The Effects of Resveratrol on Prostate Cancer through Targeting the Tumor Microenvironment


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
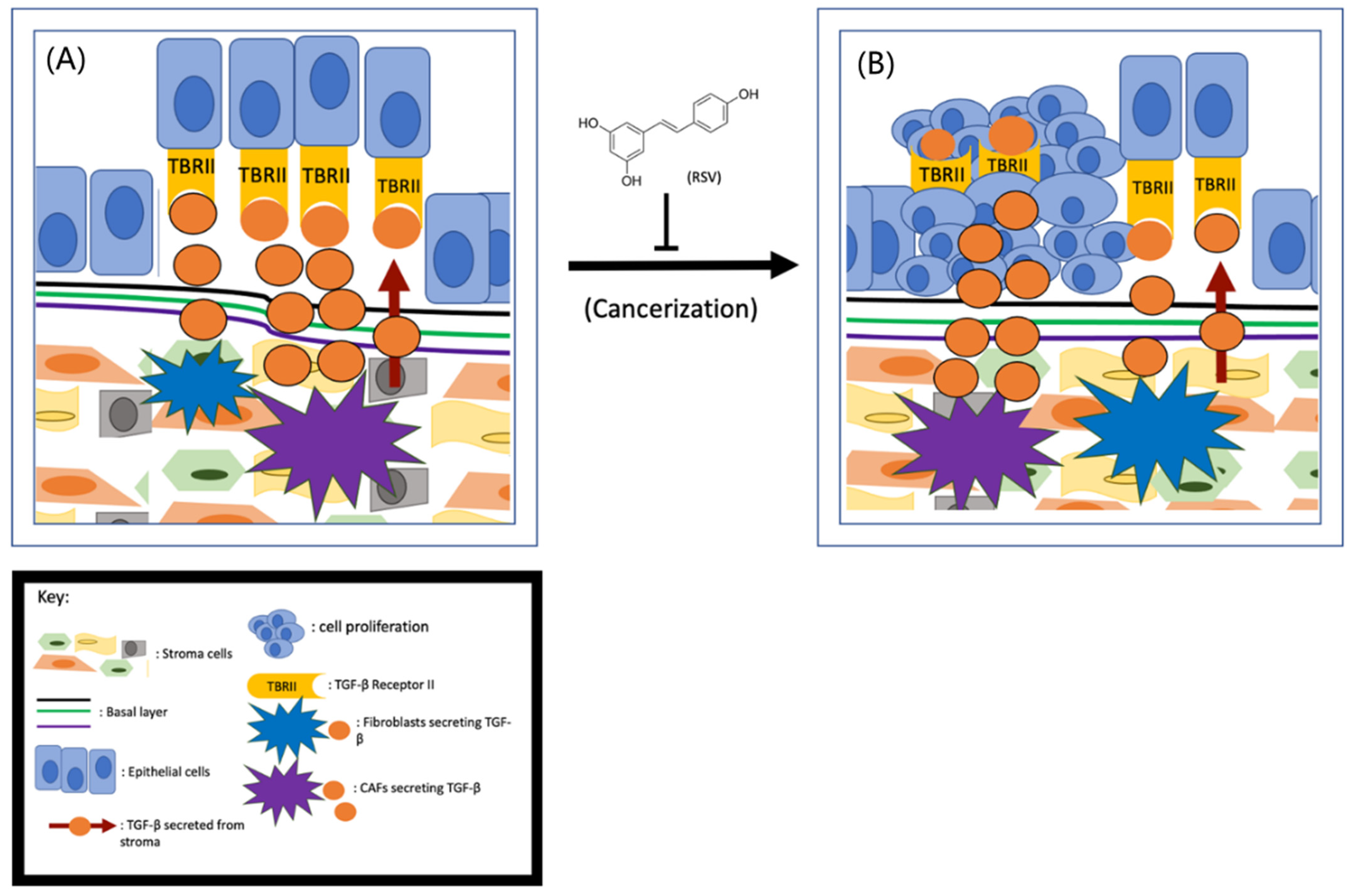
2. RSV Inhibits Prostate Cancer Initiation by Targeting the TME
3. RSV Inhibits Prostate Cancer Cell Proliferation by Targeting the TME
3.1. RSV Inhibits Prostate Cancer Proliferation by Interrupting Stroma–Tumor Cell Communication
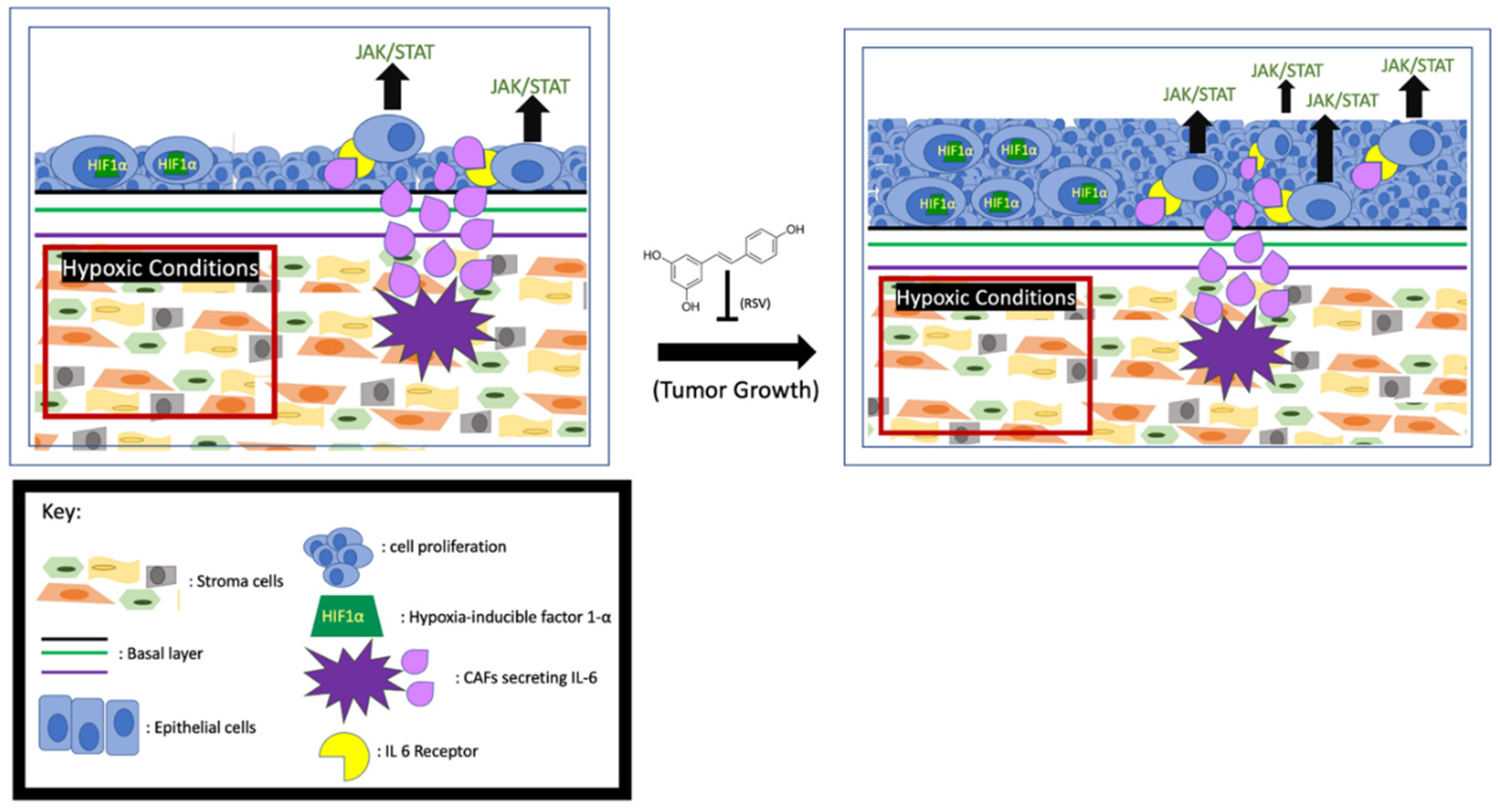
3.2. RSV Inhibits Cancer Cell Proliferation by Affecting Hypoxic Conditions of the TME
4. RSV Inhibits Prostate Cancer Metastasis by Targeting the TME
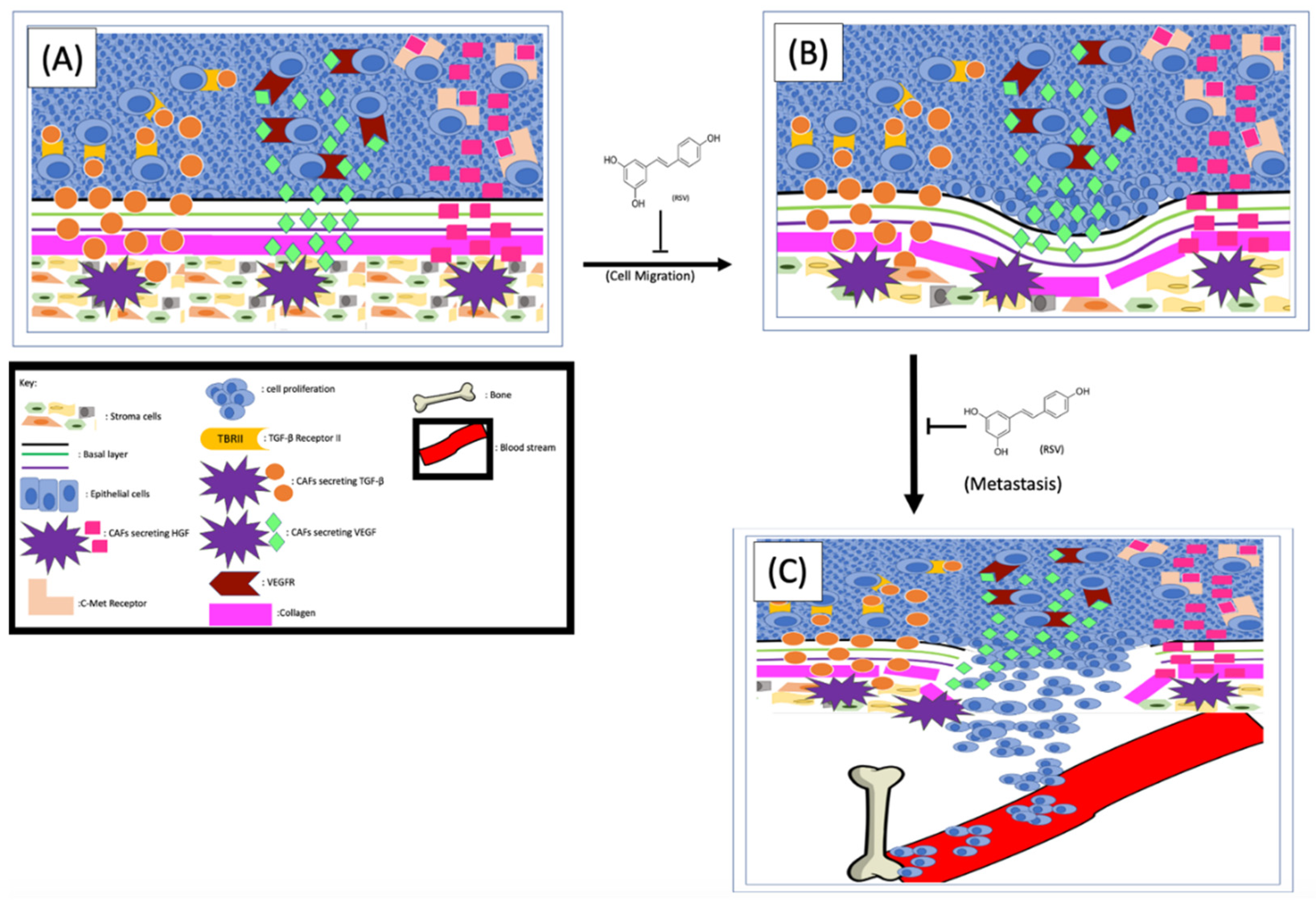
4.1. Resveratrol Interrupts the Communication between Stromal and Cancer Cells
4.2. Resveratrol Inhibits Prostate Cancer Metastasis by Targeting the Extracellular Matrix
4.3. Inhibitory Effects of RSV on Prostate Cancer Cell Bone Metastasis
4.4. RSV Inhibits Immune Cell-Mediated Prostate Cancer Metastasis
5. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Ferlay, J.; Soejomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, P.A. Histopathology of prostate cancer. Cold Spring Harbor Perspect. Med. 2017, 7, a030411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu-Yao, G.L.; Albertsen, P.C.; Moore, D.F.; Shih, W.; Lin, Y.; DiPaola, R.S.; Barry, M.J.; Zietman, A.; O’Leary, M.; Walker-Corkery, E.; et al. Outcomes of localized prostate cancer following conservative management. JAMA J. Am. Med Assoc. 2009, 302, 1202–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Docherty, F.E.; Brown, H.K.; Reeves, K.J.; Fowles, A.C.M.; Ottewell, P.D.; Dear, T.N.; Holen, I.; Croucher, P.I.; Eaton, C.L. Prostate cancer cells preferentially home to osteoblast rich areas in the early stages of bone metastasis: Evidence from in vivo models. J. Bone Miner. Res. 2014, 29, 2688–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef]
- Pandini, G.; Mineo, R.; Frasca, F.; Roberts, C.T., Jr.; Marcelli, M.; Vigneri, R.; Belfiore, A. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res. 2005, 65, 1849–1857. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Ke, M.; Wu, Q.; Tian, Q.; Liu, L.; Dai, Z.; Lu, S.; Liu, P. AZGP1 is androgen responsive and involved in AR induced prostate cancer cell proliferation and metastasis. J. Cell Physiol. 2019, 234, 17444–17458. [Google Scholar] [CrossRef]
- Wen, S.; Niu, Y.; Lee, S.O.; Chang, C. Androgen receptor (AR) positive vs negative roles in prostate cancer cell deaths including apoptosis, anoikis, entosis, necrosis and autophagic cell death. Cancer Treat Rev. 2014, 40, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Thalmann, G.N.; Rhee, H.; Sikes, R.A.; Pathak, S.; Multani, A.; Zhau, H.E.; Marshall, F.F.; Chunge, L.W.K. Human prostate fibroblasts induce growth and confer castration resistance and metastatic potential in LNCaP cells. Eur. Urol. 2009, 58, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karkampouna, S. The role of cancer-associated fibroblasts in prostate cancer tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef]
- Cioni, B.; Zwart, W.; Bergman, A.M. Androgen receptor moonlighting in the prostate cancer microenvironment. Endocr. Relat. Cancer 2018, 25, R331–R349. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar]
- Lai, K.; Yamashita, S.; Huang, C.; Yeh, S.; Chang, C. Loss of stromal androgen receptor leads to suppressed prostate tumourigenesis via modulation of pro-inflammatory cytokines/chemokines. EMBO Mol. Med. 2012, 4, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Wikström, P.; Marusic, J.; Stattin, P.; Bergh, A. Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate 2009, 69, 799–809. [Google Scholar] [CrossRef]
- Herden, J.; Weissbach, L. Utilization of active surveillance and watchful waiting for localized prostate cancer in the daily practice. World J. Urol. 2018, 36, 383–391. [Google Scholar] [CrossRef]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2018, 22, 24–38. [Google Scholar] [CrossRef] [Green Version]
- De Amicis, F.; Chimento, A.; Montalto, F.I.; Casaburi, I.; Sirianni, R.; Pezzi, V. Steroid receptor signallings as targets for resveratrol actions in breast and prostate cancer. Int. J. Mol. Sci. 2019, 20, 1087. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Auchus, R.J. The backdoor pathway to dihydrotestosterone. Trends Endocrinol. Metab. 2004, 15, 432–438. [Google Scholar] [CrossRef]
- Guerrero, J.; Alfaro, I.E.; Gómez, F.; Protter, A.A.; Bernales, S. Enzalutamide, an androgen receptor signaling inhibitor, induces tumor regression in a mouse model of castration-resistant prostate cancer. Prostate 2013, 73, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.W.; Fong, H.H.S.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Kumar, A.; Butt, N.A.; Zhang, L.; Williams, R.; Rimando, A.M.; Biswas, P.K.; Levenson, A.S. Molecular insight into the differential anti-androgenic activity of resveratrol and its natural analogs: In silico approach to understand biological actions. Mol. Biosyst. 2016, 12, 1702–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.H.; Kundu, J.K.; Keum, Y.S.; Cho, Y.Y.; Surh, Y.J.; Choi, B.Y. Resveratrol inhibits IL-6-induced transcriptional activity of AR and STAT3 in human prostate cancer LNCaP-FGC cells. Biomol. Ther. 2014, 22, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Vancauwenberghe, E.; Noyer, L.; Derouiche, S.; Lemonnier, L.; Gosset, P.; Sadofsky, L.R.; Mariot, P.; Warnier, M.; Bokhobza, A.; Slomianny, C.; et al. Activation of mutated TRPA1 ion channel by resveratrol in human prostate cancer associated fibroblasts (CAF). Mol. Carcinog. 2017, 56, 1851–1867. [Google Scholar] [CrossRef]
- Martínez-Martínez, D.; Soto, A.; Gil-Araujo, B.; Gallego, B.; Chiloeches, A.; Lasa, M. Resveratrol promotes apoptosis through the induction of dual specificity phosphatase 1 and sensitizes prostate cancer cells to cisplatin. Food Chem. Toxicol. 2019, 124, 273–279. [Google Scholar] [CrossRef]
- Kai, L.; Levenson, A.S. Combination of resveratrol and antiandrogen flutamide has synergistic effect on androgen receptor inhibition in prostate cancer cells. Anticancer Res. 2011, 31, 3323. [Google Scholar]
- Franks, L.M.; Riddle, P.N.; Carbonell, A.W.; Gey, G.O. A comparative study of the ultrastructure and lack of growth capacity of adult human prostate epithelium mechanically separated from its stroma. J. Pathol. 1970, 100, 113–119. [Google Scholar] [CrossRef]
- Chung, L.W.; Chang, S.M.; Bell, C.; Zhau, H.E.; Ro, J.Y.; von Eschenbach, A.C. Co-inoculation of tumorigenic rat prostate mesenchymal cells with non-tumorigenic epithelial cells results in the development of carcinosarcoma in syngeneic and athymic animals. Int. J. Cancer 1989, 43, 1179–1187. [Google Scholar] [CrossRef]
- Marker, P.C.; Donjacour, A.A.; Dahiya, R.; Cunha, G.R. Hormonal, cellular, and molecular control of prostatic development. Dev. Biol. 2003, 253, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Miah, S.; Catto, J. BPH and prostate cancer risk. Indian J. Urol. 2014, 30, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Ao, M.; Franco, O.E.; Park, D.; Raman, D.; Williams, K.; Hayward, S.W. Cross-talk between paracrine-acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer Res. 2007, 67, 4244–4253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, D.; Buchanan, G. Stromal androgen receptor in prostate cancer development and progression. Cancers 2017, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Barron, D.A.; Rowley, D.R. The reactive stroma microenvironment and prostate cancer progression. Endocr. Relat. Cancer 2012, 19, R187–R204. [Google Scholar] [CrossRef] [Green Version]
- Cunha, G.R.; Hayward, S.W.; Wang, Y.Z. Role of stroma in carcinogenesis of the prostate. Differentiation 2002, 70, 473–485. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Singh, U.P.; Grizzle, W.E.; Lillard, J.W., Jr. CXCL12-CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab. Investig. 2004, 84, 1666–1676. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Huang, G.; Song, H.; Chen, Y.; Chen, L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 2017, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.K.; Pickup, M.W.; Chytil, A.; Aakre, M.; Owens, P.; Moses, H.L.; Novitskiy, S.V. TGFβ signaling in myeloid cells regulates mammary carcinoma cell invasion through fibroblast interactions. PLoS ONE 2015, 10, e0117908. [Google Scholar] [CrossRef]
- Schoonen, W.M.; Salinas, C.A.; Kiemeney, L.A.; Stanford, J.L. Alcohol consumption and risk of prostate cancer in middle-aged men. Int. J. Cancer 2005, 113, 133–140. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, H.; Yu, Y.; Zhang, J.; Liu, Q.; Yang, B. Resveratrol improved detrusor fibrosis induced by mast cells during progression of chronic prostatitis in rats. Eur. J. Pharmacol. 2017, 815, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhou, J.; Zhao, Y.; Liu, P.Y.; Yao, H.J.; Da, J.; Zhang, M.; Zhou, Z.; Chen, Q.; Peng, Y.B.; et al. Normal peripheral prostate stromal cells stimulate prostate cancer development: Roles of c-kit signal. Am. J. Transl. Res. 2015, 7, 502–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, S.; Chung, L.W.K. Prostate tumor-stroma interaction: Molecular mechanisms and opportunities for therapeutic targeting. Differentiation 2002, 70, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Liyanarachchi, S.; Davuluri, R.V.; Auer, H.; Martin Jr, E.W.; de la Chapelle, A.; Frankel, W.L. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene 2004, 23, 7366–7377. [Google Scholar] [CrossRef] [Green Version]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Wang, T.T.; Hudson, T.S.; Wang, T.C.; Remsberg, C.M.; Davies, N.M.; Takahashi, Y.; Kim, Y.S.; Seifried, H.; Vinyard, B.T.; Perkins, S.N.; et al. Differential effects of resveratrol on androgen-responsive LNCaP human prostate cancer cells in vitro and in vivo. Carcinogenesis 2008, 29, 2001–2010. [Google Scholar] [CrossRef]
- Harper, C.E.; Patel, B.B.; Wang, J.; Arabshahi, A.; Eltoum, I.A.; Lamartiniere, C.A. Resveratrol suppresses prostate cancer progression in transgenic mice. Carcinogenesis 2007, 28, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.W. STAT3 target genes relevant to human cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.C.; Zha, H.; Aime-Sempe, C.; Takayama, S.; Wang, H.G. Structure-function analysis of bcl-2 family proteins. regulators of programmed cell death. Adv. Exp. Med. Biol. 1996, 406, 99–112. [Google Scholar] [PubMed]
- Twillie, D.A.; Eisenberger, M.A.; Carducci, M.A.; Hseih, W.S.; Kim, W.Y.; Simons, J.W. Interleukin-6: A candidate mediator of human prostate cancer morbidity. Urology 1995, 45, 542–549. [Google Scholar] [CrossRef]
- Chung, L.W.; Baseman, A.; Assikis, V.; Zhau, H.E. Molecular insights into prostate cancer progression: The missing link of tumor microenvironment. J. Urol. 2005, 173, 10–20. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Sarne, V.; Ceder, S.; Bianchi, J.; Augsten, M.; Rundqvist, H.; Egevad, L.; Östman, A.; Wiman, K.G. Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells. Cell Death Discov. 2020, 6, 42–45. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef]
- Hobisch, A.; Eder, I.E.; Putz, T.; Horninger, W.; Bartsch, G.; Klocker, H.; Culig, Z. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998, 58, 4640–4645. [Google Scholar]
- van der Poel, H.G.; Zevenhoven, J.; Bergman, A.M. Pim1 regulates androgen-dependent survival signaling in prostate cancer cells. Urol. Int. 2010, 84, 212–220. [Google Scholar] [CrossRef]
- Ha, S.; Iqbal, N.J.; Mita, P.; Ruoff, R.; Gerald, W.L.; Lepor, H.; Taneja, S.S.; Lee, P.; Melamed, J.; Garabedian, M.J.; et al. Phosphorylation of the androgen receptor by PIM1 in hormone refractory prostate cancer. Oncogene 2013, 32, 3992–4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemskova, M.; Sahakian, E.; Bashkirova, S.; Lilly, M. The PIM1 kinase is a critical component of a survival pathway activated by docetaxel and promotes survival of docetaxel-treated prostate cancer cells. J. Biol. Chem. 2008, 283, 20635–20644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampan, N.C.; Xiang, S.D.; McNally, O.M.; Stephens, A.N.; Quinn, M.A.; Plebanski, M. Immunotherapeutic interleukin-6 or interleukin-6 receptor blockade in cancer: Challenges and opportunities. Curr. Med. Chem. 2018, 25, 4785–4806. [Google Scholar] [CrossRef] [PubMed]
- Gharaee-Kermani, M.; Moore, B.B.; Macoska, J.A. Resveratrol-mediated repression and reversion of prostatic myofibroblast phenoconversion. PLoS ONE 2016, 11, e0158357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Yen, M.L.; Lin, C.Y.; Kuo, M.L. Inhibition of vascular endothelial growth factor-induced angiogenesis by resveratrol through interruption of src-dependent vascular endothelial cadherin tyrosine phosphorylation. Mol. Pharmacol. 2003, 64, 1029–1036. [Google Scholar] [CrossRef]
- Al Aameri, R.F.H.; Sheth, S.; Alanisi, E.M.A.; Borse, V.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Tonic suppression of PCAT29 by the IL-6 signaling pathway in prostate cancer: Reversal by resveratrol. PLoS ONE 2017, 12, e0177198. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.-M.; Yang-Yen, H.-F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar] [CrossRef]
- Sinibaldi, D.; Wharton, W.; Turkson, J.; Bowman, T.; Pledger, W.J.; Jove, R. Induction of p21WAF1/CIP1 and cyclin D1 expression by the src oncoprotein in mouse fibroblasts: Role of activated STAT3 signaling. Oncogene 2000, 19, 5419–5427. [Google Scholar] [CrossRef] [Green Version]
- Parkins, C.S.; Stratford, M.R.; Dennis, M.F.; Stubbs, M.; Chaplin, D.J. The relationship between extracellular lactate and tumour pH in a murine tumour model of ischaemia-reperfusion. Br. J. Cancer 1997, 75, 319–323. [Google Scholar] [CrossRef] [Green Version]
- Deep, G.; Panigrahi, G.K. Hypoxia-induced signaling promotes prostate cancer progression: Exosomes role as messenger of hypoxic response in tumor microenvironment. Crit. Rev. Oncog. 2015, 20, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Fraga, A.; Ribeiro, R.; Principe, P.; Lopes, C.; Medeiros, R. Hypoxia and prostate cancer aggressiveness: A tale with many endings. Clin. Genitourin Cancer 2015, 13, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. The mitochondrial production of reactive oxygen species: Mechanisms and implications in human pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.L.; Salter, J.D.; Brookes, P.S. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: Implications for hypoxic cell signaling. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, 101. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorlach, A.; Kietzmann, T. Superoxide and derived reactive oxygen species in the regulation of hypoxia-inducible factors. Methods Enzymol. 2007, 435, 421–446. [Google Scholar] [CrossRef]
- Rodriguez-Enriquez, S.; Pacheco-Velazquez, S.C.; Marin-Hernandez, A.; Gallardo-Pérez, J.C.; Robledo-Cadena, D.X.; Hernández-Reséndiz, I.; García-García, J.D.; Belmont-Díaz, J.; López-Marure, R.; Hernández-Esquivel, L.; et al. Resveratrol inhibits cancer cell proliferation by impairing oxidative phosphorylation and inducing oxidative stress. Toxicol. Appl. Pharmacol. 2019, 370, 65–77. [Google Scholar] [CrossRef]
- Kovacic, P.; Somanathan, R. Multifaceted approach to resveratrol bioactivity: Focus on antioxidant action, cell signaling and safety. Oxid. Med. Cell Longev. 2010, 3, 86–100. [Google Scholar] [CrossRef]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Zhou, J.; Jiang, B.; Miao, M. Resveratrol and inflammatory bowel disease. Ann. N. Y. Acad. Sci. 2017, 1403, 38–47. [Google Scholar] [CrossRef]
- Zhang, J.; Patel, L.; Pienta, K.J. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev. 2010, 21, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Shamim, U.; Hanif, S.; Albanyan, A.; Beck, F.W.J.; Bao, B.; Wang, Z.; Banerjee, S.; Sarkar, F.H.; Mohammad, R.M.; Hadi, S.M.; et al. Resveratrol-induced apoptosis is enhanced in low pH environments associated with cancer. J. Cell Physiol. 2012, 227, 1493–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muqbil, I.; Beck, F.W.; Bao, B.; Sarkar, F.H.; Mohammad, R.M.; Hadi, S.M.; Azmi, A.S. Old wine in a new bottle: The warburg effect and anticancer mechanisms of resveratrol. Curr. Pharm. Des. 2012, 18, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.F.; Wei, Q.Y.; Cai, Y.J.; Fang, J.G.; Zhou, B.; Yang, L.; Liu, Z.-L. DNA damage induced by resveratrol and its synthetic analogues in the presence of cu (II) ions: Mechanism and structure-activity relationship. Free Radic. Biol. Med. 2006, 41, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, Z.; Zhang, X. Resveratrol induces apoptosis in murine prostate cancer cells via hypoxia-inducible factor 1-alpha (HIF-1alpha)/reactive oxygen species (ROS)/P53 signaling. Med. Sci. Monit. 2018, 24, 8970–8976. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Low, I.C.; Chen, Z.X.; Pervaiz, S. Bcl-2 modulates resveratrol-induced ROS production by regulating mitochondrial respiration in tumor cells. Antioxid. Redox. Signal. 2010, 13, 807–819. [Google Scholar] [CrossRef]
- Niu, Y.N.; Xia, S.J. Stroma-epithelium crosstalk in prostate cancer. Asian J. Androl. 2009, 11, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Xia, N.; Daiber, A.; Forstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chong, T.; Wang, Z.; Chen, H.; Li, H.; Cao, J.; Zhang, P.; Li, H. A novel anti-cancer effect of resveratrol: Reversal of epithelial-mesenchymal transition in prostate cancer cells. Mol. Med. Rep. 2014, 10, 1717–1724. [Google Scholar] [CrossRef] [Green Version]
- Varkaris, A.; Corn, P.G.; Gaur, S.; Dayyani, F.; Logothetis, C.J.; Gallick, G.E. The role of HGF/c-met signaling in prostate cancer progression and c-met inhibitors in clinical trials. Expert Opin. Investig. Drugs 2011, 20, 1677–1684. [Google Scholar] [CrossRef] [Green Version]
- Gmyrek, G.A.; Walburg, M.; Webb, C.P.; Yu, H.M.; You, X.; Vaughan, E.D.; Vande Woude, G.F.; Knudsen, B.S. Normal and malignant prostate epithelial cells differ in their response to hepatocyte growth factor/scatter factor. Am. J. Pathol. 2001, 159, 579–590. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, T.C.; Wu, J.M. Resveratrol suppresses prostate cancer epithelial cell scatter/invasion by targeting inhibition of hepatocyte growth factor (HGF) secretion by prostate stromal cells and upregulation of E-cadherin by prostate cancer epithelial cells. Int. J. Mol. Sci. 2020, 21, 1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Wang, S.; Kogure, Y.; Yamamoto, S.; Noguchi, K.; Dai, Y. Modulation of TRP channels by resveratrol and other stilbenoids. Mol. Pain 2013, 9, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalli, M.; Ortar, G.; Moriello, A.S.; Morera, E.; Di Marzo, V.; De Petrocellis, L. TRPA1 channels as targets for resveratrol and related stilbenoids. Bioorganic Med. Chem. Lett. 2016, 26, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: Induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Putzke, A.P.; Ventura, A.P.; Bailey, A.M.; Akture, C.; Opoku-Ansah, J.; Çeliktaş, M.; Hwang, M.S.; Darling, D.S.; Coleman, I.M.; Nelson, P.S.; et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and SRC family kinases. Am. J. Pathol. 2011, 179, 400–410. [Google Scholar] [CrossRef]
- Jennbacken, K.; Tesan, T.; Wang, W.; Gustavsson, H.; Damber, J.E.; Welén, K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr. Relat. Cancer 2010, 17, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Wegner, K.A.; Mueller, B.R.; Unterberger, C.J.; Avila, E.J.; Ruetten, H.; Turco, A.E.; Oakes, S.R.; Girardi, N.M.; Halberg, R.B.; Swanson, S.M.; et al. Prostate epithelial-specific expression of activated PI3K drives stromal collagen production and accumulation. J. Pathol. 2020, 250, 231–242. [Google Scholar] [CrossRef]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 115–116. [Google Scholar] [CrossRef] [Green Version]
- Phan, S.H. Biology of fibroblasts and myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Lichtinghagen, R.; Musholt, P.B.; Lein, M.; Römer, A.; Rudolph, B.; Kristiansen, G.; Hauptmann, S.; Schnorr, D.; ALoening, S.; Jung, K. Different mRNA and protein expression of matrix metalloproteinases 2 and 9 and tissue inhibitor of metalloproteinases 1 in benign and malignant prostate tissue. Eur. Urol. 2002, 42, 398–406. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Stetler-Stevenson, W.G. Matrix metalloproteinases and metastasis. Cancer Chemother Pharmacol. 1999, 43, S42–S51. [Google Scholar] [CrossRef] [PubMed]
- PLoS ONE Editors. Expression of concern: Resveratrol enhances antitumor activity of TRAIL in prostate cancer xenografts through activation of FOXO transcription factor. PLoS ONE 2019, 14, e0223138. [Google Scholar] [CrossRef]
- Jang, Y.G.; Go, R.E.; Hwang, K.A.; Choi, K.C. Resveratrol inhibits DHT-induced progression of prostate cancer cell line through interfering with the AR and CXCR4 pathway. J. Steroid. Biochem. Mol. Biol. 2019, 192, 105406. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Sung, S.Y.; Hsieh, C.L.; Law, A.; Zhau, H.E.; Pathak, S.; Multani, A.S.; Lim, S.; Coleman, I.M.; Wu, L.-C.; Figg, W.D. Coevolution of prostate cancer and bone stroma in three-dimensional coculture: Implications for cancer growth and metastasis. Cancer Res. 2008, 68, 9996–10003. [Google Scholar] [CrossRef] [Green Version]
- Dubrovska, A.; Elliott, J.; Salamone, R.J.; Telegeev, G.; Stakhovsky, A.E.; Schepotin, I.B.; Yan, F.; Wang, Y.; Bouchez, L.; Kularatne, S.A.; et al. CXCR4 expression in prostate cancer progenitor cells. PLoS ONE 2012, 7, e31226. [Google Scholar] [CrossRef]
- Conley-LaComb, M.K.; Semaan, L.; Singareddy, R.; Li, Y.; Heath, E.I.; Kim, S.; Cher, M.L.; Chinni, S.R. Pharmacological targeting of CXCL12/CXCR4 signaling in prostate cancer bone metastasis. Mol. Cancer 2016, 15, 68. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Sun, Y.X.; Schneider, A.; Jung, Y.; Wang, J.; Dai, J.; Wang, J.; Cook, K.; Osman, N.I.; Koh-Paige, A.J.; Shim, H.; et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J. Bone Miner. Res. 2005, 20, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Pérez, J.; Monter-Vera, M.D.R.; Barrientos-Alvarado, C.; Toscano-Garibay, J.D.; Cuesta-Mejías, T.; Flores-Estrada, J. Evaluation of VEGF and PEDF in prostate cancer: A preliminary study in serum and biopsies. Oncol. Lett. 2018, 15, 1072–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.L.; Zhao, H.; Ren, X.B. Relationship of VEGF/VEGFR with immune and cancer cells: Staggering or forward? Cancer Biol. Med. 2016, 13, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Hossain, M.A.; Kim, M.Y.; Kim, J.-A.; Yoon, J.-H.; Suh, H.S.; Kim, G.-Y.; Choi, Y.H.; Chung, H.Y.; Kim, N.D. A novel resveratrol analogue, HS-1793, inhibits hypoxia-induced HIF-1α and VEGF expression, and migration in human prostate cancer cells. Int. J. Oncol. 2013, 43, 1915–1924. [Google Scholar] [CrossRef] [Green Version]
- Walsh, K.; Sriprasad, S.; Hopster, D.; Codd, J.; Mulvin, D. Distribution of vascular endothelial growth factor (VEGF) in prostate disease. Prostate Cancer Prostatic Dis. 2002, 5, 119–122. [Google Scholar] [CrossRef]
- Chen, J.; De, S.; Brainard, J.; Byzova, T.V. Metastatic properties of prostate cancer cells are controlled by VEGF. Cell Commun. Adhes. 2004, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.R.; Kyprianou, N. Growth factor signalling in prostatic growth: Significance in tumour development and therapeutic targeting. Br. J. Pharmacol. 2006, 147 (Suppl. 2), 144. [Google Scholar] [CrossRef] [Green Version]
- Stephens, T.C.; Currie, G.A.; Peacock, J.H. Repopulation of gamma-irradiated lewis lung carcinoma by malignant cells and host macrophage progenitors. Br. J. Cancer 1978, 38, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Lissbrant, I.F.; Stattin, P.; Wikstrom, P.; Damber, J.E.; Egevad, L.; Bergh, A. Tumor associated macrophages in human prostate cancer: Relation to clinicopathological variables and survival. Int. J. Oncol. 2000, 17, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Shimura, S.; Yang, G.; Ebara, S.; Wheeler, T.M.; Frolov, A.; Thompson, T.C. Reduced infiltration of tumor-associated macrophages in human prostate cancer: Association with cancer progression. Cancer Res. 2000, 60, 5857–5861. [Google Scholar] [PubMed]
- Mantovani, A.; Germano, G.; Marchesi, F.; Locatelli, M.; Biswas, S.K. Cancer-promoting tumor-associated macrophages: New vistas and open questions. Eur. J. Immunol. 2011, 41, 2522–2525. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Kimura, Y.; Sumiyoshi, M. Resveratrol prevents tumor growth and metastasis by inhibiting lymphangiogenesis and M2 macrophage activation and differentiation in tumor-associated macrophages. Nutr. Cancer 2016, 68, 667–678. [Google Scholar] [CrossRef]
- Solís-Martínez, R.; Cancino-Marentes, M.; Hernández-Flores, G.; Ortiz-Lazareno, P.; Mandujano-Álvarez, G.; Cruz-Gálvez, C.; Sierra-Díaz, E.; Rodríguez-Padilla, C.; Jave-Suárez, L.F.; Aguilar-Lemarroy, A.; et al. Regulation of immunophenotype modulation of monocytes-macrophages from M1 into M2 by prostate cancer cell-culture supernatant via transcription factor STAT3. Immunol. Lett. 2018, 196, 140–148. [Google Scholar] [CrossRef]
- Tomé-Carneiro, J.; Larrosa, M.; González-Sarrías, A.; Tomás-Barberán, F.A.; García-Conesa, M.T.; Espín, J.C. Resveratrol and clinical trials: The crossroad from in vitro studies to human evidence. Curr. Pharm. Des. 2013, 19, 6064–6093. [Google Scholar] [CrossRef] [Green Version]
- Kotha, A.; Sekharam, M.; Cilenti, L.; Siddiquee, K.; Khaled, A.; Zervos, A.S.; Carter, B.; Turkson, J.; Jove, R. Resveratrol inhibits src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol. Cancer Ther. 2006, 5, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Sobhani, N.; Generali, D.; D’Angelo, A.; Aieta, M.; Roviello, G. Current status of androgen receptor-splice variant 7 inhibitor niclosamide in castrate-resistant prostate-cancer. Investig. New Drugs 2018, 36, 1133–1137. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Wu, J.M. Grape-derived chemopreventive agent resveratrol decreases prostate-specific antigen (PSA) expression in LNCaP cells by an androgen receptor (AR)-independent mechanism. Anticancer Res. 2000, 20, 225–228. [Google Scholar]
- Benitez, D.A.; Pozo-Guisado, E.; Clementi, M.; Castellón, E.; Fernandez-Salguero, P.M. Non-genomic action of resveratrol on androgen and oestrogen receptors in prostate cancer: Modulation of the phosphoinositide 3-kinase pathway. Br. J. Cancer 2007, 96, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.B.; DePrimo, S.E.; Whitfield, M.L.; Brooks, J.D. Resveratrol-induced gene expression profiles in human prostate cancer cells. Cancer Epidemiol. Biomark. Prev. 2005, 14, 596–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeni, A.; Takahashi, S.; Takeshita, K.; Tang, M.; Sugiura, S.; Sato, S.; Shirai, T. Suppression of prostate cancer growth by resveratrol in the transgenic rat for adenocarcinoma of prostate (TRAP) model. Asian Pac. J. Cancer Prev. 2008, 9, 7–14. [Google Scholar]
- Harada, N.; Murata, Y.; Yamaji, R.; Miura, T.; Inui, H.; Nakano, Y. Resveratrol down-regulates the androgen receptor at the post-translational level in prostate cancer cells. J. Nutr. Sci. Vitaminol. 2007, 53, 556–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.F.; Leong, M.; Cho, E.; Farrell, J.; Chen, H.; Tian, J.; Zhang, D. Repressive effects of resveratrol on androgen receptor transcriptional activity. PLoS ONE 2009, 4, e7398. [Google Scholar] [CrossRef]
- Izumi, K.; Mizokami, A. Suppressive role of androgen/androgen receptor signaling via chemokines on prostate cancer cells. J. Clin. Med. 2019, 8, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, L.C.; Lu, C.; Antonarakis, E.S.; Luo, J.; Armstrong, A.J. Androgen receptor variant-driven prostate cancer II: Advances in clinical investigation. Prostate Cancer Prostatic Dis. 2020, 23, 367–380. [Google Scholar] [CrossRef]
- Wilson, S.; Cavero, L.; Tong, D.; Liu, Q.; Geary, K.; Talamonti, N.; Xu, J.; Fu, J.; Jiang, J.; Zhang, D. Resveratrol enhances polyubiquitination-mediated ARV7 degradation in prostate cancer cells. Oncotarget 2017, 8, 54683–54693. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silk, N.; Reich, J.; Sinha, R.; Chawla, S.; Geary, K.; Zhang, D. The Effects of Resveratrol on Prostate Cancer through Targeting the Tumor Microenvironment. J. Xenobiot. 2021, 11, 16-32. https://0-doi-org.brum.beds.ac.uk/10.3390/jox11010002
Silk N, Reich J, Sinha R, Chawla S, Geary K, Zhang D. The Effects of Resveratrol on Prostate Cancer through Targeting the Tumor Microenvironment. Journal of Xenobiotics. 2021; 11(1):16-32. https://0-doi-org.brum.beds.ac.uk/10.3390/jox11010002
Chicago/Turabian StyleSilk, Natalie, Jeremy Reich, Rahul Sinha, Shivansh Chawla, Kyla Geary, and Dianzheng Zhang. 2021. "The Effects of Resveratrol on Prostate Cancer through Targeting the Tumor Microenvironment" Journal of Xenobiotics 11, no. 1: 16-32. https://0-doi-org.brum.beds.ac.uk/10.3390/jox11010002