
Biological Activity Characterization of the Diagnostically Relevant Human Papillomavirus 16 E1C RNA

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of E1C Monoclonal Antibody (mAb)
2.2. Cell Culture
2.3. Transient Transfections
2.4. Luciferase Reporter Assay
2.5. Lentiviral Transduction
2.6. Western Blot
2.7. HPV16 E1C Immunofluorescence
2.8. Mass Spectrometry
2.9. RNA Extraction and RT-qPCR
2.10. Statistical Analysis
3. Results
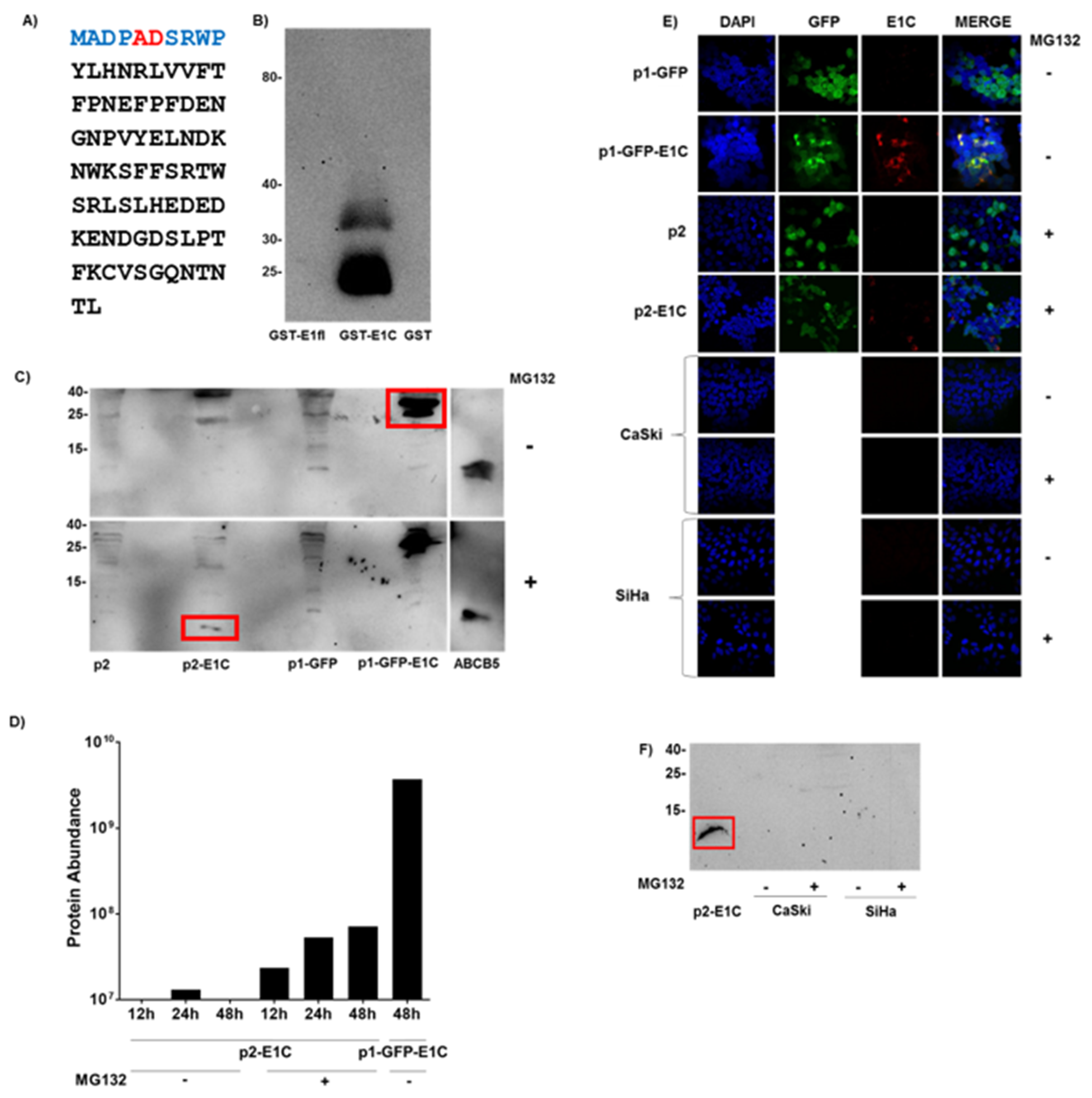
3.1. Detection of E1C Protein
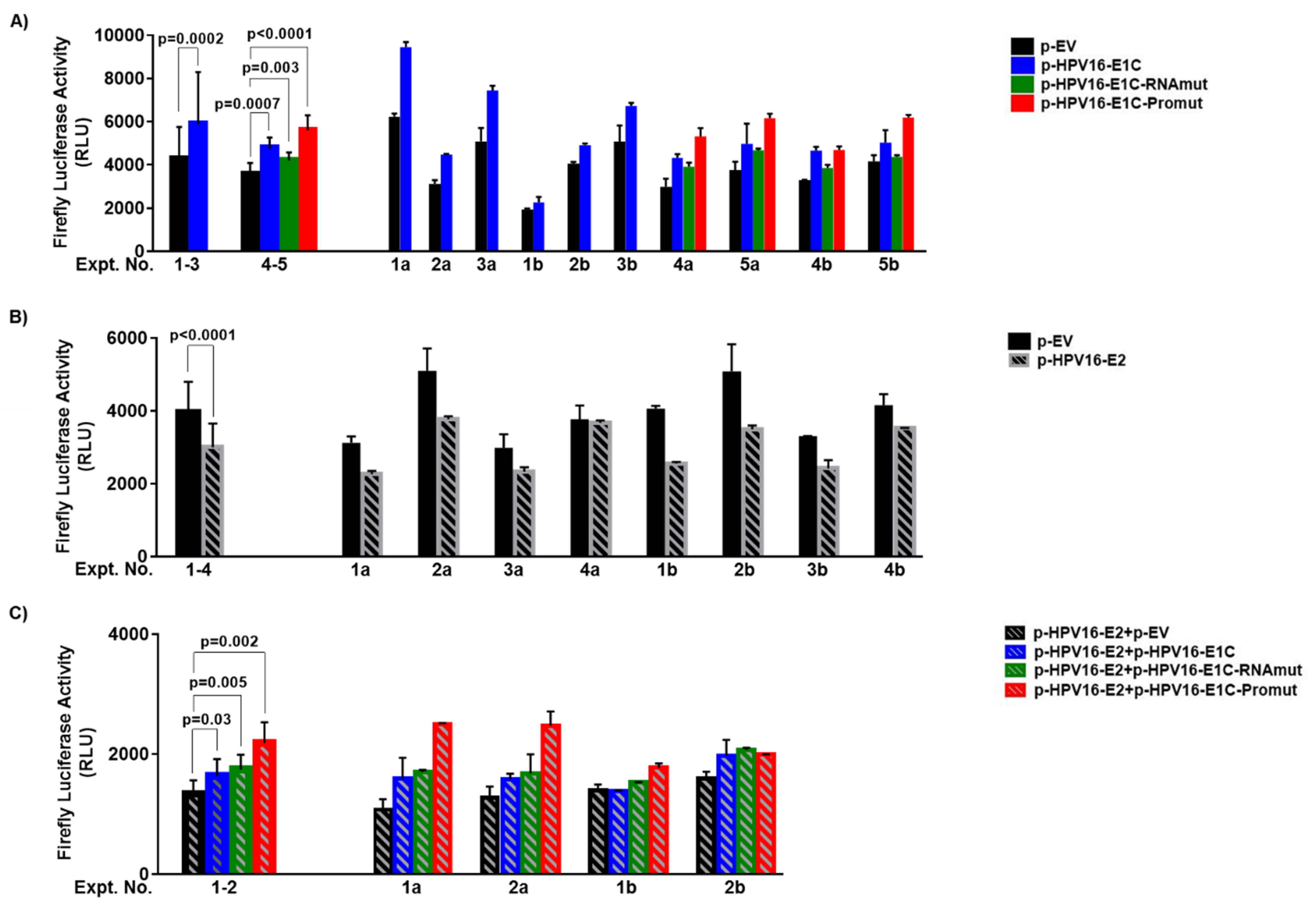
3.2. URR Activation Is Not Mediated by E1C Protein
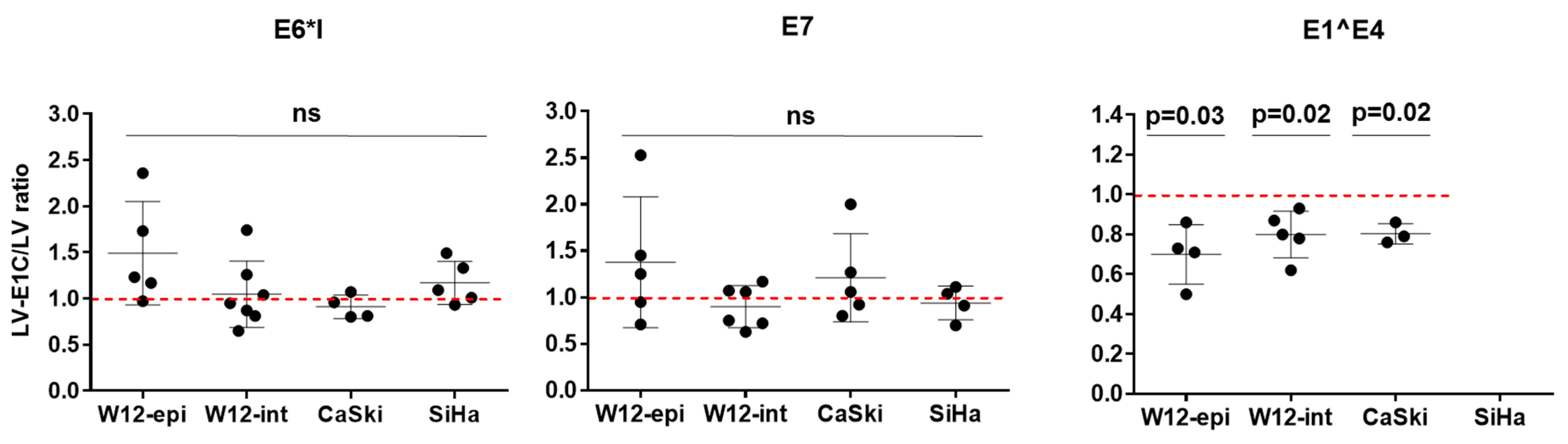
3.3. Overexpression of E1C in HPV16-Positive Cell Lines Decreases E1^E4 RNA Expression
3.4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Guan, P.; Howell-Jones, R.; Li, N.; Bruni, L.; de Sanjosé, S.; Franceschi, S.; Clifford, G.M. Human papillomavirus types in 115,789 HPV-positive women: A meta-analysis from cervical infection to cancer. Int. J. Cancer 2012, 131, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef] [Green Version]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; De Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef]
- Schmitt, M.; Pawlita, M. The HPV transcriptome in HPV16 positive cell lines. Mol. Cell Probes 2011, 25, 108–113. [Google Scholar] [CrossRef]
- Burley, M.; Roberts, S.; Parish, J.L. Epigenetic regulation of human papillomavirus transcription in the productive virus life cycle. Semin. Immunopathol. 2020, 42, 159–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Dalstein, V.; Waterboer, T.; Clavel, C.; Gissmann, L.; Pawlita, M. Diagnosing cervical cancer and high-grade precursors by HPV16 transcription patterns. Cancer Res. 2010, 70, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höfler, D.; Böhmer, G.; Von Wasielewski, R.; Neumann, H.; Halec, G.; Holzinger, D.; Dondog, B.; Gissmann, L.; Pawlita, M.; Schmitt, M. HPV16 RNA patterns defined by novel high-throughput RT-qPCR as triage marker in HPV-based cervical cancer precursor screening. Gynecol. Oncol. 2015, 138, 676–682. [Google Scholar] [CrossRef]
- Alloul, N.; Sherman, L. Transcription-modulatory activities of differentially spliced cDNAs encoding the E2 protein of human papillomavirus type 16. J. Gen. Virol. 1999, 80 Pt 9, 2461–2470. [Google Scholar] [CrossRef]
- Alloul, N.; Sherman, L. The E2 protein of human papillomavirus type 16 is translated from a variety of differentially spliced polycistronic mRNAs. J. Gen. Virol. 1999, 80 Pt 1, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Sehr, P.; Zumbach, K.; Pawlita, M. A generic capture ELISA for recombinant proteins fused to glutathione S-transferase: Validation for HPV serology. J. Immunol. Methods 2001, 253, 153–162. [Google Scholar] [CrossRef]
- Combes, J.D.; Pawlita, M.; Waterboer, T.; Hammouda, D.; Rajkumar, T.; Vanhems, P.; Snijders, P.; Herrero, R.; Franceschi, S.; Clifford, G. Antibodies against high-risk human papillomavirus proteins as markers for invasive cervical cancer. Int. J. Cancer 2014, 135, 2453–2461. [Google Scholar] [CrossRef] [PubMed]
- EH, D.L. Antibodies: A Laboratory Manual, 1st ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1988. [Google Scholar]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Norby, K.; Hayes, M.; Chiu, Y.F.; Sugden, B.; Lambert, P.F. Using Organotypic Epithelial Tissue Culture to Study the Human Papillomavirus Life Cycle. Curr. Protoc. Microbiol. 2016, 41, 14B-8. [Google Scholar] [CrossRef] [PubMed]
- Pattillo, R.A.; Hussa, R.O.; Story, M.T.; Ruckert, A.C.; Shalaby, M.R.; Mattingly, R.F. Tumor antigen and human chorionic gonadotropin in CaSki cells: A new epidermoid cervical cancer cell line. Science 1977, 196, 1456–1458. [Google Scholar] [CrossRef]
- Friedl, F.; Kimura, I.; Osato, T.; Ito, Y. Studies on a new human cell line (SiHa) derived from carcinoma of uterus. I. Its establishment and morphology. Proc. Soc. Exp. Biol. Med. 1970, 135, 543–545. [Google Scholar] [CrossRef]
- Pear, W.S.; Nolan, G.P.; Scott, M.L.; Baltimore, D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA 1993, 90, 8392–8396. [Google Scholar] [CrossRef] [Green Version]
- Castro, F.; Dirks, W.G.; Fahnrich, S.; Hotz-Wagenblatt, A.; Pawlita, M.; Schmitt, M. High-throughput SNP-based authentication of human cell lines. Int. J. Cancer 2013, 132, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.; Pawlita, M. High-throughput detection and multiplex identification of cell contaminations. Nucleic Acids Res. 2009, 37, e119. [Google Scholar] [CrossRef] [PubMed]
- Sehr, P.; Rubio, I.; Seitz, H.; Putzker, K.; Ribeiro-Müller, L.; Pawlita, M.; Müller, M. High-throughput pseudovirion-based neutralization assay for analysis of natural and vaccine-induced antibodies against human papillomaviruses. PLoS ONE 2013, 8, e75677. [Google Scholar] [CrossRef]
- Zentgraf, H.; Frey, M.; Schwinn, S.; Tessmer, C.; Willemann, B.; Samstag, Y.; Velhagen, I. Detection of histidine-tagged fusion proteins by using a high-specific mouse monoclonal anti-histidine tag antibody. Nucleic Acids Res. 1995, 23, 3347–3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De-Castro Arce, J.; Gockel-Krzikalla, E.; Rosl, F. Silencing of multi-copy HPV16 by viral self-methylation and chromatin occlusion: A model for epigenetic virus-host interaction. Hum. Mol. Genet. 2012, 21, 1693–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Boehm, J.S.; Yang, X.; Salehi-Ashtiani, K.; Hao, T.; Shen, Y.; Lubonja, R.; Thomas, S.R.; Alkan, O.; Bhimdi, T.; et al. A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 2011, 8, 659–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, F.; Bieg, M.; Will, R.; Körner, C.; Weichenhan, D.; Bott, A.; Ishaque, N.; Lutsik, P.; Moskalev, E.A.; Mueller, S.K.; et al. Enhancer hijacking activates oncogenic transcription factor NR4A3 in acinic cell carcinomas of the salivary glands. Nat. Commun. 2019, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Pendse, S.S.; Lapchak, P.H.; Margaryan, A.; Shlain, D.; Doeing, C.; Sayegh, M.H.; Frank, M.H. Regulation of progenitor cell fusion by ABCB5 P-glycoprotein, a novel human ATP-binding cassette transporter. J. Biol. Chem. 2003, 278, 47156–47165. [Google Scholar] [CrossRef] [Green Version]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Chotewutmontri, S.; Wolf, S.; Klos, U.; Schmitz, M.; Dürst, M.; Schwarz, E. Multiplex Identification of Human Papillomavirus 16 DNA Integration Sites in Cervical Carcinomas. PLoS ONE 2013, 8, e66693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsell, D.A.; Sauer, R.T. The structural stability of a protein is an important determinant of its proteolytic susceptibility in Escherichia coli. J. Biol. Chem. 1989, 264, 7590–7595. [Google Scholar] [CrossRef]
- Chang, S.W.; Liu, W.C.; Liao, K.Y.; Tsao, Y.P.; Hsu, P.H.; Chen, S.L. Phosphorylation of HPV-16 E2 at serine 243 enables binding to Brd4 and mitotic chromosomes. PLoS ONE 2014, 9, e110882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvey, L.A.; Dunn, L.A.; Tindle, R.W.; Park, D.S.; Frazer, I.H. Human papillomavirus (HPV) type 18 E7 protein is a short-lived steroid-inducible phosphoprotein in HPV-transformed cell lines. J. Gen. Virol. 1994, 75, 1647–1653. [Google Scholar] [CrossRef]
- Schrader, E.K.; Harstad, K.G.; Matouschek, A. Targeting proteins for degradation. Nat. Chem. Biol. 2009, 5, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Sherman, L.; Jackman, A.; Itzhaki, H.; Stoppler, M.C.; Koval, D.; Schlegel, R. Inhibition of serum- and calcium-induced differentiation of human keratinocytes by HPV16 E6 oncoprotein: Role of p53 inactivation. Virology 1997, 237, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.W.; Choi, S.W.; You, B.H. Incredible RNA: Dual Functions of Coding and Noncoding. Mol. Cells 2016, 39, 367–374. [Google Scholar]
- Rossetto, C.C.; Tarrant-Elorza, M.; Verma, S.; Purushothaman, P.; Pari, G.S. Regulation of viral and cellular gene expression by Kaposi’s sarcoma-associated herpesvirus polyadenylated nuclear RNA. J. Virol. 2013, 87, 5540–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, P.; Sampath, K. cncRNAs: Bi-functional RNAs with protein coding and non-coding functions. Semin. Cell Dev. Biol. 2015, 47–48, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Tao, M.; McCoy, J.P.; Jr Zheng, Z.M. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J. Virol. 2006, 80, 4249–4263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippova, M.; Evans, W.; Aragon, R.; Filippov, V.; Williams, V.M.; Hong, L.; Reeves, M.E.; Duerksen-Hughes, P. The small splice variant of HPV16 E6, E6, reduces tumor formation in cervical carcinoma xenografts. Virology 2014, 450–451, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davy, C.E.; Jackson, D.J.; Wang, Q.; Raj, K.; Masterson, P.J.; Fenner, N.F. Identification of a G(2) arrest domain in the E1 wedge E4 protein of human papillomavirus type 16. J. Virol. 2002, 76, 9806–9818. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varghese, C.S.; Will, R.; Tessmer, C.; Hofmann, I.; Hessling, B.; Pawlita, M.; Höfler, D. Biological Activity Characterization of the Diagnostically Relevant Human Papillomavirus 16 E1C RNA. Microbiol. Res. 2021, 12, 539-552. https://0-doi-org.brum.beds.ac.uk/10.3390/microbiolres12030038
Varghese CS, Will R, Tessmer C, Hofmann I, Hessling B, Pawlita M, Höfler D. Biological Activity Characterization of the Diagnostically Relevant Human Papillomavirus 16 E1C RNA. Microbiology Research. 2021; 12(3):539-552. https://0-doi-org.brum.beds.ac.uk/10.3390/microbiolres12030038
Chicago/Turabian StyleVarghese, Christy Susan, Rainer Will, Claudia Tessmer, Ilse Hofmann, Bernd Hessling, Michael Pawlita, and Daniela Höfler. 2021. "Biological Activity Characterization of the Diagnostically Relevant Human Papillomavirus 16 E1C RNA" Microbiology Research 12, no. 3: 539-552. https://0-doi-org.brum.beds.ac.uk/10.3390/microbiolres12030038