1. Introduction
The global community is under siege due to the on-going devastating pandemic by one of the most serious zoonotic coronavirus lineages of all times. In a matter of months, Covid-19, the disease caused by the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV2), has reached every corner on earth dramatically changing the way humans live. As of January 12, 2021, 88,387,352 cases and about 2 million deaths were documented in the WHO situation report [
https://www.who.int/publications/m/item/weekly-epidemiological-update---12-january-2021] accessed on 12 January 2021. The unprecedented speed in the spread of SARS-CoV2 and the subsequent evolution of mutants have resulted in increased hospitalizations, mortality are rates, and paralyzed global healthcare systems. Significant gaps have been created in our knowledge of evolutionary dynamics, epidemicity, and adaptive transcriptomic of coronavirus pandemics. It is not fully clear how the recurring zoonotic coronavirus lineages of including SARS-CoV2, successfully adapted, evolved, host-jumped, and maintained transmissions into humans.
Coronaviruses are enveloped, positive-sense, single-stranded RNA viruses that belong to the family Coronaviridae. Based on phylogenetic relationships and genomic structures, there are four genera of CoVs, namely,
Alphacoronavirus (αCoV),
Betacoronavirus (βCoV),
Deltacoronavirus (δCoV), and
Gammacoronavirus (γCoV) [
1]. It has been widely established that most α- and βCoVs infect mammals, bats and rodents, where they usually cause respiratory illness in humans and gastroenteritis in animals, while avian species carry δCoVs and γCoVs. There are four known human coronaviruses (HCoV-NL63, HCoV-229E, HCoV-OC43 and HKU1). These were mostly known to cause mild infections in immunocompetent people until the outbreak of severe acute respiratory syndrome (SARS) in 2002 and 2003 in Guangdong province, China [
2,
3,
4,
5]. A decade later, another highly pathogenic coronavirus, the Middle East respiratory syndrome coronavirus (MERSCoV) emerged in Middle Eastern countries [
6]. The current emergence of the SARS-CoV2 implies occurrence of significant changes in the evolutionary dynamics of the virus-host interactions in coronaviruses.
The molecular mechanisms underlying evolution of new coronavirus lineages with enhanced ability in cross-host transmissions is not well understood. Domain variations and receptor binding specificities were reported. All of the three viruses—SARS-CoV, MERS-CoV, and SARS-CoV2—showed frequent crosses to species barriers leading to rapid adaptation and human to human transmissions [
7,
8,
9]. The massive number of coronaviruses carried by different bat species and the increasing human exploitation of the wild implied continued transmissions from bats to animals to humans. SARS-CoV and MERS-CoV were transmitted directly to humans from market civets and dromedary camels, respectively [
10,
11]; however both viruses were thought to have originated in bats [
12,
13]. In human, transmission was found facilitated by the SARS-CoV high affinity for angiotensin-converting enzyme 2 (ACE2) [
14,
15]. However, variations in binding affinities of SARS CoV for human ACE2 affected infectivity of human cells [
16]. In the 2002–2003 outbreak strain hTor02 had a high affinity for ACE2 and efficiently transmitted between humans. However, palm civets strains cSz02 and cHb05 showed low affinity and low infectivity in human cells [
10,
17]. On the other hand, MERS-CoV uses dipeptidyl peptidase 4 (DPP4; also known as CD26) as a receptor and infects unciliated bronchial epithelial cells and type II pneumocytes [
18,
19]. While the S1-CTD is common in RBD of both MERS-CoV and SARS-CoV, the latter uses ACE2 and the former uses DPP4. These observations indicate potential changes in subtle translation and functional mechanisms, rather than sequence variations of accessory genes.
Domain variations within the binding structures of previous coronaviruses have been reported. Although SARS-CoVs and bat SARSr-CoVs share high sequence identity in the S2 domain, they mainly vary in three regions: (1) the spike protein (S) (both the S1 amino-terminal domain (S1-NTD) and the S1 receptor binding domain [
20] (2) ORF8 (8a and b) and (3) ORF3 (3a and 3b). Compared with human and civet SARS-CoV, bat SARSr-CoV S1 can be separated out into two clades. Clade 1, Yunnan province specific and has the same size S protein as human and civet isolates and clade 2 which is universal in many locations has a shorter size S protein due to deletions of 5, 12 or 13 amino acid residues [
21]. The second variation occurs in the 366 or 369 nucleotides
orf8 locus retained intact in all bat SARSr-CoVs and shared among themselves and with civet and human SARS CoVs. Analysis of the 2002–2003 outbreak transmission pattern explained how the
orf8 gene underwent phases of adaptations during transmission from animals to humans. The outbreak occurred in three phases; in the early-phase (localized limited cases) patients had two genotypes of
orf8 on their viral genomes; (one with a complete
orf8 of 369 nucleotides, and the other with 82-nucleotide deletion}. Whereas, in late-phase (pandemic) and most middle-phase (super-reader) patients had a split
orf8 (
orf8a and
orf8b) on their viral genomes because of a 29-nucleotide deletion [
17]. However, two exceptions were found in the middle-phase genomes, one that had an 82-nucleotide deletion in
orf8 and the other with the whole
orf8 deleted. The human isolates from 2004 and all civet SARS-CoV genomes had a complete
orf8 except one civet strain with an 82-nucleotide deletion. Thus,
orf8 genes are accessory adaptation markers that aid in transmission from animals to humans and thus, are potential targets as gene candidates. Interestingly, the European bat SARSr-CoV has completely lost
orf8 [
22] implying that the
orf8 accessory genes in bat SARSrCoVs are potential targets for continuous evolution in their natural hosts like bats, civets and humans and warranting further investigations for diagnosis and therapeutics. The third variable locus is in ORF3. On SARS CoV genome, this is a 154-amino acid that codes for an interferon antagonist. While the ORF3a locus is highly similar in both SARSr-CoVs and SARS-CoV (96.4–98.9% amino acid identity), the SARSr-CoVs have different sizes of ORF3b [
21]. These observations indicate functional changes in addition to sequence variations. Thus, there seems to be a significant knowledge gap in how coronaviruses exploit functional mechanisms, RNA modifications, and coding potentials to adapt to different hosts, despite RNA stability.
RNA modifications and signature sequences play important roles in prokaryotic adaptive expressions. Differential over expression of chromosomal genes, mutation in accessory genes, slippage on signature sequences, and repeat based hot spots that serve as ON/OFF switches are some of the mechanisms involved. In bacteria, these are mostly found associated to successfully re-emerging and outbreak pathogens such as
Hemophilus influenza, streptococcus, and methicillin-resistant
Staphylococcus aureus. In E. coli prfB mRNA for instance, the U CUU UGA slippery site contains the in frame UGA termination codon which is recognized by RF2 [
23]. Although adaptive replication and transcription mechanisms are common in prokaryotes, it is still unclear whether the general rule applies to SARS-CoV-2. The translational reading frame establishment and maintenance with a universal start AUG codon (encoding methionine), has become a more complicated scenario in polycistronic prokaryotes mRNA translation processes. Relatively recent data on ribosome profiling suggest large amounts of translation initiation events occur at only a subset of non-AUG codons [
24]. Enabling multiple coding mechanism to manipulate RNA structure-based strategies for translation is a selective advantage for rapid adaptive expressions [
25]. An example of these are production of overlapping Open Reading Frames (ORFs) through Programmed Ribosomal Frameshifting (PRF) recoding, ON/OFF molecular switches, and differential expressions, to enhance the coding capacity of a single RNA template. These mechanisms are all ‘programmed’ at specific cis-acting elements on mRNAs, and at higher orders of magnitude more frequent than non-programmed events. Some of the cis-acting signals located in mRNA sequences are known regulatory loci that improve gene expression. It involves classes of loci that direct elongating ribosomes shifting the reading frame by one (or more) base in either directions; the 5′ (−1) or 3′ (+1). The process is largely programed for over expression to compensate for the compact genomes size and hence it is called Programmed Ribosomal Frameshifting (PRF). Its common functional property is to stimulate ribosomes to pause at specific ‘slippery’ sequences. These PRF events were found to operate at efficiencies as high as 80% increasing the coding capacity of genomes at the same time regulating mRNA stability [
26]. However, ribosome can also shift by –2, –4, +2, +5, or +6 nucleotides in rare events [
27,
28]. In coronaviruses, alterations in triplet decoding of the messenger RNA by the elongating ribosome is influenced by many factors including RNA dimerization [
29]. As a result, protein synthesis does not proceed through the usual translation steps. Instead, while the first major polyprotein produced by the ORF1a that encodes non-structural proteins is translated normally, PRF acts on the elongation process. Slippery signals just before the termination codon of ORF1a redirect some species of translating ribosomes making them to bypass the stop codon. This allows translation to continue in the −1 reading frame thereby creating the larger ORF1ab polyprotein [
30,
31,
32]. As a result, the ORF1a produces polypeptide 440–500 kDa protein that is cut into ~11 non structural proteins (nsps). The -1 PRF occurs right upstream of the ORF1a stop codon allowing translation to continue giving the large polypeptide ORF1b measuring 740–810 kDa that is cut into 16 nsps. Nevertheless, it is not yet clear what are the types, numbers, and locations of specific protective modification events that stabilize RNA and the types of changes that occur in the protein. In eukaryotic viruses, the slippery site has the heptameric motif N NNW WWH, where the incoming reading frame is indicated, and N = any three identical nucleotides, W = AAA or UUU, and N ≠ G [
33]. The slippery site can also be presented as: X XXY YYZ, where the underlined codons denote the 0 reading frame. The ribosome slips on these sites to change register by 1 nucleotide in the 5′ direction, followed by a pseudoknot. DNA nanoball sequencing has revealed that the transcriptome of SARS-CoV2 is highly complex due to numerous discontinuous transcription events [
34]. However, it is known that RNA is protected through series of mechanisms such as 5′ cap, a highly methylated modification, and a Poly A tail to each end of a pre-mRNA molecule to protects the new mRNA. While genomic RNA is used to assemble progeny virions, shorter sgRNAs encode conserved structural proteins. These are namely: the spike surface glycoprotein (S), small envelope protein (E), matrix protein (M), and nucleocapsid protein (N). The S protein is involved in binding to receptors on the host cell and determines host tropism [
35,
36]. Although SARS-CoV and MERS-CoV are related, their spike proteins bind different receptors indicating different mechanisms under common genomic backgrounds. In addition, SARS-CoV-2 is known to have at least eight accessory proteins (3a, 3b, p6, 7a, 7b, 8b, 9b, and orf14) [
37] (GenBank: NC_045512.2). However, not all ORFs are experimentally verified for expression.
Regular update on the evolutionary process and phylogenetics of SARS-CoV2 is imperative due to the changing epidemiology of the virus. Early genomic analysis suggested evolutionary association of 2019-nCoV and SARS like bat coronaviruses and MERS [
38]. In depth comparative analysis of the first determined genomes of the novel coronavirus 2019-nCoV with comparison to related coronaviruses strains determined that the three strains were almost identical. Comparisons included Wuhan/IVDC-HB-01/2019 (GISAID accession ID:EPI_ISL_402119) (HB01), Wuhan/IVDCHB-04/2019(EPI_ISL_402120) (HB04), and Wuhan/IVDC-HB-05/2019 (EPI_ISL_402121) (HB05). Analysis included 1008 human SARS CoV, 338 bat SARS-like CoV, and 3131 human MERS-CoV, whose genomes were published before 12 January 2020 [
37]. The notable difference are the 8a protein is present in SARS-CoV and absent in 2019-nCoV; the 8b protein is 84 amino acids in SARS-CoV, but longer in 2019-nCoV, with 121 amino acids; the 3b protein is 154 amino acids in SARS-CoV, but shorter in 2019-nCoV, with only 22 amino acids [
37]. It is still not clear how these differences affect the functionality and pathogenesis of 2019-nCoV. Similarly, recent genomic characterization and evolutionary origins of SARS-CoV-2 viruses isolated from a number of patients indicated sequence identity of about 99.9%, potentially implying a very recent host jump into humans [
39,
40,
41]. Based on phylogenetic and evolutionary dynamics, the novel coronavirus Covid 19 was found to belong to genus
Betacoronavirus, subgenus
Sarbecovirus (previously lineage 2b of Group 2 coronavirus) with 86% similarity to SARSr-CoV [
42]. According to International Committee on Taxonomy of Viruses (ICTV) criteria, only the strains found in
Rhinolophus bats in European countries, Southeast Asian countries and China are SARSr-CoV variants. These data indicate that SARSr-CoVs have wide geographical spread and might have been prevalent in bats for a very long time. In the absence of a direct ancestor of SARS-CoV in bat populations despite nearly two decades of research and since RNA coronaviruses recombination are frequent, there is a high potentiality that SARS-CoV2 have newly emerged from bat SARSr-CoVs by recombination. Recombination of two existing bat strains, WIV16 and Rf4092 giving rise to the civet SARS-CoV strain SZ3 have been reported [
21]. Furthermore, because α- and β-CoVs infect livestock animals such as porcine transmissible gastroenteritis virus, porcine enteric diarrhoea virus (PEDV) and the recently emerged swine acute diarrhea syndrome coronavirus (SADS-CoV), they pose significant risk to human. Selection pressures and high adaptability in humans might be driving further adaptive mutations in this strain. Population genetic analyses of 103 SARS-CoV-2 genomes nucleotides have shown SARS-CoV2 evolution into two major types. These include a more prevalent and aggressive L type that evolved during the outbreak from the ancestral less aggressive clone S [
43].
Thus, the main objectives of this work were to understand how SARS-CoV2 quickly adapts and evolves in humans, despite the genome stability. How the knowledge gap in pathogenicity, epidemicity, and evolutionary dynamics created by the unprecedented behavior of this virus, were different from earlier outbreaks. What were the successful mechanisms of and strategies underlying the success of the lineage in causing the pandemic. How successful bioinformatics programs in providing in silico evidences in the light of demands for higher biosafety level protocols. Thus, sequence based comparative genomics and functional analysis were successful in identifying abundance of signature-sequences as subtle expression strategies and provided evidences for sub-clonal differentiations at different regions despite genome stability. We have simultaneously examined sequence stability indicators including tandem-repeats, restriction site, and 3′UTR, 5′ UTR caps and stem-loop locations in addition to stringent alignment parameters for 100% identity, which all confirmed stability. Nevertheless, two extremely rare events were detected; one involved a segment inversion against betacoronavirus ancestor and the second was a significant polymorphism in the S gene. Furthermore, abundance of transversions, slippery sequences, and ON/OFF molecular structures, implied adaptive expressions had occurred in a clonal background. Future analysis of new isolates and experimental validation of these findings will provide more insights into the adaptive evolution of SARS-CoV2.
2. Materials and Methods
The dataset comprised of all currently available (
n = 6314) full genome sequences from the current (2019–2020) SARS-CoV2 pandemic, as well as closely related MERS (
n = 35), SARS-CoV (
n = 220 of which 32 were selected for alignments), and SARS CoV related bat strains (SARS-like CoV) (
n = 29). These were retrieved from NCBI (
http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/genbank/) and also confirmed in the GISAID (
https://www.gisaid.org/) databases. Before genomic analysis, all sequences were subjected to proofing and validations for integrity of sequences using available software and programs.
Pan-genome Sequence Analysis and phylogenetic analysis was performed using Clustal Omega 1.2.2 in Geneious Bioinformatics Package using Clustal size set at 180 in mBed Algorithm and Guide Tree and validated by MUSCLE 3.8.425, and Geneious Alignment in Geneious primer 2020.2.3 Java version 11.0.4+11 (64 bit). In alignment and guide phylogenetic tree view, strains showing 100% identity within the same clusters were dimmed in color and only one or a few representative strains were highlighted to show genomic structural details to allow maximum visibility and to accommodate all strains within different clusters tested. Analysis of the alignments was done in Geneious Bioinformatics Package (Geneious Primer 2020.2.3 Biomatters Ltd., Biomatters, New Zealand). Consensus sequence threshold was set at 100% and highlighted agreement to consensus. The following analysis were done within Geneious and in standalone programs:
In the whole genome multiple alignment view the following analysis were shown out: annotations, tracks of repeats, and slippery sequences were shown as color-coded. The following tracks of slippery sequences were identified by the search criteria: 100% match, zero mismatches, and both directions (double sided arrow indicate both directions), at approximate intervals and locations shown on all genomes on number alignment window: As an example, tracks of Strain NC_014470 will be described:
Geneious annotations and tracks: (details and locations of all of these are found in Additional Files attached) gene annotations and names (green color bars) were set to place on the sequence lines. 3′UTR, 5′ UTR, and Stem loops were shown alignments.
Slippery sequences (Geneious primer software) (brown arrows, one and two sided): slippery sequence 1: 7 nt (UUUAAC, TTTAAAC); slippery sequence 2: 7 nt (UUUAAAA, TTTAAAA); slippery sequence 3: 7 nt (UUUAAAU, TTTAAAT); slippery sequence 4: 5 nt (AAGAA); slippery sequence 5: 7 nt (UUUUUUA, TTTTTTA)
Tandem repeat tracks (using Phobos Tandem repeat finder 3.3.11 2006–2010) (each of the red color vertical arrows along genome sequence indicated a tandemly repeat unit track. The number of vertical arrows in a location indicated the number of times repeat unit copies are repeated). The program was set to “Perfect Repeats” with unit length (1–500 bp), “100% identity”, “No gaps” “high score”, “high index values”
Restriction sites: Geneious program was used to identify restriction enzyme sites on the genomes. Commonly available enzymes were selected in addition to a long list sites. Restriction site cleavage abilities were validated against methylation or variability, ambiguity, or deletion of targets.
Phylogenetic guide-trees were built by standalone Clustal Omega 1.2.2 and Geneious Tree Builder using Tamura-Nei genetic distance model and Neighbor-Joining method of tree building with and without outgroups (consensus method used for clustering was Majority Greedy clustering with (%) consensus support at nodes. Tree branches were cladogram based in a decreasing order with a line weight of 2 and tip labeled with standard strain names displaying country of isolation. The output trees were repeated several times using different programs to confirm the topology. Circular tree layout showing the “origins” of evolutionary lines consistent with the original bat strains from Europe, Bulgaria (NC_014470; GU190215; as well as a Chinese strain MT084071) from reference strains indicated by bold blue nodes were selected as the best output. All identical clusters within lineages were color coded. Similar nucleotide substitutions within clusters were also shown.
Whole genome alignment for pairwise alignment of specific genomes of interest was performed using progressive Mauve algorithm [
44] updated versions. Mauve Genome Alignment had a special viewer which enabled detection of genome rearrangements and locally aligned blocks at a glance. Each sequence was represented by one horizontal panel of blocks. Each colored block represented a region of sequence that aligned to part of another genome, and was presumably homologous and free from internal rearrangements. These were called locally collinear blocks (LCBs). The alignment display was organized into one horizontal “panel” per input genome sequence. Each genome’s panel contained the name of the genome sequence, a scale showing the sequence coordinates for that genome, and a single black horizontal center line. Colored block outlines appeared above and possibly below the center line. Each of these block outlines surrounded a region of the genome sequence that aligned to part of another genome, and was presumably homologous and internally free from genomic rearrangement. When a block lay above the center line the aligned region was in the forward orientation relative to the first genome sequence. Blocks below the center line indicated regions that aligned in the reverse complement (inverse) orientation. Regions outside blocks lacked detectable homology among the input genomes. Inside each block Mauve drew a similarity profile of the genome sequence. The height of the similarity profile corresponded to the average level of conservation in that region of the genome sequence. Areas that were completely white were not aligned and probably contained sequence elements specific to a particular genome. The height of the similarity profile was calculated to be inversely proportional to the average alignment column entropy over a region of the alignment.
The backbone color scheme in Mauve: colors regions conserved among all genomes differently than regions conserved among subsets of the genomes. Conserved regions among all genomes are termed “backbone,” which are drawn in mauve color. The colored blocks in each genome are connected by vertical lines.
The Tandem Repeat Finder program available at [
https://tandem.bu.edu/trf/trf.html] accessed on 25 September 2020, was used to confirm repeat unit sizes, copies, consensus sequences, scores, and flanking sequences.
Map to references is used for the 6312 genomes was used with the default setting except for the following criteria that were selected: Consensus set to “100% identity” Seq Coverage = 29.89, Genenious Mapper with “High Sensitivity”, five times “Fine Tuning” after alignment, saved “assembly report”, and “Do not trim” before mapping. Due to the alignment size produced, only visible regions were saved and presented in this study. Extensive analysis were carried out to understand the extent of transitions and transversions. A total of 95 seqs that showed increased transversions were extracted and used for independent analysis to identify common features, if any. The reference assembly algorithm used is a seed and expand style mapper followed by an optional fine tuning step to better align reads around indels to each other rather than the reference sequence. Various optimizations and heuristics are applied at each of analysis. The final optional fine tuning step at the end, shuffles the gaps around so that they reads better align to each other rather than the reference sequence.
Dot Plot analysis integrated in Geneious Package was used to detect and identify significantly long repeats. In addition, authenticity of the repeats was confirmed by this program. In addition, chromosomal rearrangements, inversions and translocations were identified.
Full genome sequences of the pandemic isolate SARS-CoV2 (7000 genomes by September–October 2020) as well as selected new isolates and closely related MERS (
n = 35), SARS-CoV (
n = 220 of which 32 were selected for alignments), and SARS CoV related bat isolates (SARS-like CoV) (
n = 29) were downloaded from public databases. These were retrieved from NCBI (
http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/genbank/, accessed on 26 May 2020) and GISAID (
https://www.gisaid.org/, accessed on 1 January 2020) databases. Before genomic analysis, all sequences were subjected to proofing and validations for sequence integrity using available software and programs.
Pan-genome sequence analysis and phylogenetics was performed using Clustal Omega 1.2.2 in Geneious Bioinformatics Package using Clustal size set at 180 in mBed Algorithm and Guide Tree and validated by MUSCLE 3.8.425, and Geneious Alignment in Geneious primer 2020.2.3 Java version 11.0.4+11 (64 bit). In alignment and guide phylogenetic tree view, isolates within a cluster were colord. Analysis of the alignments was done in Geneious Bioinformatics Package (Geneious Primer 2020.2.3 Biomatters Ltd.). Consensus sequence threshold was set at 100%. Two operations executed using two settings: in one setting “agreement to consensus” employed to highlight nucleotide agreements amongst all genomes; in another, the disagreements to consensus was used to highlight locations of genomic disagreements between the aligned genomes.
Phylogenetic guide-trees were built by standalone Clustal Omega 1.2.2 and Geneious Tree Builder using Tamura–Nei genetic distance model and Neighbor-Joining method of tree building with and without outgroups (consensus method used for clustering was Majority Greedy clustering with (%) consensus support at nodes. Tree branches were cladogram based in a decreasing order with a line weight of 1 and tip labeled with standard isolate names displaying country of isolation. The output trees were repeated several times using different programs to confirm the topology. Circular tree layout showing the “origins” of evolutionary lines consistent with the original bat isolates from Europe, Bulgaria (NC_014470; GU190215; as well as a Chinese isolate MT084071) from reference isolates indicated by green nodes was selected as the best output. All identical clusters within lineages were color coded. Similar nucleotide substitutions within clusters were also shown. SARS-CoV2 genomic identity and alignments were further confirmed by using Geneious Prime Dotplot; visual genome-wide sequence comparisons using Dotplot High Sensitivity settings; probabilistic score matrix and adjusting window size and threshold and a smaller tile size at 1000.
4. Discussion
It has been quite puzzling how SARS-CoV2 undergoes rapid paradigm shifts between clonal emergence and adaptational changes, despite the stable genome. To understand, we examined genomes sequences for genetic distance similarities, evolutionary phylogenomic, and
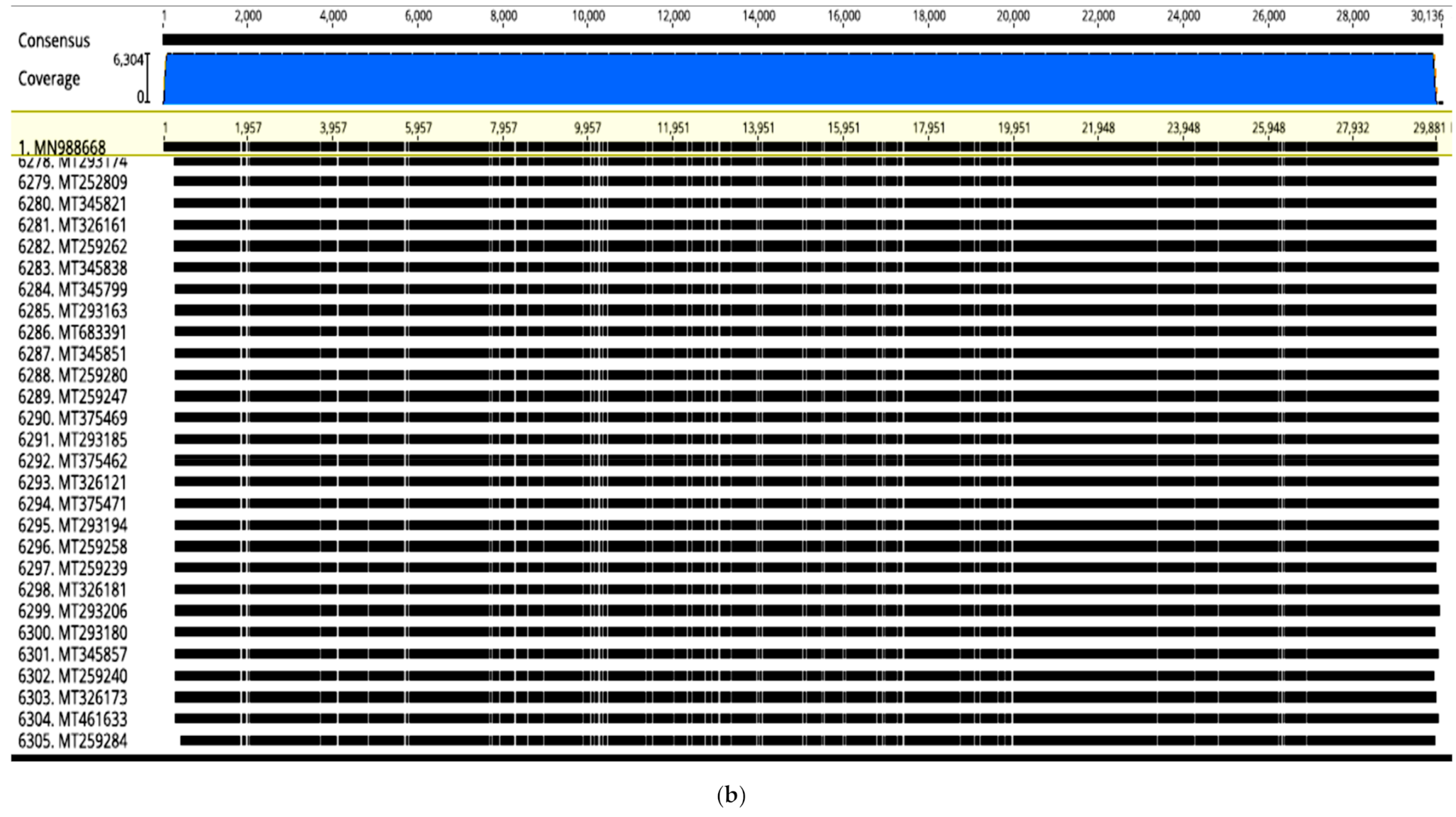
in-silico functional predictions. In this study, selected whole genome sequence alignment of 6305 SARS-CoV2 from different countries yielded a high degree of nucleotide identity (99.99%) (
Figure 1a). However, in spite of the high sequence homology, abundant transversions were detected throughout the genome sequences that slightly changed the topology (
Figure 1b). These results were in agreement with earlier finding [
39,
40,
41] implying that SARS-CoV2 lineage continues to have a clonal evolution and genome as it spreads throughout the world, despite identifications of recent point mutations (Mercatelli and Giorgi 2020;
https://0-www-bmj-com.brum.beds.ac.uk/content/bmj/371/bmj.m4857.full.pdf), accessed on 12 February 2021]. Coronavirus core genome integrity is not affected by point mutations due to the protective proof-reading activity of the non-structural protein nsp14 [
45,
46]. Differential expression of genes as a means for adaptation while maintaining genome stability was the mechanisms used during decades of pandemic outbreaks by the community acquired
Staphylococcus aureus [
47,
48].
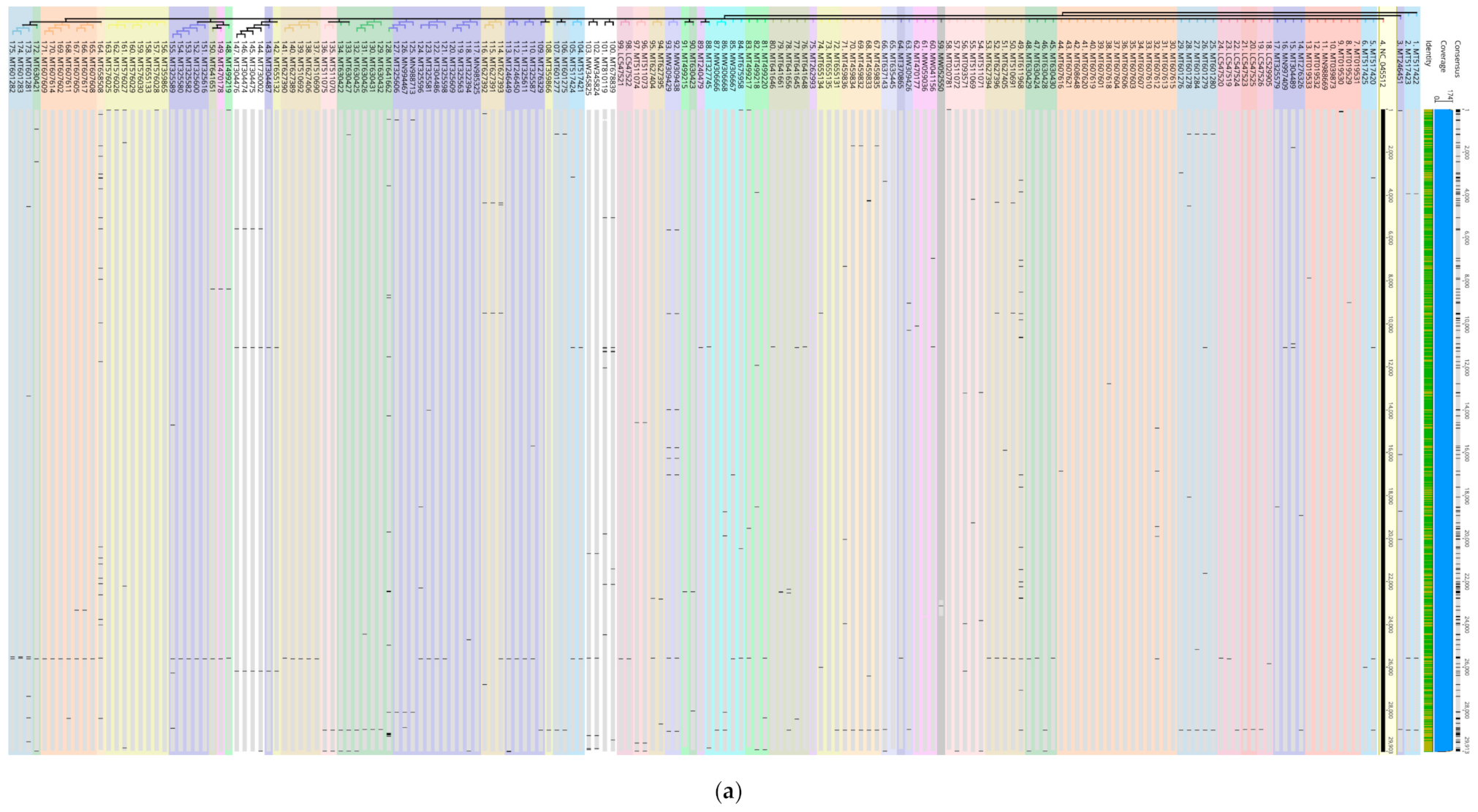
In this study, vertical analysis of the transversion loci and potential functional mutations was verified by extracting and analyzing the 92 genomes with most transversion loci amongst the 6305 genomes as well as in 174 genomes from different geographic regions against the reference NC_045512 (
Figure 2a). Interestingly, isolates within country-specific sub-clusters showed identical transversion patterns. In addition, all genomes maintained high level of sequence identity further supporting clonal evolution and genome. An example on abundance of point mutations is shown within a 342 bp region (13,093 > 13,434) in ORF1ab implying potential functional changes (
Figure 2b). Interestingly, these were of two different types of substitutions (transversion and transitions) affecting the same amino acids in all instances tested. In this representative example (
Figure 2b), four transitions (Ser, Cys, Thr, and Val) and three transversions (Thr, Ser, and Val) affected the same amino acids. Throughout the sequences, including S protein, M, S, K transversions and Y transitions were common. (Anahina for new paper on point mutations) Compensations have been experimentally confirmed to occur either for recovery [
49,
50] or to reduce deleterious effects [
51,
52,
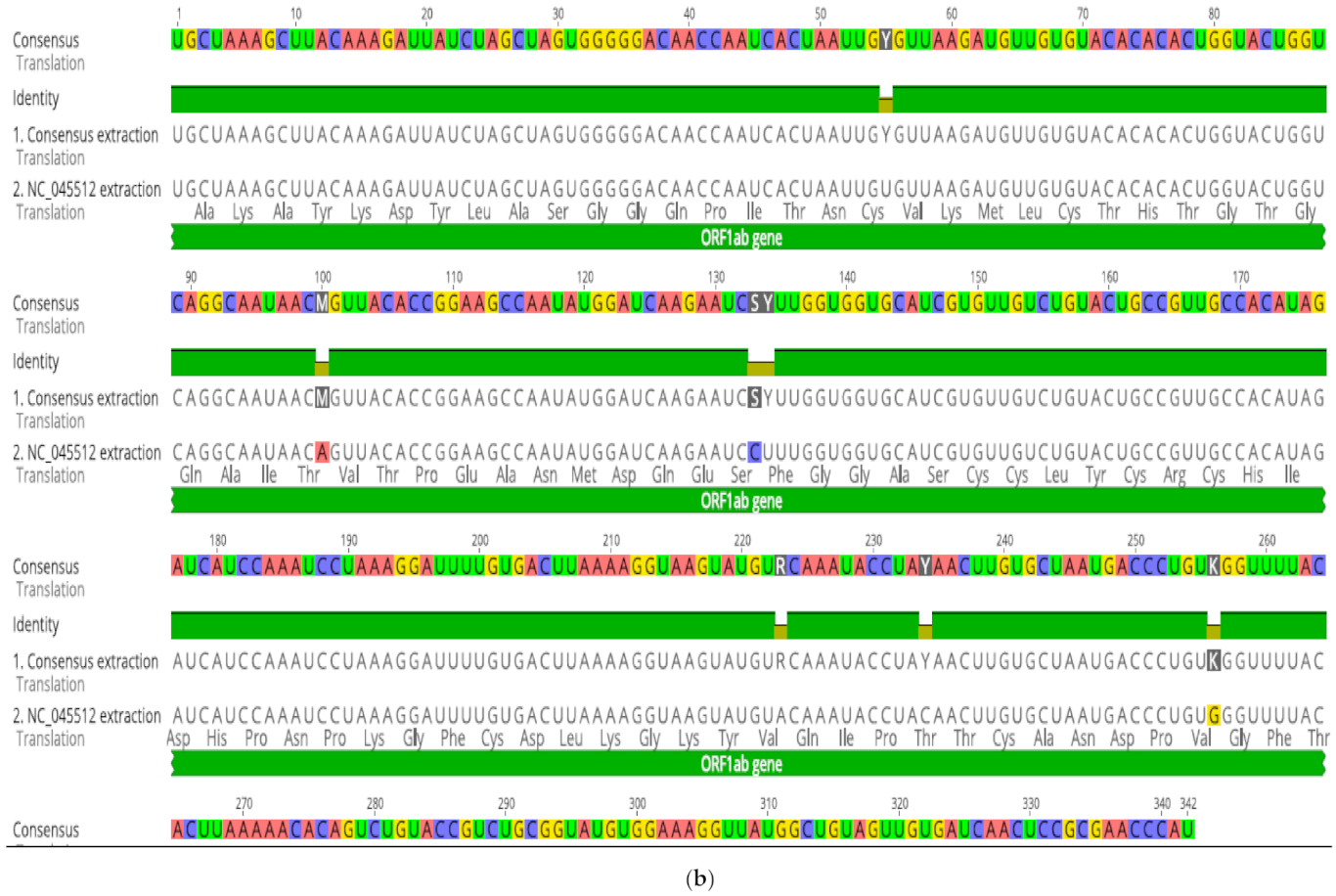
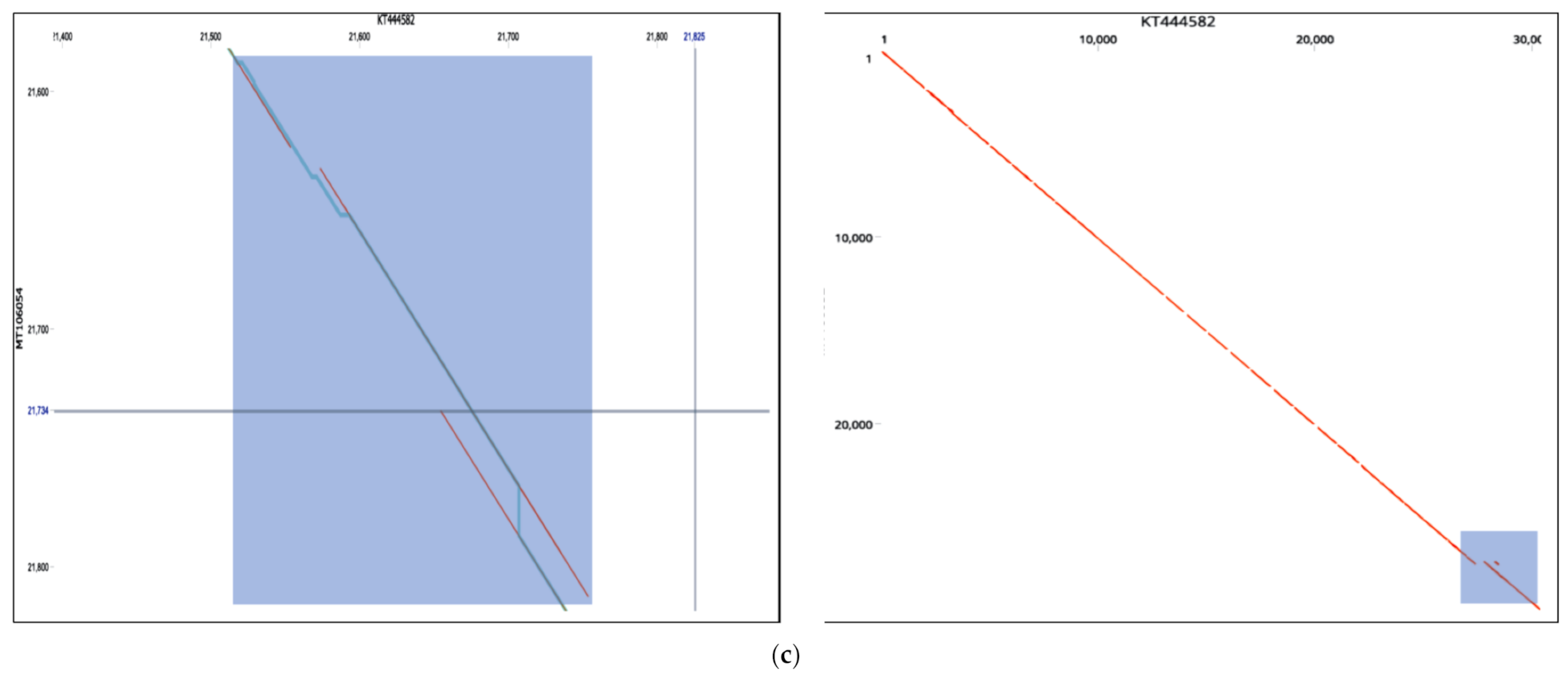
53]. Our findings remain to be verified experimentally as to whether they were compensatory or were induced by selective pressure. Deleterious or genomic rearrangements affecting sequence homology or clustering patterns were rare in this study, except for two novel events. Strains MT375469 isolate (Connecticut CT-UW) with a sum of deletions measuring up to 431 bp in the S gene (
Figure 2c,d), was the only isolate with a significant distance mapped to the reference isolates (MN988668). It was also the only isolate clustered in Australian group, in contrast to country-specific grouping. The S protein was thought to be highly conserved among SARS-CoV2 isolates and thereby of a therapeutic importance and a vaccine candidate. The S protein length polymorphism is often helpful in dictating ancestral origins. For instance, clade 2 bat SARSr-CoV S protein which is universally present in many locations has a shorter size S protein due to deletions of 5, 12 or 13 amino acid residues [
21]. While the overwhelming majority of genomes we analyzed had conserved regions, these findings might be of considerations in therapeutics applications [
54]. Another novel inversion was detected in the SARS-CoV2 isolates MT106052 (USA-CA7) and MT044257 (USA-IL2) against the reference NC_025517 betacoronavirus isolate. ProgressiveMauve, other programs, and manual analysis revealed an inversion at position (~4000 bp) in ORF1ab of the two isolates (
Figure 3a–c). Examination of Locally Collinear Blocks (LCBs) identified a 221 bp segment inversion despite high genome conservation indicated by native color codes (
Figure 3a). Dot-plot and nucleotide analysis of the region revealed a unique hot-spot with abundance of point mutations prone to genomic rearrangement (
Figure 3c). Finally, it has been found that coronaviruses maintain greater RNA structuredness across their genomes compared to MERS-CoV and high nucleotide variability had no impact on RNA secondary structure stability which suggested selective pressures against its disruption [
55].
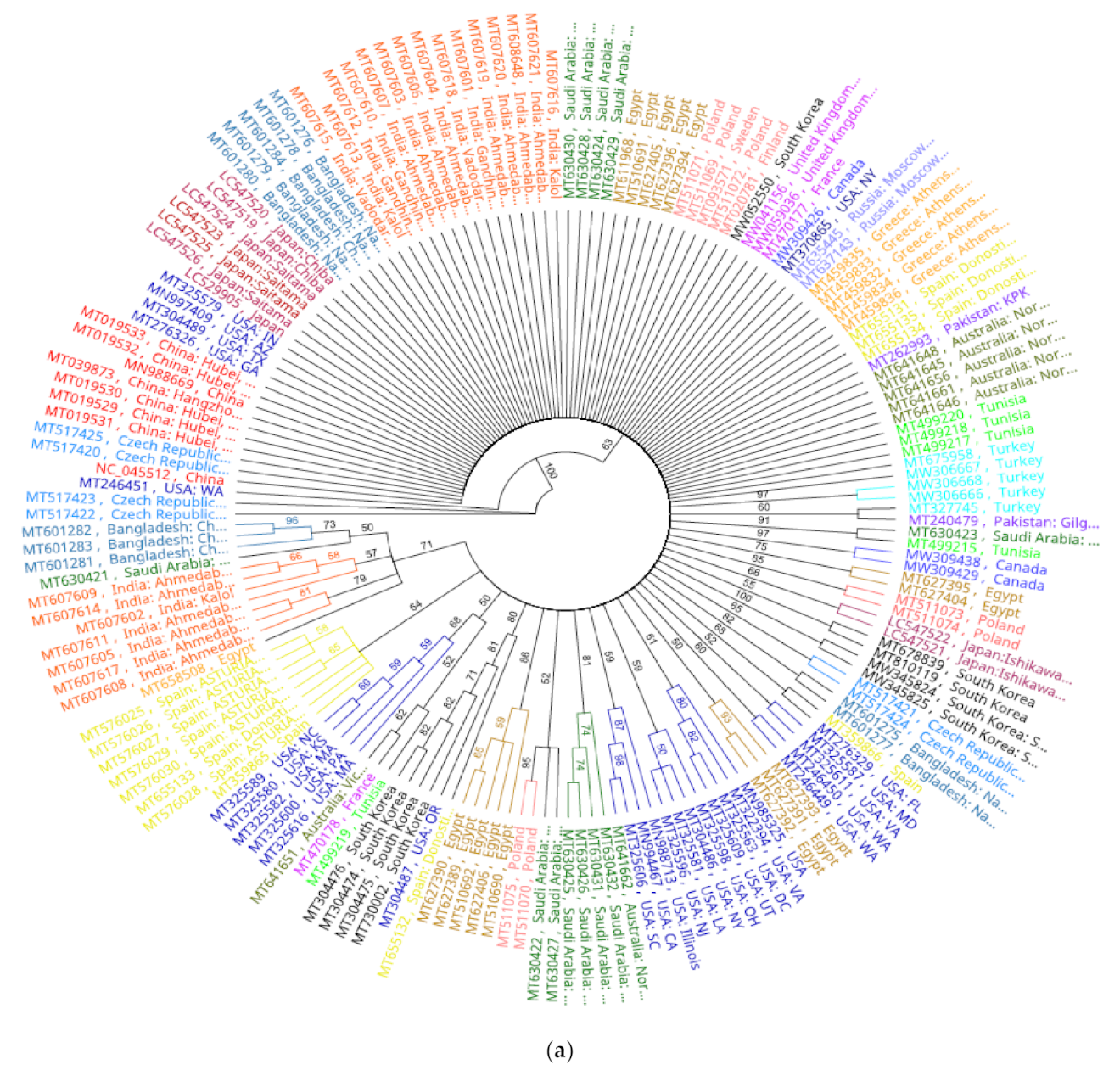
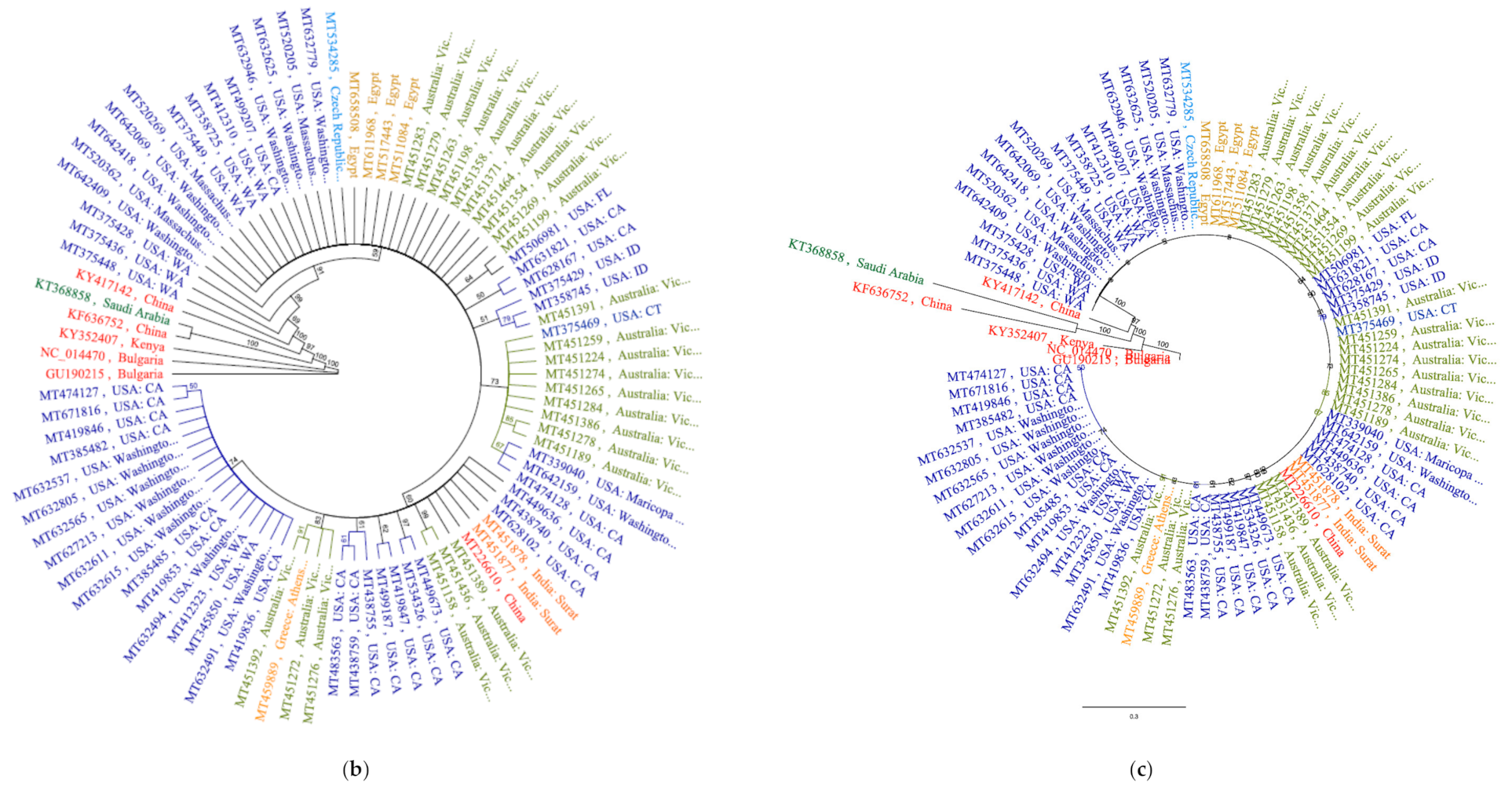
Analysis of phylogenomic trees with cladogram and genetic distance similarity revealed identical tree topologies. All SARS-CoV2 isolates were separated as a single clone in a large cluster supported by 100% bootstrap value at the branch node. Within this mega cluster, SARS-CoV2 isolates from countries were of two categories; independent lineages and country-specific sub-clusters, potentially implying subclonal evolution (
Figure 4a). Construction of a more concise tree with lesser number of genomes representing different geographic regions and different substitution patterns also yielded the same identical country-specific topology, except for isolate MT375469 that clustered with Australian cluster (
Figure 4b,c). The recent finding of re-infection a patient by two SARS-CoV2 strains with significant genetic differences and varying disease severities, supported the notion of sub-clonal evolutions under clonal a background [
56]. The latter study confirmed re-infection and ruled out in vivo evolution in the patient, owing to the greater genetic discordances between the two strains. This indicates that the variant, even though not evolved in the same patient, originated remotely supporting the country- or region-specific sub-clonal evolutions obtained in this study. It is possible that regional sub-clusters could have been due to multiple introductions. However, for this to be true, isolates from unique super-spreader region(s) or epicenters ought to be identified that shared in all local clusters. For instance, clues to multiple introductions events, before strict lockdowns were enforced, were verified by sharing epicenter isolates with Europeans [
57]. However, in this study all groups mostly contained local isolates only. In addition, the possibility that all sub-clones simultaneous originated in the epicenter in Wuhan, China, is remote.
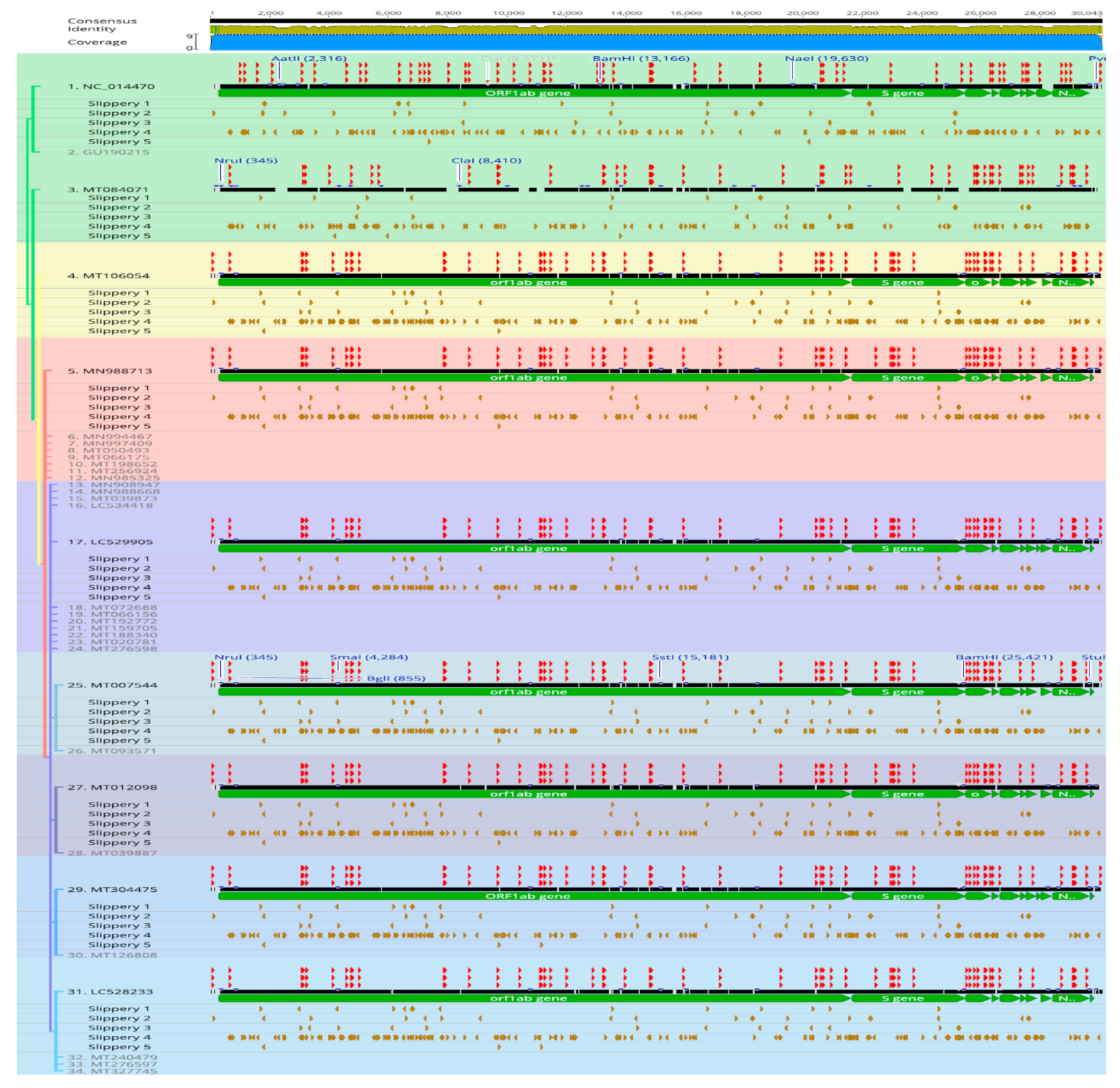
In this study, we report on in-silico evidences for adaptive overexpression strategies under a clonal SARS-CoV2 lineage background. We tested regulatory events that are prone to induce over-expression of viral proteins in SARS-CoV2 lineage. These included indels, recombination and translocations, restriction sites, AU-rich transcription regulating sequences, tandem repeats, signature sequences, and stem-loops (
Figure 5). Except for the reference and two events described below, almost all genomes showed identical patterns in these criteria in addition to sub-cluster-specific patterns of each criteria shared within phylogenetic groups consistent with clonal evolution. This supports the notion that the remarkable coronavirus core-genome conservation is independent of replication and adaptive expressions in infectious variants. It has been experimentally verified that translocations of S, M, E, and N, viral proteins in murine coronaviruses mutants had no effect in genome stability and that mutants variants remained viable in cell cultures [
58]. The inversion over the 5′ terminus region in ORF1ab gene, identified in this study, would indicated adaptive strategy rather than lethal mutation. We did not find large recombination events similar to that occurred in the two existing bat strains, WIV16 and Rf4092, giving rise to the civet SARS-CoV strain SZ3 [
21]. In the aforementioned inversion detected in this study, sites for HindIII (AꜜAGCUU) and Alul (AGCU) and Hinf1 (GAUUC) lied only a few stretches apart. However, a strong effect of
recA independent homologous recombination event was reported to occur even at distantly separated loci [
59]. Interesting, in contrast to stabilizing GC-rich regions, abundance of AT-rich tandem repeats and intervening slippery sequences were seen at the inversion locus (
Figure 5). These included multiple copies of repeating units (ATC, AAG, AATGTC, AAAAGT, AAG, AAACAG, AAAG, ATCC). The full list of repeats is found in the Additional
Supplementary Material Files (attached). Recent studies indicated that
recA-independent recombination can be as efficient as
recA-dependent in the absence of exonucleases activity [
60]. Future analysis of more newly sequenced isolates and experimental validations would reveal more insights into the evolutionary adaptive mechanisms of SARS-CoV2. Regular updating and sequence analysis of SARS-CoV2 has become imperative as frequent evolution of mutants with particular signatures some of which is associated to patient mortality, is on the rise [
61,
62]. As the time of writing the manuscript, evolution of the following variants is being monitored; B.1.1.7 lineage originated in the UK, B.1.351 lineage in South Africa, and P.1 lineage in Brazil are being monitored, in addition to some new variants being reported in the USA at the time of writing the manuscript, (available at:
https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-research/scientific-brief-emerging-variants.html accessed on 2 February 2021).
Thus, in this study we present evidences for subtle adaptive strategies that occurred during the rapid evolution of SARS-CoV2 pandemic. Comparative genomics of whole genome alignments, tandem repeat screening, restriction site mapping, and pairwise genomic comparisons indicated recent evolution and clonality of SARS-CoV2 lineage. However, despite the high degree of core genome conservation and strong evidence for clonal emergence, limited diversifications were believed to occur during outbreak resulting in sub-clonal populations at different regions. Validation of an extremely rare event of adaptive rearrangement in SARS-CoV2 against remote betacoronavirus ancestor would provide details of evolutionary mechanisms. In addition, detection of an extremely rare but a substantial difference in the S protein due to potential deletions would be of therapeutic importance. More importantly, abundance of transversions, slippery sequences, and ON/OFF molecular structures implied subtle adaptive expressions had occurred in a clonal background. Future analysis of additional newly sequenced isolates and experimental validation of selected in-silico findings will provide more insights into the adaptive evolution of SARS-CoV2.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











