7, 2968–2979. [Google Scholar] [CrossRef]
- Nilsson, H.O.; Taneera, J.; Castedal, M.; Glatz, E.; Olsson, R.; Wadstrom, T. Identification of Helicobacter pylori and other Helicobacter species by PCR, hybridization, and partial DNA sequencing in human liver samples from patients with primary sclerosing cholangitis or primary biliary cirrhosis. J. Clin. Microbiol. 2000, 38, 1072–1076. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.G.; Dewhirst, F.E.; Shen, Z.; Feng, Y.; Taylor, N.S.; Paster, B.J.; Ericson, R.L.; Lau, C.N.; Correa, P.; Araya, J.C.; et al. Hepatic Helicobacter speciesia identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology 1998, 114, 755–763. [Google Scholar] [CrossRef]
- Leong, R.W.; Sung, J.J. Review article: Helicobacter species and hepatobiliary diseases. Aliment. Pharmacol. Ther. 2002, 16, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Bottalico, L.; Castellaneta, F.; Charitos, I.A. From hydrotherapy to the discovery of the gut microbiota: The historical gastrointestinal health concept. Pharmacophore 2020, 11, 82–90. [Google Scholar]
- Santacroce, L.; Charitos, I.A.; Ballini, A.; Inchingolo, F.; Luperto, P.; De Nitto, E.; Topi, S. The Human Respiratory System and its Microbiome at a Glimpse. Biology 2020, 9, 318. [Google Scholar] [CrossRef]
- Man, A.; Ciurea, C.N.; Pasaroiu, D.; Savin, A.I.; Toma, F.; Sular, F.; Santacroce, L.; Mare, A. New perspectives on the nutritional factors influencing growth rate of Candida albicans in diabetics. An in vitro study. Mem. Inst. Oswaldo Cruz 2017, 112, 587–592. [Google Scholar] [CrossRef]
- Ozbey, G.; Sproston, E.; Hanafiah, A. Helicobacter pylori Infection and Gastric Microbiota. Euroasian J. Hepatogastroenterol. 2020, 10, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Charitos, I.A.; Castellaneta, F.; Santacroce, L.; Bottalico, L. Historical anecdotes and breakthroughs of histamine: From discovery to date. Endocr. Metab. Immune Disord. Drug Targets 2020. Epub ahead of print. [Google Scholar] [CrossRef]
- Santacroce, L.; Inchingolo, F.; Topi, S.; Del Prete, R.; Di Cosola, M.; Charitos, I.A.; Montagnani, M. Potential beneficial role of probiotics on the outcome of COVID-19 patients: An evolving perspective. Diabetes Metab. Syndr. 2021, 15, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, L.; Sardaro, N.; Topi, S.; Pettini, F.; Bottalico, L.; Cantore, S.; Cascella, G.; Del Prete, R.; Dipalma, G.; Inchingolo, F. The pivotal role of oral microbiota in health and disease. J. Biol. Regul. Homeost. Agents 2020, 34, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, L.; Mavaddati, S.; Hamedi, J.; Zeinali, B.; Ballini, A.; Bilancia, M. Expressive Analysis of Gut Microbiota in Pre- and Post- Solid Organ Transplantation Using Bayesian Topic Models Computational Science and Its Applications—ICCSA 2020; Springer: Cham, Switzerland, 2020; pp. 150–165. [Google Scholar] [CrossRef]
- Polimeno, L.; Barone, M.; Mosca, A.; Viggiani, M.T.; Leo, A.; Debellis, L.; Troisi, M.; Daniele, A.; Santacroce, L. Gut Microbiota Imbalance is Related to Sporadic Colorectal Neoplasms. A Pilot Study. Appl. Sci. 2019, 9, 5491. [Google Scholar] [CrossRef] [Green Version]
- Ballini, A.; Scacco, S.; Boccellino, M.; Santacroce, L.; Arrigoni, R. Microbiota and Obesity: Where Are We Now? Biology 2020, 9, 415. [Google Scholar] [CrossRef] [PubMed]
- Di Serio, F.; Lovero, R.; D’Agostino, D.; Nisi, L.; Miragliotta, G.; Contino, R.; Man, A.; Ciccone, M.M.; Santacroce, L. Evaluation of procalcitonin, Vitamin D and C-reactive protein levels in septic patients with positive emocoltures. Our preliminary experience. Acta Med. Mediterr. 2016, 32, 1911–1914. [Google Scholar] [CrossRef]
- Santacroce, L.; D’agostino, D.; Charitos, I.A.; Bottalico, L.; Ballini, A. A short review about electrophysiology and bioimpedance: History and perspectives. Indian J. Public Health Res. Dev. 2018, 9, 577–591. [Google Scholar] [CrossRef]
- Isacco, C.G.; Ballini, A.; De Vito, D.; Nguyen, K.C.D.; Cantore, S.; Bottalico, L.; Quagliuolo, L.; Boccellino, M.; Di Domenico, M.; Santacroce, L.; et al. Rebalance the oral microbiota as efficacy tool in endocrine, metabolic, and immune disorders. Endocr. Metab. Immune Disord. Drug Targets 2020. Epub ahead of print. [Google Scholar] [CrossRef]
- Polimeno, L.; Barone, M.; Mosca, A.; Viggiani, M.T.; Joukar, F.; Mansour-Ghanaei, F.; Mavaddati, S.; Daniele, A.; Debellis, L.; Bilancia, M.; et al. Soy Metabolism by Gut Microbiota from Patients with Precancerous Intestinal Lesions. Microorganisms 2020, 8, 469. [Google Scholar] [CrossRef]
- Frost, F.; Kacprowski, T.; Rühlemann, M.; Bang, C.; Franke, A.; Zimmermann, K.; Nauck, M.; Völker, U.; Völzke, H.; Biffar, R.; et al. Helicobacter pylori infection associates with fecal microbiota composition and diversity. Sci. Rep. 2019, 9, 20100. [Google Scholar] [CrossRef]
- Dash, N.R.; Khoder, G.; Nada, A.M.; Al Bataineh, M.T. Exploring the impact of Helicobacter pylori on gut microbiome composition. PLoS ONE 2019, 14, e0218274. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.M.; Chen, C.C.; Chang, C.M.; Fang, Y.J.; Bair, M.J.; Chen, P.Y.; Chang, C.Y.; Hsu, Y.C.; Chen, M.J.; Chen, C.C.; et al. Taiwan Gastrointestinal Disease and Helicobacter Consortium. Long-term changes of gut microbiota, antibiotic resistance, and metabolic parameters after Helicobacter pylori eradication: A multicentre, open-label, randomised trial. Lancet Infect. Dis. 2019, 19, 1109–1120. [Google Scholar] [CrossRef]
- Schulz, C.; Schütte, K.; Mayerle, J.; Malfertheiner, P. The role of the gastric bacterial microbiome in gastric cancer: Helicobacter pylori and beyond. Therap. Adv. Gastroenterol. 2019, 12, 1756284819894062. [Google Scholar] [CrossRef] [Green Version]
- Vazirzadeh, J.; Falahi, J.; Moghim, S.; Narimani, T.; Rafiei, R.; Karbasizadeh, V. Molecular Assessment of Resistance to Clarithromycin in Helicobacter pylori Strains Isolated from Patients with Dyspepsia by Fluorescent In Situ Hybridization in the Center of Iran. BioMed Res. Int. 2020, 2020, 2304173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajbakhsh, S.; Samarbaf-Zadeh, A.R.; Moosavian, M. Comparison of fluorescent in situ hybridization and histological method for the diagnosis of Helicobacter pylori in gastric biopsy samples. Med. Sci. Monit. 2008, 14, BR183–BR187. [Google Scholar]
- Makristathis, A.; Hirschl, A.M.; Mégraud, F.; Bessède, E. Review: Diagnosis of Helicobacter pylori infection. Helicobacter 2019, 24 (Suppl. 1), e12641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atherton, J.C. Non-endoscopic tests in the diagnosis of Helicobacter pylori infection Aliment. Pharmacol. Ther. 1997, 11 (Suppl. 1), 11–20. [Google Scholar] [CrossRef]
- Miftahussurur, M.; Yamaoka, Y. Diagnostic Methods of Helicobacter pylori Infection for Epidemiological Studies: Critical Importance of Indirect Test Validation. BioMed Res. Int. 2016, 2016, 4819423. [Google Scholar] [CrossRef] [Green Version]
- Garza-Gonzalez, E.; Bosques-Padilla, F.J.; Tijerina-Menchaca, R.; Flores-Gutierrez, J.P.; Maldo-nado-Garza, H.J.; Perez-Perez, G.I. Comparison of endoscopy-basedand serum-based methods for the diagnosis ofHel icobacterpylori. Can. J. Gastroenterol. 2003, 17, 101–642. [Google Scholar] [CrossRef] [Green Version]
- Cutler, A.F.; Prasad, V.M. Long-term follow-up of Helicobacter pylori serology after successful eradication. Am. J. Gastroenterol. 1996, 91, 85–843. [Google Scholar]
- Fallone, C.A.; Moss, S.F.; Malfertheiner, P. Reconciliation of Recent Helicobacter pylori Treatment Guidelines in a Time of Increasing Resistance to Antibiotics. Gastroenterology 2019, 157, 44–53. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Megraud, F.; O’Morain, C.A.; Gisbert, J.P.; Kuipers, E.J.; Axon, A.T.; Bazzoli, F.; Gasbarrini, A.; Atherton, J.; Graham, D.Y.; et al. European Helicobacter and Microbiota Study Group and Consensus panel. Management of Helicobacter pylori infection-the Maastricht V/Florence Consensus Report. Gut 2017, 66, 6–30. [Google Scholar] [CrossRef] [Green Version]
- Saleem, N.; Howden, C.W. Update on the Management of Helicobacter pylori Infection. Curr. Treat. Options Gastroenterol. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, L.; Charitos, I.A.; Bottalico, L. A successful history: Probiotics and their potential as antimicrobials. Expert Rev. Anti Infect. Ther. 2019, 17, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Signorini, L.; De Leonardis, F.; Santacroce, L.; Haxhirexha, K.; Topi, S.; Fumarola, L.; Dipalma, G.; Coscia, M.F.; Inchingolo, F. Probiotics may modulate the impact of aging on adults. J. Biol. Regul. Homeost. Agents 2020, 34, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Ballini, A.; Santacroce, L.; Cantore, S.; Bottalico, L.; Dipalma, G.; Topi, S.; Saini, R.; De Vito, D.; Inchingolo, F. Probiotics Efficacy on Oxidative Stress Values in Inflammatory Bowel Disease: A Randomized Double-Blinded Placebo-Controlled Pilot Study. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Inchingolo, F.; Santacroce, L.; Cantore, S.; Ballini, A.; Del Prete, R.; Topi, S.; Saini, R.; Dipalma, G.; Arrigoni, R. Probiotics and EpiCor® in human health. J. Biol. Regul. Homeost. Agents 2019, 33, 1973–1979. [Google Scholar] [CrossRef]
- Inchingolo, F.; Dipalma, G.; Cirulli, N.; Cantore, S.; Saini, R.S.; Altini, V.; Santacroce, L.; Ballini, A.; Saini, R. Microbiological results of improvement in periodontal condition by administration of oral probiotics. J. Biol. Regul. Homeost. Agents 2018, 32, 1323–1328. [Google Scholar]
- Ballini, A.; Gnoni, A.; De Vito, D.; Dipalma, G.; Cantore, S.; Gargiulo Isacco, C.; Saini, R.; Santacroce, L.; Topi, S.; Scarano, A.; et al. Effect of probiotics on the occurrence of nutrition absorption capacities in healthy children: A randomized double-blinded placebo-controlled pilot study. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8645–8657. [Google Scholar] [CrossRef]
- Pacifici, L.; Santacroce, L.; Dipalma, G.; Haxhirexha, K.; Topi, S.; Cantore, S.; Altini, V.; Pacifici, A.; De Vito, D.; Pettini, F.; et al. Gender medicine: The impact of probiotics on male patients. Clin. Ter. 2021, 171, e8–e15. [Google Scholar] [CrossRef] [PubMed]
- Signorini, L.; Ballini, A.; Arrigoni, R.; De Leonardis, F.; Saini, R.; Cantore, S.; De Vito, D.; Coscia, M.F.; Dipalma, G.; Santacroce, L.; et al. Evaluation of a nutraceutical product with probiotics, vitamin d, plus banaba leaf extracts (Lagerstroemia speciosa) in glycemic control. Endocr. Metab. Immune Disord. Drug Targets 2020. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Leone, D.; Valenzano, A.; Grande, G.; Santacroce, L. Interazioni farmaco-alimentari: Un problema terapeutico sempre più attuale [Drug/food interactions: An actual therapeutic outcome]. Clin. Ter. 2004, 155, 139–147. [Google Scholar]
- Lv, Z.; Wang, B.; Zhou, X.; Wang, F.; Xie, Y.; Zheng, H.; Lv, N. Efficacy and safety of probiotics as adjuvant agents for Helicobacter pylori infection: A meta-analysis. Exp. Ther. Med. 2015, 9, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesbros-Pantoflickova, D.; Corthésy-Theulaz, I.; Blum, A.L. Helicobacter pylori and probiotics. J. Nutr. 2007, 137 (Suppl. 2), 812S–818S. [Google Scholar] [CrossRef] [Green Version]
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter pylori Virulence Factors-Mechanisms of Bacterial Pathogenicity in the Gastric Microenvironment. Cells 2020, 10, 27. [Google Scholar] [CrossRef]
- Larussa, T.; Leone, I.; Suraci, E.; Imeneo, M.; Luzza, F. Helicobacter pylori and T Helper Cells: Mechanisms of Immune Escape and Tolerance. J. Immunol. Res. 2015, 2015, 981328. [Google Scholar] [CrossRef] [Green Version]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [Green Version]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K.O. The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef] [Green Version]
- Santacroce, L.; Bufo, P.; Latorre, V.; Losacco, T. Ruolo dei mastociti nella fisiopatologia delle lesioni gastriche indotte da Helicobacter pylori [Role of mast cells in the physiopathology of gastric lesions caused by Helicobacter pylori]. Chir. Ital. 2000, 52, 527–531. [Google Scholar]
- De Luca, A.; Baldi, A.; Russo, P.; Todisco, A.; Altucci, L.; Giardullo, N.; Pasquale, L.; Iaquinto, S.; D’Onofrio, V.; Parodi, M.C.; et al. Coexpression of Helicobacter pylori’s proteins CagA and HspB induces cell proliferation in AGS gastric epithelial cells, independently from the bacterial infection. Cancer Res. 2003, 63, 6350–6356. [Google Scholar] [PubMed]
- Loguercio, S.; Dian, C.; Flagiello, A.; Scannella, A.; Pucci, P.; Terradot, L.; Zagari, A. In HspA from Helicobacter pylori vicinal disulfide bridges are a key determinant of domain B structure. FEBS Lett. 2008, 582, 3537–3541. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yokota, K.; Ayada, K.; Yamamoto, Y.; Okada, T.; Shen, L.; Oguma, K. Helicobacter pylori heat-shock protein 60 induces interleukin-8 via a Toll-like receptor (TLR)2 and mitogen-activated protein (MAP) kinase pathway in human monocytes. J. Med. Microbiol. 2007, 56 Pt 2, 154–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, Y.; Yokota, K.; Mizuno, M.; Yunoki, N.; Uesu, T.; Okada, H.; Kobayashi, K.; Hirai, Y.; Oguma, K.; Tsuji, T. Antibodies to human gastric epithelial cells and heat shock protein 60 in Helicobacter pylori positive mucosa associated lymphoid tissue lymphoma. Gut 1999, 45, 20–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.S.; He, P.J.; Tsai, N.M.; Li, C.H.; Yang, S.C.; Hsu, W.T.; Wu, M.S.; Wu, C.J.; Cheng, T.L.; Liao, K.W. A potential role for Helicobacter pylori heat shock protein 60 in gastric tumorigenesis. Biochem. Biophys. Res. Commun. 2010, 392, 183–189. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kita, M.; Kodama, T.; Sawai, N.; Tanahashi, T.; Kashima, K.; Imanishi, J. Chemokines in the gastric mucosa in Helicobacter pylori infection. Gut 1998, 42, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, B.; Chen, C.; Yi, Q.; Zhang, X.; Wu, X.; Zheng, S.; Li, N.; She, F. N-terminal region of Helicobacter pylori CagA induces IL-8 production in gastric epithelial cells via the β1 integrin receptor. J. Med. Microbiol. 2020, 69, 457–464. [Google Scholar] [CrossRef]
- Naito, M.; Eguchi, H.; Goto, Y.; Kondo, T.; Nishio, K.; Ishida, Y.; Kawai, S.; Okada, R.; Hishida, A.; Wakai, K.; et al. Associations of plasma IL-8 levels with Helicobacter pylori seropositivity, gastric atrophy, and IL-8 T-251A genotypes. Epidemiol. Infect. 2010, 138, 512–518. [Google Scholar] [CrossRef]
- Mazzucchelli, L.; Blaser, A.; Kappeler, A.; Schärli, P.; Laissue, J.A.; Baggiolini, M.; Uguccioni, M. BCA-1 is highly expressed in Helicobacter pylori-induced mucosa-associated lymphoid tissue and gastric lymphoma. J. Clin. Investig. 1999, 104, R49–R54. [Google Scholar] [CrossRef] [Green Version]
- Michelis, R.; Sela, S.; Sbeit, W.; Cohen, H.I.; Reshef, R. Decreased TFF2 expression in the gastric antrum in patients infected with CagA-positive Helicobacter pylori. Isr. Med. Assoc. J. 2009, 11, 11–15. [Google Scholar] [PubMed]
- Peterson, A.J.; Menheniott, T.R.; O’Connor, L.; Walduck, A.K.; Fox, J.G.; Kawakami, K.; Minamoto, T.; Ong, E.K.; Wang, T.C.; Judd, L.M.; et al. Helicobacter pylori infection promotes methylation and silencing of trefoil factor 2, leading to gastric tumor development in mice and humans. Gastroenterology 2010, 139, 2005–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, H.; Takaishi, S.; Menheniott, T.R.; Yang, X.; Shibata, W.; Jin, G.; Betz, K.S.; Kawakami, K.; Minamoto, T.; Tomasetto, C.; et al. Inhibition of gastric carcinogenesis by the hormone gastrin is mediated by suppression of TFF1 epigenetic silencing. Gastroenterology 2011, 140, 879–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, H.; Liu, J.; Lin, B.; Wang, Z.; Sun, J.; Huang, G. A meta-analysis of interleukin8- 251 promoter polymorphism associated with gastric cancer risk. PLoS ONE 2012, 7, e28083. [Google Scholar] [CrossRef] [Green Version]
- Sukri, A.; Hanafiah, A.; Mohamad Zin, N.; Kosai, N.R. Epidemiology and role of Helicobacter pylori virulence factors in gastric cancer carcinogenesis. APMIS 2020, 128, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Toh, J.W.T.; Wilson, R.B. Pathways of Gastric Carcinogenesis, Helicobacter pylori Virulence and Interactions with Antioxidant Systems, Vitamin C and Phytochemicals. Int. J. Mol. Sci. 2020, 21, 6451. [Google Scholar] [CrossRef]
Figure 1.
(a) Anthony van Leeuwenhoek’s microscope; (b) Robert Hooke’s observations under his microscope: teeth of the snail description (from his book “Micrographia”, p. 180, schem. XXV, fig.1, National Library of Wales, UK).
Figure 1.
(a) Anthony van Leeuwenhoek’s microscope; (b) Robert Hooke’s observations under his microscope: teeth of the snail description (from his book “Micrographia”, p. 180, schem. XXV, fig.1, National Library of Wales, UK).
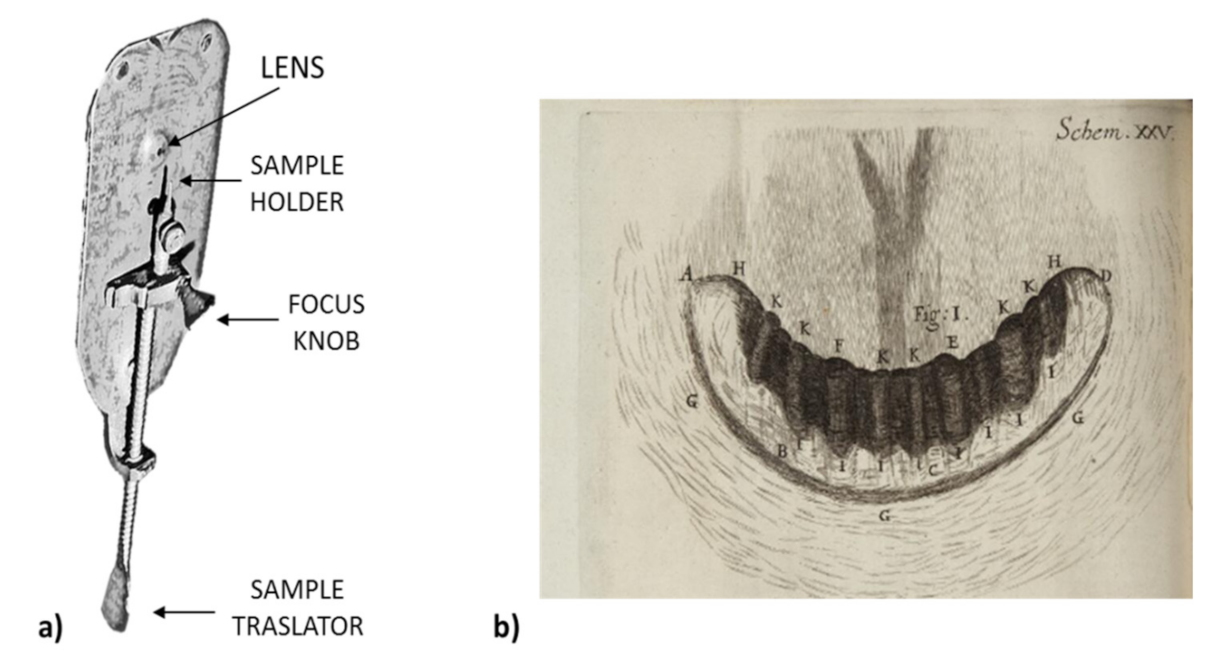
Figure 2.
Partial illustrations: (a) the spiral microorganisms (c-spirilla) in the mucus layer and gastric gland, drawn by the Italian Helicobacter pioneer G. Bizzozero, the first that detected Helicobacter spp. by observing the stomachs of dogs and cats with a microscope (1893). Later, in 1996, the renaming of Helicobacter heilmannii in Helicobacter bizzozeronii reports his last name; (b) spiral microorganisms in the protoplasm, drawn by H. Salomon (1896) [25].
Figure 2.
Partial illustrations: (a) the spiral microorganisms (c-spirilla) in the mucus layer and gastric gland, drawn by the Italian Helicobacter pioneer G. Bizzozero, the first that detected Helicobacter spp. by observing the stomachs of dogs and cats with a microscope (1893). Later, in 1996, the renaming of Helicobacter heilmannii in Helicobacter bizzozeronii reports his last name; (b) spiral microorganisms in the protoplasm, drawn by H. Salomon (1896) [25].
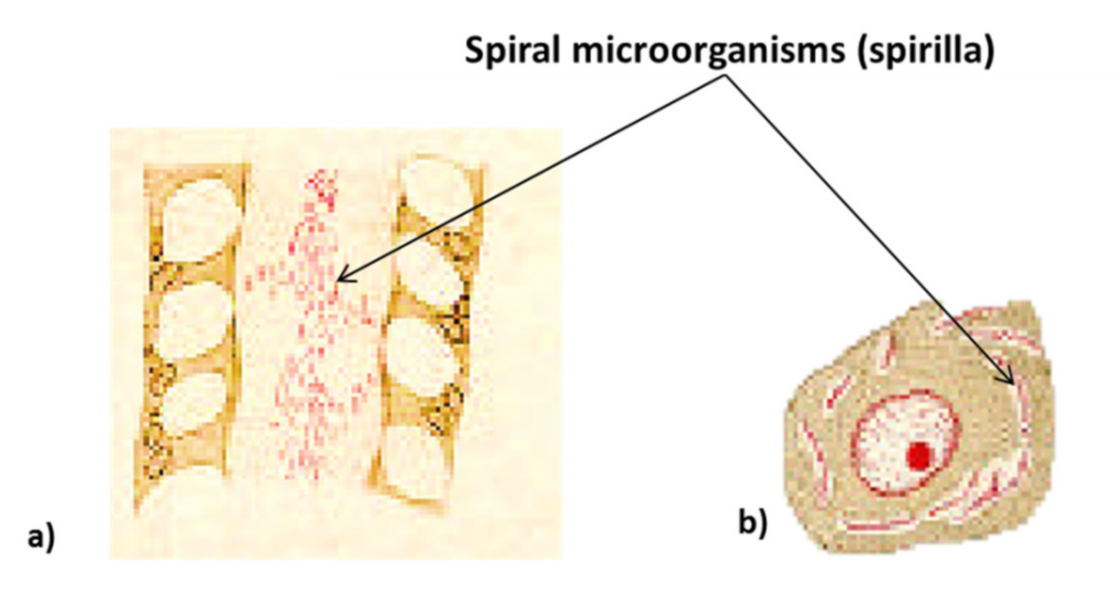
Figure 3.
The spread of H. pylori in the continents over time.

Figure 4.
H. pylori has the ability to detect the pH level of the stomach environment and, thanks to its flagella, to go to the region with the least acidic pH (chemotaxis). H. pylori moves rapidly with the help of its flagella ending in a swollen extremity, consisting of two structural subunits, FlaA and FlaB, and a hook (FlgE) [46]. Other flagella proteins include the flagellar biosynthetic protein FliP (FliP), which influences mobility through slipped-strand mispairing-mediated frameshifting of DNA. In addition, they have a distinctive shell, made up of the HpaA protein, which protects the bacterium from the harmful effects of stomach acid [47]. It also neutralizes the strong pH by promoting a rich production of urease (it can be located on the surface and inside the bacterium). The bacterium accumulates CO(NH2)3 and H2O from the vessels and from the stomach itself, and the enzyme converts them by chemical reaction in 2NH3 and CO2 which neutralize gastric acid, creating a buffer barrier around the H. pylori, surrounding the bacteria to protect them from strong acids in the environment. This also makes urease necessary to maintain a chronic infection. In addition, the H. pylori arginase enzyme captures the L-arginine reaction to produce L-ornithine and CO(NH2)3. This helps in adaptation and resistance to the acidic environment of the stomach and for the colonization of the bacterium towards the epithelium. Furthermore, the subsequent conversion of L-ornithine in polyamines contributes to the dysregulation of the host’s immune response to infection [48].
Figure 4.
H. pylori has the ability to detect the pH level of the stomach environment and, thanks to its flagella, to go to the region with the least acidic pH (chemotaxis). H. pylori moves rapidly with the help of its flagella ending in a swollen extremity, consisting of two structural subunits, FlaA and FlaB, and a hook (FlgE) [46]. Other flagella proteins include the flagellar biosynthetic protein FliP (FliP), which influences mobility through slipped-strand mispairing-mediated frameshifting of DNA. In addition, they have a distinctive shell, made up of the HpaA protein, which protects the bacterium from the harmful effects of stomach acid [47]. It also neutralizes the strong pH by promoting a rich production of urease (it can be located on the surface and inside the bacterium). The bacterium accumulates CO(NH2)3 and H2O from the vessels and from the stomach itself, and the enzyme converts them by chemical reaction in 2NH3 and CO2 which neutralize gastric acid, creating a buffer barrier around the H. pylori, surrounding the bacteria to protect them from strong acids in the environment. This also makes urease necessary to maintain a chronic infection. In addition, the H. pylori arginase enzyme captures the L-arginine reaction to produce L-ornithine and CO(NH2)3. This helps in adaptation and resistance to the acidic environment of the stomach and for the colonization of the bacterium towards the epithelium. Furthermore, the subsequent conversion of L-ornithine in polyamines contributes to the dysregulation of the host’s immune response to infection [48].

Figure 5.
H. pylori adheres to the lipids and carbohydrates of the stomach epithelium through the production of adhesins. The adhesin BabA will bind with the Lewis b antigen of the epithelial cell. Another adhesin, SabA, binds with the epithelial cell and stimulates the increase in sialyl-Lewis x antigen on the gastric mucosa. The bacterium is usually found in the mucus area or on the epithelial cells, but rarely within them [50,51,52].
Figure 5.
H. pylori adheres to the lipids and carbohydrates of the stomach epithelium through the production of adhesins. The adhesin BabA will bind with the Lewis b antigen of the epithelial cell. Another adhesin, SabA, binds with the epithelial cell and stimulates the increase in sialyl-Lewis x antigen on the gastric mucosa. The bacterium is usually found in the mucus area or on the epithelial cells, but rarely within them [50,51,52].

Figure 6.
The toxigenic gene vacuolating cytotoxin A (vacA) and the synergy with cytotoxin-associated protein A (CagA) (released by the type IV secretory system—T4SS) effects on the cell.
Figure 6.
The toxigenic gene vacuolating cytotoxin A (vacA) and the synergy with cytotoxin-associated protein A (CagA) (released by the type IV secretory system—T4SS) effects on the cell.

Figure 7.
The cag pathogenicity island (cag-PAI) of the genome is composed of two main sections: cag I and cag II. Its genes encode the type IV secretory system (T4SS) necessary for the intracellular transport (from the bacterium to the host cell) of the CagA protein and peptidoglycan (iE-DAP). After the entry of CagA into the host cell, its Src-Abl-dependent phosphorylation occurs, which activates the RAP1A-BRAF-ERK (Ras-related protein 1-BRaf proto-oncogene serine/threonine kinase - Extracellular signal-regulated kinase) signaling pathway. This leads to the hummingbird phenotype (rearrangement of the actin-based cytoskeletal phenotype characterized by long, thin cellular extensions regulated by Rho GTPases and the Actin Related Protein (Arp) 2/3 complex) and the deregulation of cell migration and proliferation. Subsequently, due to the rupture of the apical junctions and the loss of membrane polarity, it leads to deregulation of cellular permeability mechanisms. Furthermore, CagA suppresses the apoptotic activity of VacA and activates the signaling pathway of the factor NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), allowing the subsequent overexpression of anti-apoptotic agents, interleukin (IL)-8, and defensins (hβDs). H. pylori causes apoptosis accompanied by increased expression of the pro-apoptotic multi-domain proteins Bax (Bcl-2-associated X protein) and Bak proteins that favor apoptosis, suggesting that Bax and Bak may be important mediators of gastric epithelial cells of apoptosis [68,69,70,71].
Figure 7.
The cag pathogenicity island (cag-PAI) of the genome is composed of two main sections: cag I and cag II. Its genes encode the type IV secretory system (T4SS) necessary for the intracellular transport (from the bacterium to the host cell) of the CagA protein and peptidoglycan (iE-DAP). After the entry of CagA into the host cell, its Src-Abl-dependent phosphorylation occurs, which activates the RAP1A-BRAF-ERK (Ras-related protein 1-BRaf proto-oncogene serine/threonine kinase - Extracellular signal-regulated kinase) signaling pathway. This leads to the hummingbird phenotype (rearrangement of the actin-based cytoskeletal phenotype characterized by long, thin cellular extensions regulated by Rho GTPases and the Actin Related Protein (Arp) 2/3 complex) and the deregulation of cell migration and proliferation. Subsequently, due to the rupture of the apical junctions and the loss of membrane polarity, it leads to deregulation of cellular permeability mechanisms. Furthermore, CagA suppresses the apoptotic activity of VacA and activates the signaling pathway of the factor NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), allowing the subsequent overexpression of anti-apoptotic agents, interleukin (IL)-8, and defensins (hβDs). H. pylori causes apoptosis accompanied by increased expression of the pro-apoptotic multi-domain proteins Bax (Bcl-2-associated X protein) and Bak proteins that favor apoptosis, suggesting that Bax and Bak may be important mediators of gastric epithelial cells of apoptosis [68,69,70,71].

Figure 8.
Pathogenetic hypothesized mechanisms on the correlation between H. pylori atherogenesis and coronary heart disease.
Figure 8.
Pathogenetic hypothesized mechanisms on the correlation between H. pylori atherogenesis and coronary heart disease.

Figure 9.
Other hypotheses about gastric oncogenesis pathways of H. pylori.


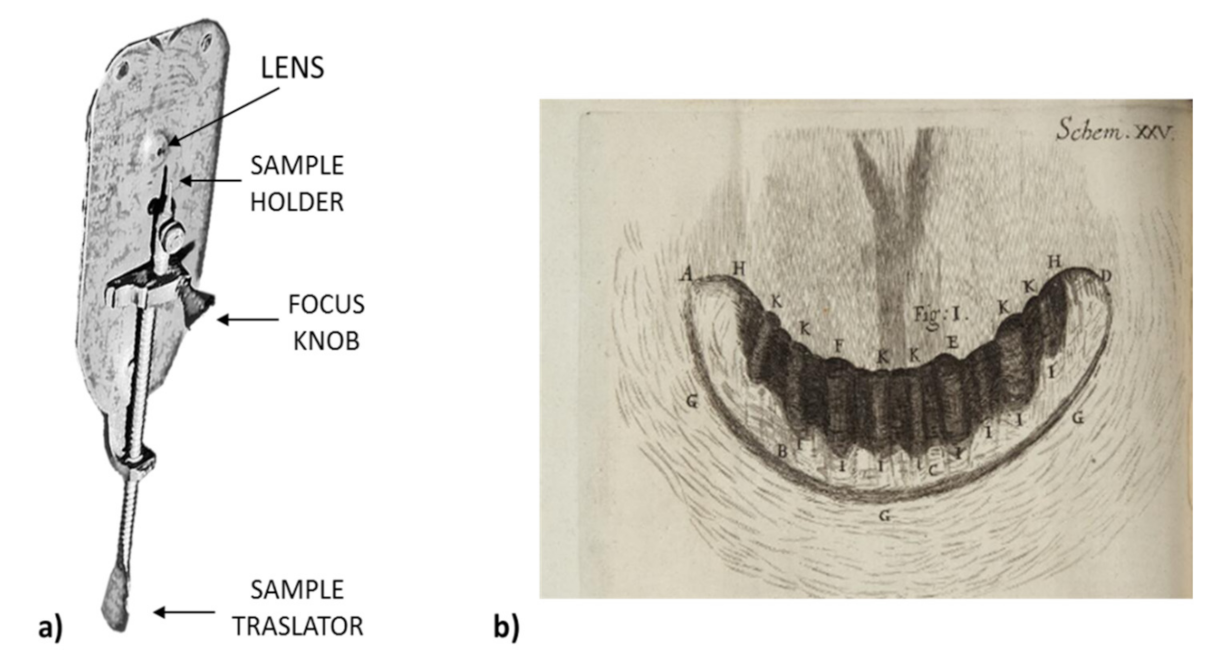{kind=link}
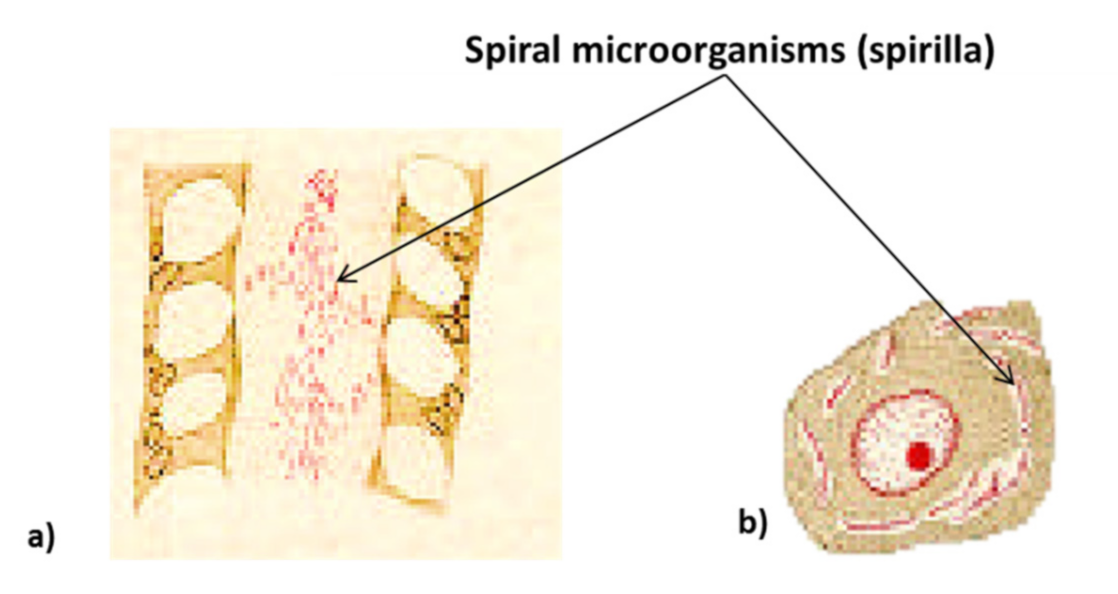{kind=link}
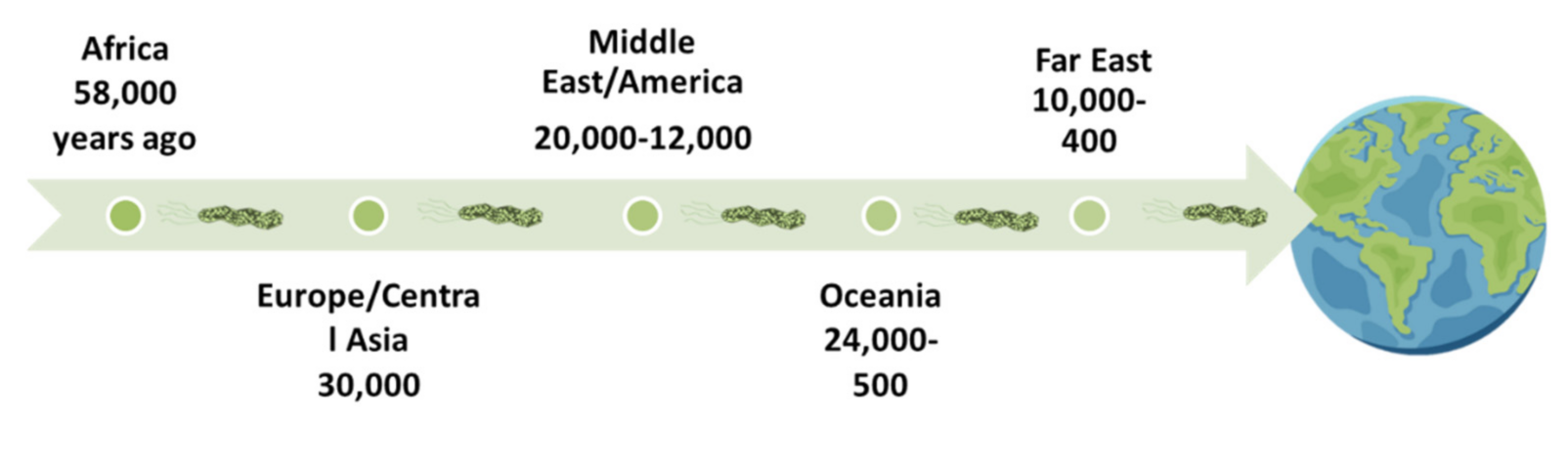{kind=link}
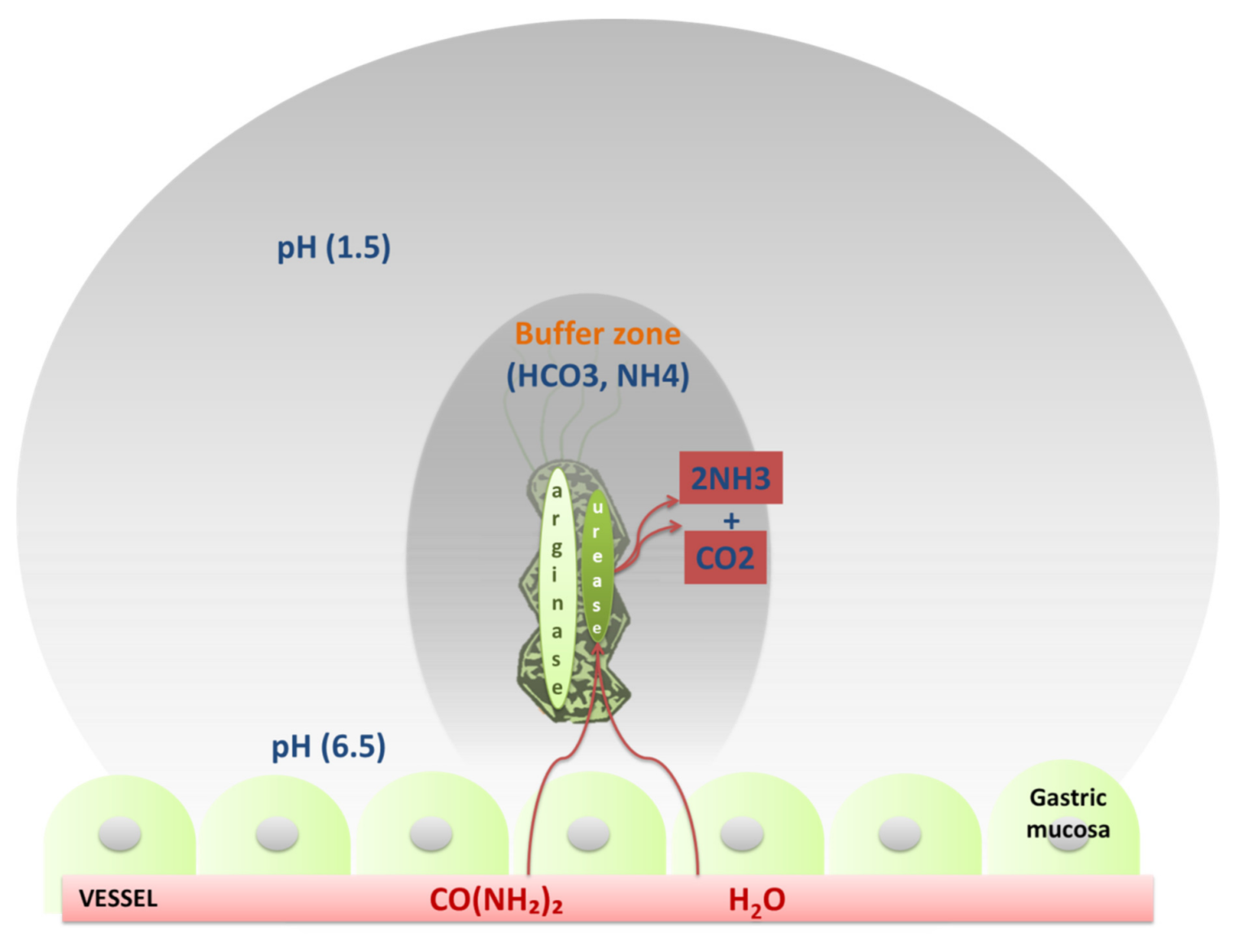{kind=link}
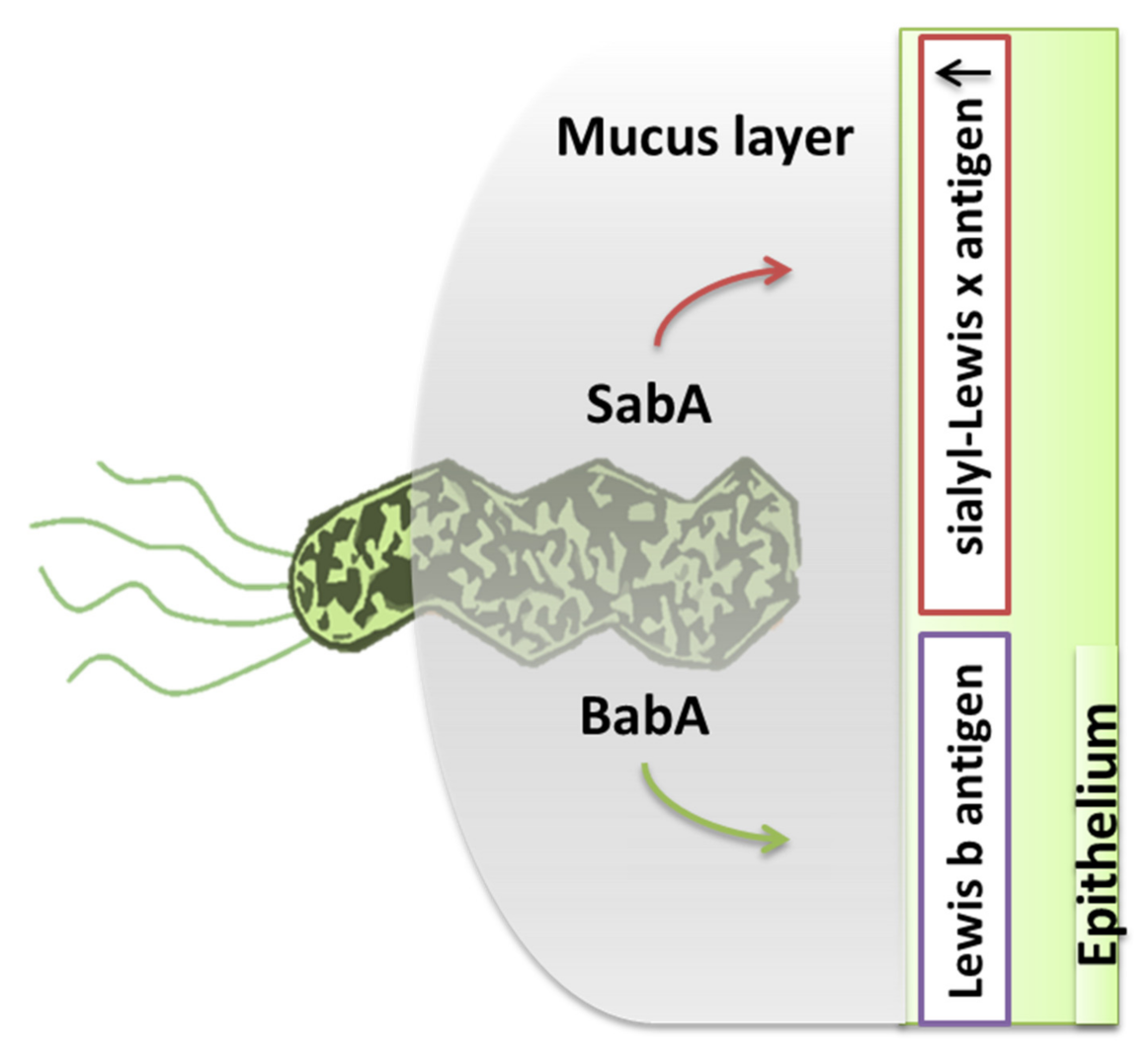{kind=link}
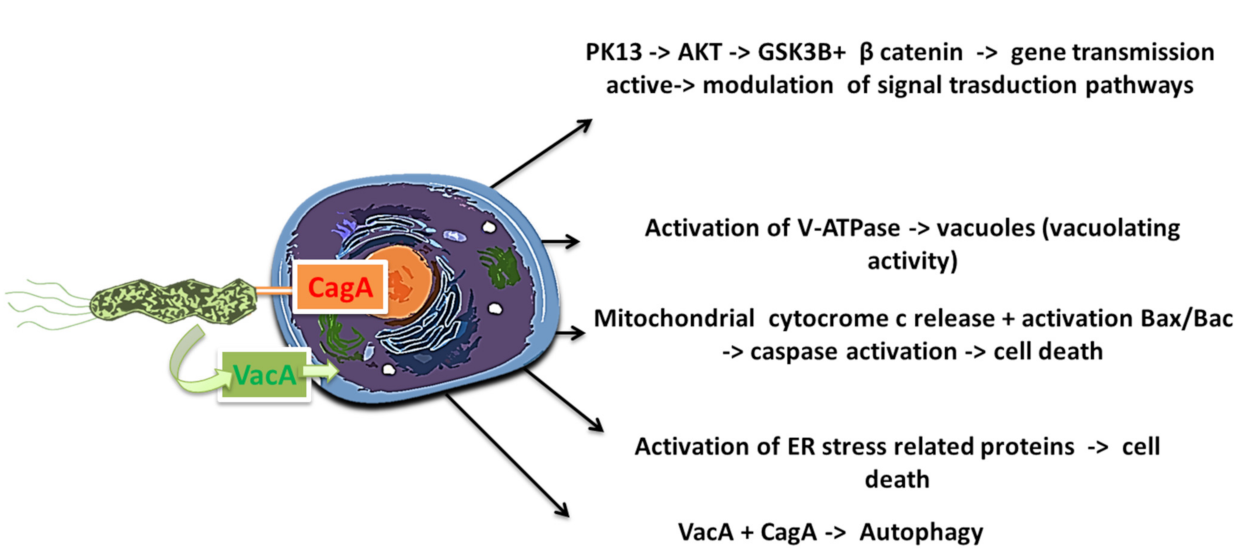{kind=link}
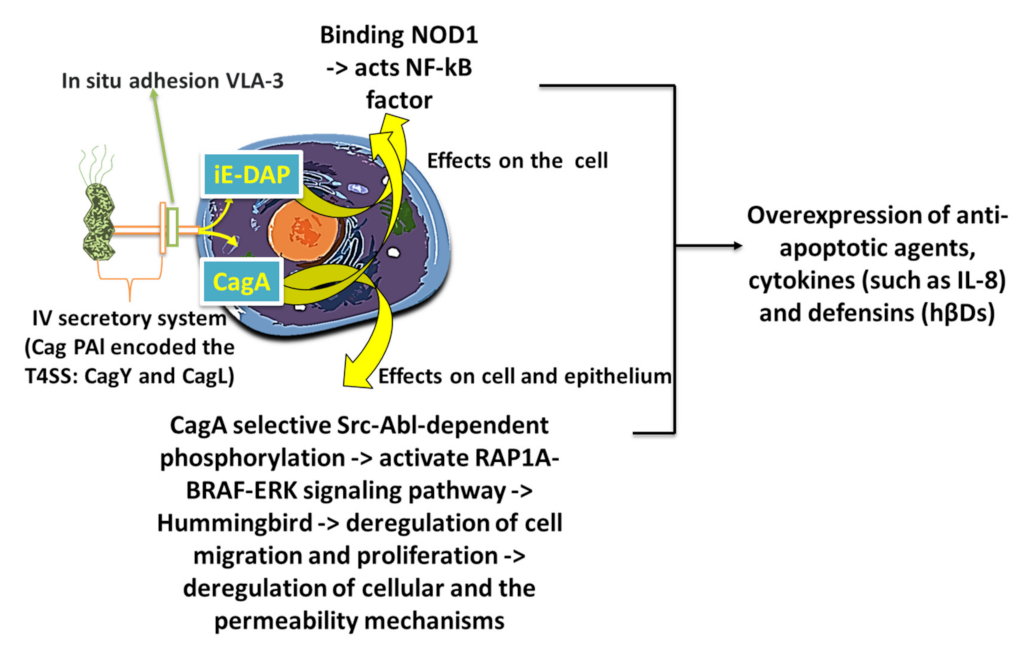{kind=link}
{kind=link}
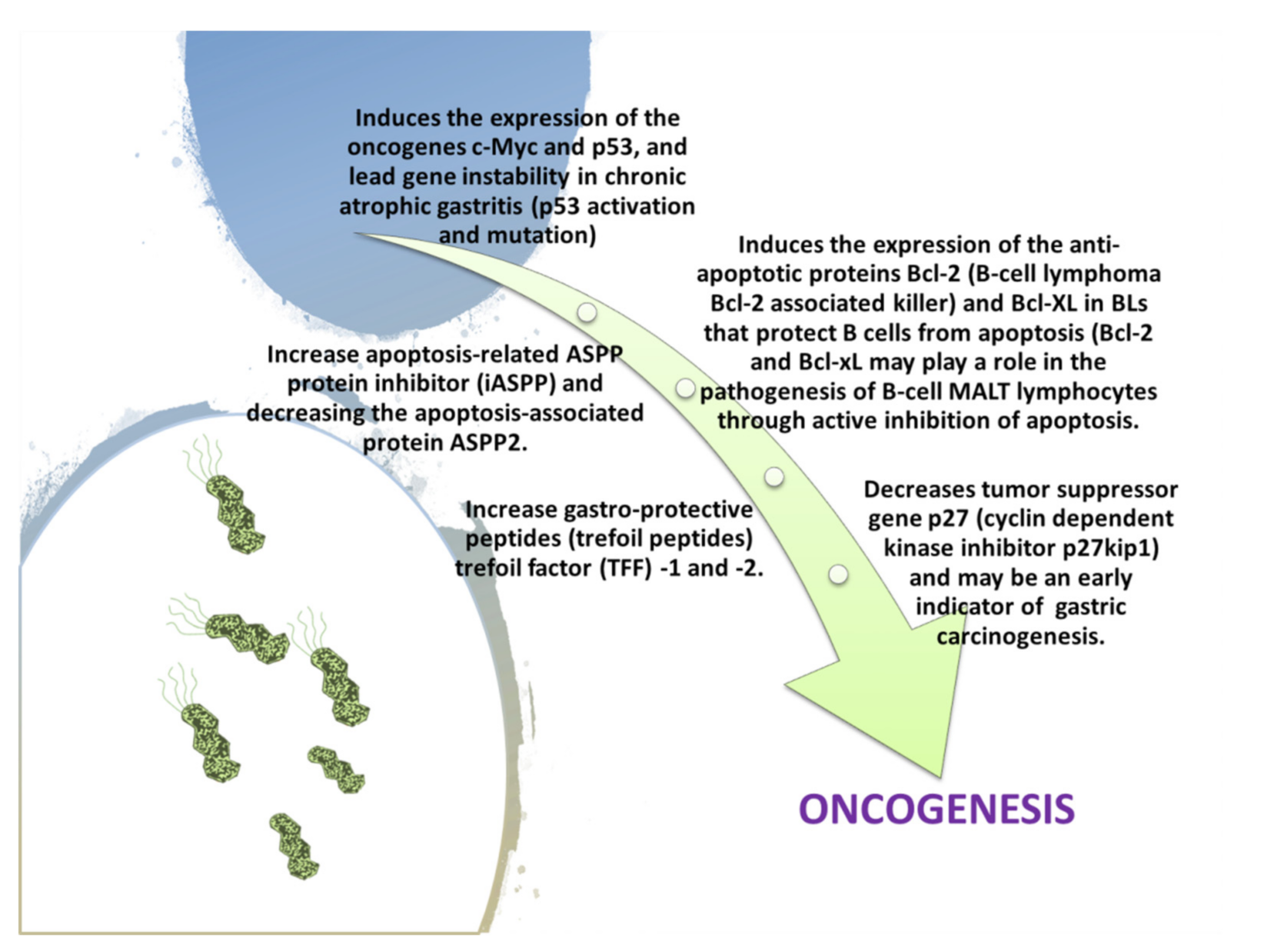{kind=link}
Table 1.
The main steps in the research for bacteria in the stomach over time (Adapted from Suzuki, H.; Warren, R.; Marshall, B. Helicobacter pylori; Springer: Berlin/Heidelberg, Germany, 2016, p. 8).
Table 1.
The main steps in the research for bacteria in the stomach over time (Adapted from Suzuki, H.; Warren, R.; Marshall, B. Helicobacter pylori; Springer: Berlin/Heidelberg, Germany, 2016, p. 8).
| Gastric Bacteria Discoveries—Historical Timeline | |
|---|---|
| Year | Researchers |
| 1875 | G. Bottcher/M. Letulle (demonstrated bacteria in ulcer margins) |
| 1881 | C. Klebs (indentified bacterial colonization and inter-glandular small cell infiltration) |
| 1889 | W. Jaworski (noticed spiral organisms, Vibrio rugula, in gastric washings) |
| 1893 | G. Bizzozero (identified spirochetes in gastric mucosa of dogs) |
| 1896 | H. Salomon (spirochetes noted in gastric mucosa and experimentally transferred to mice) |
| 1906 | W.Krienitz (noticed spirochetes in gastric contents of a patient with gastric carcinoma) |
| 1916 | E.C. Rosenow (described Streptococcus-induced gastric ulcers) |
| 1917 | L.R. Dragstedt (bacteria in experimental ulcers, no significant role identified) |
| 1924 | J.M. Luck (discovered gastric mucosal urease-1 in dogs) |
| 1925 | B. Hoffma (described B. hoffmani as a putative ulcerous agent) |
| 1938 | J.L. Doenge (Spirochetes in gastric glands of Macacus rhesus and humans) |
| 1940 | S.Freedberg (note spirochaetes in biopsies of patients operated on for ulcer or stomach cancer) and F.D. Gorham (postulated gastric acidophilic bacteria as etiologic agents in ulcer disease) |
| 1983 | J.R. Warren (identified Campylobacter pylori in human gastritis) |
| 1967 | S.Ito (in his “Handbook of physiology” reported an image of a spiral bacterium with flagella such as the Helicobacter pylori from samples taken from own stomach) |
| 1981 | R.Warren (after biopsy noticed curved bacteria in the stomach of some patients) and B.Marshall (begins the pilot study for this) |
| 1982 | B. Marshall (isolated and cultured Helicobacter pylori) |
| 1983 | B. Marshall and R. Warren (they describe H. pylori as a new species on Lancet) |
| 1985/1987 | B. Marshall/A. Morris (ingested and proved the infectivity of H. pylori according to the Koch’s 3rd criterium postulate) |
Table 2.
The actions of the virulence factors of H. pylori.
| Virulence Factors in H. pylori | |||||||
|---|---|---|---|---|---|---|---|
| Flagella | Urease | Lipopolysaccharides | Adhesins | Type IV Secretion System | Exotoxins | Lytic Enzymes | Heat Shock Proteins (Hsps) |
| Chemotaxis | Buffer the gastric acidic pH (creates mucosal damage through ammonia production) | Cell adhesion andinflammation | Cell adhesion, inflammation, stimulates the increase of sialyl-Lewis x antigen on the gastric mucosa. | Inject CagA and other factors. It causes the production of pro-inflammatory factors with subsequent cell apoptosis | VacA causes mucosal damage | Mucinases, proteases, lipases that cause damage to the mucosa | HSP-A and HSP-B proteins lead to inflammation |
Table 3.
The main H. pylori-associated diseases.
| H. pylori Infection Associated Diseases | |
|---|---|
| Digestive Tract | Extra-Digestive |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Charitos, I.A.; D’Agostino, D.; Topi, S.; Bottalico, L. 40 Years of Helicobacter pylori: A Revolution in Biomedical Thought. Gastroenterol. Insights 2021, 12, 111-135. https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12020011
AMA Style
Charitos IA, D’Agostino D, Topi S, Bottalico L. 40 Years of Helicobacter pylori: A Revolution in Biomedical Thought. Gastroenterology Insights. 2021; 12(2):111-135. https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12020011
Chicago/Turabian StyleCharitos, Ioannis Alexandros, Donato D’Agostino, Skender Topi, and Lucrezia Bottalico. 2021. "40 Years of Helicobacter pylori: A Revolution in Biomedical Thought" Gastroenterology Insights 12, no. 2: 111-135. https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12020011