
The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family

Center for Toxicogenomics and Human Health (ToxOmics), Genetics, Oncology and Huma Toxicology, NOVA Medical School/Faculty of Medical Sciences, Universidade NOVA de Lisboa, 1169-056 Lisboa, Portugal
*
Author to whom correspondence should be addressed.
J. Xenobiot. 2021, 11(3), 94-114; https://0-doi-org.brum.beds.ac.uk/10.3390/jox11030007
Submission received: 10 May 2021
/
Revised: 14 June 2021
/
Accepted: 16 June 2021
/
Published: 22 June 2021
Abstract
:Human Cytochrome P450 (CYP) enzymes constitute a superfamily of membrane-bound hemoproteins that are responsible for the metabolism of a wide variety of clinically, physiologically, and toxicologically important compounds. These heme-thiolate monooxygenases play a pivotal role in the detoxification of xenobiotics, participating in the metabolism of many structurally diverge compounds. This short-review is intended to provide a summary on the major roles of CYPs in Phase I xenobiotic metabolism. The manuscript is focused on eight main topics that include the most relevant aspects of past and current CYP research. Initially, (I) a general overview of the main aspects of absorption, distribution, metabolism, and excretion (ADME) of xenobiotics are presented. This is followed by (II) a background overview on major achievements in the past of the CYP research field. (III) Classification and nomenclature of CYPs is briefly reviewed, followed by (IV) a summary description on CYP’s location and function in mammals. Subsequently, (V) the physiological relevance of CYP as the cornerstone of Phase I xenobiotic metabolism is highlighted, followed by (VI) reviewing both genetic determinants and (VI) nongenetic factors in CYP function and activity. The last topic of the review (VIII) is focused on the current challenges of the CYP research field.
1. Xenobiotics Disposition and Excretion
Humans are continuously exposed to a wide variety of chemicals. An important portion of these compounds is not essential for maintenance of normal homeostasis. They are neither nutrients nor intermediate metabolites, produced from nutritional metabolism. Drugs, environmental pollutants, cosmetics, and even components present in our diet, such as food additives, form this extended group of xenobiotics in general harmless, but potentially toxic [1,2,3,4]. In a human lifetime, one might be exposed to 1–3 million different foreign compounds, which can accumulate within a variety of different organs and tissues [4]. Storage of xenobiotics can function as either a protective mechanism or as a mean by which bioaccumulation can trigger toxic effects. This potential toxic route depends on the physiologic relationship between the storage depot and the target tissues for a specific toxicant [1,3].
Xenobiotics are metabolized and ultimately eliminated through the urine, bile, and feces, with minor routes through expiration and sweat. However, without effective detoxification and subsequent excretion, many compounds may reach toxic levels and interfere with cellular homeostasis, leading to cellular and tissue damage, with detrimental effects on health [1,2,4]. Harmful cellular and tissue concentrations can be prevented by direct elimination of xenobiotics, whose mechanisms are dependent on the physico-chemical proprieties of the compounds. Yet, the vast majority undergo biotransformation by complex metabolic mechanisms, resulting in the formation of numerous metabolites, some of which have the potential to cause unintendedly, more toxic effects, i.e., bioactivation [1,4,5,6]. Detoxification routes comprise enzymatic functionalization and/or conjugation reactions that facilitate elimination and excretion. All combined, these pathways act jointly to detoxify xenobiotics and remove them from cells and tissues [1,3,4,6,7,8,9]. In chemical toxicology, it is therefore of great interest to have comprehensive and integrated knowledge of in vivo xenobiotic metabolism, to understand, predict and prevent potential health hazards through bioavailability, bioaccumulation, or generation of harmful reactive metabolites, after chemical exposure.
Four stages can be distinguished in the processes of absortion, metabolism and cellular excretion of xenobiotics, namely (i) influx by transporter enzymes, biotransformation in (ii) Phases I and (iii) II, mediated by drug-metabolizing enzymes (DMEs), followed by (iv) Phase III, the excretion mediated mostly through transporter enzymes [1,6,9,10,11,12] (Figure 1). Organic anion transporting polypeptides (OATP), organic anion transporters (OAT) and sodium taurocholate cotransporting polypeptide (NTCP), are involved in the influx of the xenobiotics (reviewed in Murray M, Zhou F, 2017) [12]. Phase I enzymes, which include the cytochrome P450s (CYPs) superfamily—the major contributer— but also flavin-containing monooxygenases (FMOs), NAD(P)H:quinone oxidoreductases (NQOs), amine oxidases, alcohol dehydrogenases, esterases and peroxidases (reviewed in Gan J, et al., 2016) catalyze the oxidation, reduction or hydrolyses of primarily lipophilic xenobiotics into more polar molecules [7,13,14,15,16]. The introduction of polar groups by Phase I reactions provides sites that enable conjugation reactions, mediated by Phase II enzymes [5,7,17,18]. Although Phase II enzymes can directly act on the parent compound [19], typically Phase I-metabolites are conjugated with charged species, such as glucuronic acid, glutathione, sulfate, amino acids (glycine, taurine, glutamic acid), methyl or acetyl groups. Addition of these large anionic groups, which may detoxify reactive electrophiles (either parent compound or Phase I metabolite), produce Phase II metabolites, with increased hydrophilicity and molecular weight, which in larger part are not able to diffuse across phospholipid membrane barrier (reviewed in Jančová P, Šiller M, 2012) [6,10,11,13,19,20]. Phase III xenobiotic transporters excrete hydrophilic conjugates, with the anionic groups acting as affinity tags for a variety of membrane carriers belonging to two main clusters: ATP binding cassette (ABC), including the multidrug resistance protein (MRP) family, and solute carrier (SLC) transporters (reviewed in Döring B, Petzinger E, 2014) [21,22,23].
There are major inter- and intra-individual variations in the capacity to metabolize, detoxify and extrude xenobiotics (see below). These are of genetic, epigenetic, environmental, and physio- or pathophysiological origin, and vary during lifetime [9,13,21,24,25,26,27,28,29]. Most xenobiotics are detoxified and cleared through an intricate network of multiple enzymes and pathways. The relationship between xenobiotic local/cellular concentration, specific enzymes affinity, tissue specific enzyme expression, stability, and cofactors availability, often determine which metabolic reactions dominate in a given individual, at one precise moment [1,7,30].
2. Historical Aspects of Cytochrome P450s Research
Cytochrome P450s comprise the major Phase I family of enzymes capable of catalyzing the oxidative biotransformation of a vast majority of lipophilic xenobiotics and are the focus of research in areas such as clinical pharmacology and toxicology [5,18,25,31,32,33]. The early reports dealing with CYP, dated back to the 1940s and were related with in vitro studies on the metabolism of steroids and xenobiotics, including drugs and carcinogens [34].
The establishment of the CYP research field goes back to the late 50s/early 60s, when several groups focused on a particular pigment in animal liver microsomes, which seemed to be directly related with specific hepatic functions [35,36]. In 1962, a major breakthrough was obtained by Drs. Tsuneo Omura and Ryo Sato, describing for the first time, the spectral observation of CYP in liver microsomes [37]. In fact, the name of CYP (P450) is derived from the characteristic absorption maximum at 450 nm, observed in differential spectra of the reduced CO-bond enzyme complex. Initially, CYP was thought to be a single enzymatic entity, which was believed to exist almost exclusively in the liver, responsible for the metabolism of drugs and other foreign chemicals [38,39,40]. Several seminal reports described the inducibility of CYP activities, of clinical relevance in pharmacology and therapeutics [40,41,42]. Subsequently, other remarkable discoveries were made, namely: the role of CYPs in steroid and fatty acid hydroxylation, both in adrenal cortex and liver (1963) [43,44]; protein separation of CYPs [45] and their purification [46,47]; identification and biochemical characterization of multiple CYP forms—evidencing the existence of numerous isoenzymes (late 60s/early 70s) [39,47,48,49,50]; biophysical studies and biochemical characterization of bacterial CYP (P450cam or CYP101A1)—of importance to establish template protein structures to be used in homology modeling (1968) [51]; and data supporting models on oxygen activation, evidencing a stepwise process involving C-H bond breaking (1978) [52]. In 1983, the first complete analysis of a CYP gene (rat) was accomplished [53]. Thereafter, the evolution of molecular genetic techniques led researchers to uncover the extensive inter- and intra-species variability of the CYP genes superfamily. Genetic studies have shown the considerable inter-individual differences in expression of CYP isoenzymes in the human population (since the 80s) [30,54,55,56,57,58,59]. In the waking of the new millennium, 57 human CYP genes (plus multiple pseudogenes) were identified by the human genome project (2003) [26,60,61]. During this period other significant contributions were the development of mammalian (including human) heterologous CYP expression, using recombinant DNA technology (early 90s)—critical for crystallography and functional studies [62,63,64,65]; application of engineered bacterial strains expressing human CYP enzymes complex in biotransformation and genotoxicity studies of xenobiotics (late 90s) [66,67]; the first CYP proteins structures solved (bacterial P450cam and P450BM3) (early 80s and 90s) [68,69,70], followed by the crystal structures of human CYPs—particularly high-resolution structures (2000s) [71,72,73,74]; and the description of determinant protein-protein interactions within CYP enzymes complex—impacting CYP activity (2000s and early 10s) [75,76,77,78,79,80,81].
3. Classification and Nomenclature of Human Cytochrome P450s
Back in the 80s, studies enabled by the big boom in molecular biology demonstrated that CYP genes are ubiquitously present in almost all life forms, from prokaryotes to humans, adding a new dimension to the complex repertoire of functions catalyzed by this super enzyme family [53,54]. The genes encoding these heme-thiolate monooxygenases capable of catalyzing the oxidative biotransformation of endogenous compounds and xenobiotics, diverged from a single ancestor in an evolutionary process started 3 billion years ago [82,83]. In contrast with “conventional” enzymes, which normally demonstrate high substrate specificity and turnover rates, human CYP isoenzymes involved in drug metabolism, evolved favoring low substrate specificity and turnover rates, characteristics of DMEs in general [30].
Although the outcome of the CYP-mediated metabolism prevents bioaccumulation by chemically transforming lipophilic compounds (readily absorbed) into hydrophilic metabolites (readily excreted) (Figure 2), CYP mediated biotransformation as well as other DMEs, evolved in a species-specific manner [54]. Historically, species of rodents, dogs, rabbits and pigs were considered to be suitable organisms for comparative drug metabolism studies. However, the variability in xenobiotic metabolism among species, in particular CYP metabolism, is evidenced by the fact that mutagenicity profiles of chemicals determined by the Ames test [84,85] when using human liver S9 fraction is significantly different from the ones observed with the rat liver S9 fraction [86]. This raises an important issue regarding the utility and reliability of results obtained from toxicological studies, using animal materials and models for risk assessments of human exposure [87]. Several factors must be carefully considered for the appropriate selection of an animal model which adequately mimics the human metabolism of a particular xenobiotic. Species-specific expression, regulation and function of CYPs have to be taken into account to avoid significant deviations. This inter-species variability is harshly exemplified by the case of thalidomide, in the late 50s/early 60s. This drug was commercialized as a non-addictive, non-barbiturate hypnotic and anti-emetic, and used for treatment of morning sickness of pregnant women. Thalidomide was tested for potential teratogenicity in mice and considered to be safe, however its use led to severe birth defects involving limb malformation. Teratogenicity is thought to occur through a reactive metabolite of thalidomide (arene oxide) (Figure 2A), formed in significantly higher proportions in human (and rabbit) liver than in mouse liver. After this occurrence, U.S. Food and Drug Administration (FDA), and medicine agencies worldwide, implemented obligatory full multi-species (in vivo) teratogenicity testing as a requisite for drug approval [88,89]. Another example is the metabolism and effect of aflatoxin B1 in trout. Aflatoxin B1, the most potent hepatic chemical carcinogen known today is activated via a CYP-dependent reaction (Figure 2G). Interestingly, and in contrast with the rat model, trout exposure to aflatoxin B1 may result in a hepatocellular carcinoma with relevant similarity to the one found in humans [90,91].
In 1987, the gene superfamily nomenclature system, based on evolutionary divergence of CYP, was proposed [54,83]. Since then, CYPs are organized into families and subfamilies, based on the percentage of amino acid sequence identity (Figure 3) [61,92,93]. Cytochrome P450s forms sharing an identity of ≥40% constitute a particular family designated by an Arabic numeral, whereas enzymes with ≥55% identity are assigned to a particular subfamily designated by a letter. Finally, the gene coding the isoenzyme is designated by an Arabic numeral (https://drnelson.uthsc.edu/) (accessed on 20 June 2021). In humans, the CYP superfamily consists of 57 genes (and 58 pseudo-genes) divided into 18 families and 44 sub-families and can be classified based on major substrate classes (Table 1) (https://cyped.biocatnet.de/) (accessed on 20 June 2021) [5,25,82,94].
4. Location and Function of Cytochrome P450s in Mammals
Cytochrome P450s are expressed virtually in all tissues, with highest concentrations found in the small intestine, but particularly in the liver. These membrane-bound hemeproteins contain more than 500 amino acid residues and a single heme prosthetic group in the active site [14,25,26,32]. CYPs are abundant in the microsomal fraction of the liver, playing a central role in bile acid biosynthesis and in the metabolism of foreign compounds [5,25,94]. CYPs are also involved in the homeostasis of steroid hormones, with relevant CYP forms present in the inner membrane of mitochondria of steroidogenic tissues, such as adrenal cortex, testis, ovary, breasts, and placenta [43,44]. Additionally, CYPs are of importance in vitamin metabolism, metabolism of unsaturated fatty acids, and in cholesterol biosynthesis [54,82,95] (Table 1).
The human gut microbiome represents a site for xenobiotic metabolism, altering the pharmacokinetics outcome of drugs, environmental toxicants, and heavy metals. Increased metabolism or biactivation of xenobiotics by the gut microbiome may occur, either through the intestinal tract or re-entering the gut via enterohepatic circulation. This is dependent on the enzymatic activity within the microbial niche [96,97]. CYP host expression in mice showed to be modulated by the collection of microorganisms in the gastrointestinal tract via altered xenobiotic nuclear receptors activity [98,99]. In a dual effect, the microbiota modulates the pharmacokinetics of the xenobiotics, while reciprocally xenobiotics can influence the viability and metabolism of the microbiota.
Cytochrome P450s catalyze a variety of oxidation and reduction reactions involving broad, and in many cases overlapping substrate specificity (Figure 2) [8,18,26,100,101,102,103,104,105,106]. In this context, a single compound can be metabolized by different CYP isoenzymes, in a complex biotransformation enabled by multiple pathways, resulting in numerous metabolites. On the other hand, a unique compound can be metabolized by a single CYP originating different metabolites. The typical CYP-mediated monooxygenation consists of the incorporation of one oxygen atom into the substrate (RH + O2 + 2e− + 2H+ → ROH + H2O), although the activated oxygen atom may not necessarily be incorporated but used in different types of reactions. Reactions catalyzed by CYPs include: aliphatic, aromatic and N-hydroxylation; epoxidation; N-, O- and S-dealkylation; N- and S-oxidation; oxidative deamination; dehalogenation; dehydrogenation; dehydration; C-C bond cleavage; isomerization; reduction; and esterase [107].
The energy required to activate oxygen is supplied to microsomal CYPs (including those of families 1–4, involved in xenobiotic metabolism, see below) by cytochrome P450 oxidoreductase (CPR) [108]. This reductase obtains two electron equivalents in the form of a hydride (H−) from NADPH, which is received by its flavin adenine dinucleotide (FAD) moiety (reductase) and subsequently donated to CPR’s second flavin prosthetic group, flavin mononucleotide (FMN) (transporter). Through an extensive open/close protein dynamics, the FMN is reduced (inter-flavin electron transfer, closed conformation) and subsequently, CPR undergoes a large rearrangement, allowing the interaction of CYPs with the FMN domain of CPR (open conformation), with electron equivalents transferred, one at a time, to the heme group of CYPs (inter-protein electron transfer) [80,81,109,110,111,112,113]. Additionally, cytochrome b5 may play an auxiliary role in sustaining CYPs in their activity, by donating the second electron, which is facultative and dependent on the CYP isoenzyme and/or substrate [75,76,77,78,79].
5. The Central Role of Cytochrome P450s as Xenobiotic-Metabolizing Enzymes
In humans, the CYP enzyme family represents the most important enzymatic system involved in Phase I drug metabolism [5,10,13,20,25]. A survey on literature databases of human oxidoreductases and CYP enzymes implicated in the Phase I metabolism evidenced that CYPs are involved in the vast majority (approx. 90–95%) of enzymatic reactions in the metabolism of xenobiotics [7]. Additional enzyme systems may play a role in hepatic Phase I metabolism, albeit to a lesser extent than CYPs, as mentioned above. These include FMOs, NQOs, amine oxidases, alcohol dehydrogenases, esterases and peroxidases [7,13,14,15,16].
Although the majority of the isoenzymes of the 18 human CYP families have specific functions in the metabolism of endobiotics, about 15 isoforms belonging to CYP families 1, 2 and 3 (Table 2) are accountable for 70–80% of all Phase I metabolism of clinically used drugs [25] and are involved in the biotransformation of a vast diversity of environmental chemicals (approx. 90%), including 66% metabolism reactions of chemical carcinogens [114]. From these, CYP1A2, CYP2C9, CYP2D6 and CYP3A4/5, are responsible for about 72% of all CYP-mediated metabolism of clinically marketed drugs [25]. Although typically contributing to the ω-oxidation of endogenous fatty acids and eicosanoids, members of the CYP4 family are additionally involved in xenobiotic metabolism (e.g., CYP4A11, CYP4F2 and CYP4F12), albeit to a much lower extend than isoforms of the CYP1–3 families [115,116,117,118].
The CYP-enzymes involved in xenobiotic metabolism have evolved to protect humans against potential toxic agents. However, CYP-mediated biotransformation may result in metabolic activation of environmental chemicals to reactive carcinogenic products, a process known as “lethal synthesis” or bioactivation [14,25,82,94,114,119,120]. While CYPs may catalyze the activation of procarcinogens to electrophilic ultimate carcinogens, the Phase II enzymes in general detoxify electrophilic intermediates into non-toxic substrates, with few exceptions of non-canonical bioactivation through conjugation reactions [19]. The potentially toxic reactive metabolites that “escape” from Phase II metabolism are able to covalently interact with nucleophilic structures such as those of nucleic acids, proteins, or lipids, causing cell damage and potentially triggering carcinogenesis, teratogenicity, and/or adverse drug reactions (ADRs) [1,3,20,58,103,104,105,114,120] (Figure 1). Several CYP-mediated oxidations contribute to the synthesis of more toxic metabolites, after exposure to specific compounds such as: (i) polycyclic aromatic hydrocarbons (PAHs) (e.g., benzo[a]pyrene, Figure 2D), including nitro-polycyclic aromatic hydrocarbons, derived from incomplete combustion; (ii) heterocyclic aromatic amines (HAAs) from charbroiled meats; (iii) aromatic amines as dyes, or present in pesticides, tobacco smoke and pharmaceuticals (e.g., paracetamol, Figure 2E); (iv) nitrosamines present in tobacco smoke (e.g., nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, NNK, Figure 2F) and diet, formed from nitrites and nitrates; (v) toxins present in food products (e.g., grains or cereals) contaminated with pathogenic microorganisms (e.g., aflatoxin B1 produced by Aspergillus flavus and A. parasiticus, Figure 2G). These xenobiotics are transformed to their toxic forms—usually electrophiles such as epoxides, hydroxylamides, acyl halides, among others—through CYP-mediated activity [5,14,106,107,121].
6. Genetic Determinants in Cytochrome P450s Expression and Activity
With the advances in research during the last 30 years, it became clear that the effects of genetic variability of DMEs, particularly those of the CYP enzymes complex, are highly relevant in terms of drug response and detoxification or bioactivation of xenobiotics in general [24,26,57,59,114]. Cytochrome P450s exhibit genetic polymorphisms with multiple allelic variants, demonstrating frequencies varying between different populations and ethnicities [57,58,59,82,106,122] (Figure 4). These include single nucleotide variants, small deletions or insertions, and copy number variants (gene deletion or duplication/amplification, the later more frequent) [55,56,57]. These genetic variants can alter structurally the CYP enzyme or its expression, resulting in normal, reduced, increased or absence of activity [58,59,82,122,123]. A nomenclature system has been set up for the CYP alleles (using the suffixes *1, *2, *3…; where *1 designates the “wild type,” or most common gene-variant) (Figure 3). Allelic variants are summarized and described on the home page of the human CYP allele nomenclature committee, at Pharmacogene Variation (PharmVar) Consortium (https://www.pharmvar.org/htdocs/archive/index_original.htm) (accessed on 20 June 2021).
Isoforms CYP2C9, 2C19 and 2D6 present the highest genetic variability in the human population, with so far 70, 38 and 145 allelic variants being identified, respectively (Table 2). These three CYPs have been estimated to account for approximately 35–40% of oxidative drug metabolism and a quarter of biotransformation of xenobiotics in general, including environmental and industrial pollutants [7,17,25,114]. The high frequencies of genetic polymorphism of these three CYPs were demonstrated to cause significant functional effects and high penetrance in individual susceptibility in the human population [4,5,56,57,58,82]. Conversely, severe loss-of-function alleles or functional gene duplications are rare in genes encoding CYP1A2 and 3A4 (21 and 32 allelic variants, respectively) [7,14,25,57,122]. These two CYPs together are responsible for 35% and 30% of drug- and of general chemical metabolism, respectively.
This extensive genetic variability, causing inter-individual differences in expression and activity of human CYPs is considered to be one of the major causes in the lack of efficacy and in ADRs of therapeutic drugs, as well as in the variability of toxic outcome after exposure to environmental compounds [55,56,58,124,125]. Pharmacogenetic testing of CYPs is gaining attention due to the possibility of developing safer drugs and patient-tailored drug therapy in precision medicine [5,25,26]. The occurrence of specific gene variants translates into four major metabolizer phenotypes: (i) ultrarapid metabolizers (UM), involving two or more active gene copies or allelic variants, encoding more efficient enzymes or over-expressed CYP isoforms; (ii) extensive metabolizers (EM), carriers of two functional CYP alleles; (iii) intermediate metabolizers (IM), heterozygous for a defect allele or carrying two alleles causing combined decreased activity of a CYP; (iv) poor metabolizers (PM), carrying two defective alleles, producing CYPs with very low or without activity/function [55,56,122,125]. A potential additional genetic variability in CYP mediated xenobiotic metabolism has recently been revealed. This regards the genetic variability of CPR (encoded by the POR gene), the obligatory redox partner of microsomal CYPs for the reception of electron equivalents to sustain their activity. Recent data suggest that natural occurring genetic variants of POR may lead to altered CYP mediated drug metabolism [126,127,128,129].
Genotyping provides sequence data allowing the estimation of the expectable CYP metabolizer phenotypes. However, additional determinants have been shown to play a role in modulating CYP enzymes function, such as those of the environment and physio-pathological conditions (discussed below) [25,130,131]. Of relevance also to mention two main epigenetic mechanisms affecting CYP gene expression: (i) altered DNA methylation —involved in biased cellular control of gene expression; and (ii) microRNA (miR) regulation—affecting expression levels of target CYPs [5,132,133]. The inhibition of methylation in hepatic cell lines has been reported to induce CYP genes expression, particularly CYP3A genes [134]. Additionally, methylation patterns in the promoters of CYP genes seem to be different in distinct physiological conditions or environmental exposures (e.g., decreased inducibility of CYP1B1 due to promoter methylation at multiple CpG sites; lower methylation in the CYP1A1 promoter found in heavy smokers) [132,135]. Direct regulation of CYPs by miRs was evidenced for CYP1B1 (miR-27b), CYP2C9 (miR-130b), CYP2C9 (miR-34a), CYP2E1 (miR-378), and CYP3A4 (miR-27b, miR148a, and miR34a) [133,136,137,138]. Additionally, histone protein modification, an epigenetic mechanism that may affect chromatin structure, impacting accessibility, have been indicated to be involved in transcriptional regulation of CYP expression. Epigenetic patterns leading to divergent CYP-mediated metabolism are normally reversible, tissue-specific and highly dependent on environmental and individual physio-pathological conditions [25,56,139].
7. Nongenetic Factors Influencing Cytochrome P450s Expression and Activity
Factors such as age, sex, hormone levels, and environment, as well as pathological conditions such as infection, inflammation, cholestasis, and cancer are aspects demonstrated to influence CYP expression and activity [25,130,131] (Figure 4). Other biochemical factors such as protein-protein interaction—involving CYP interaction with redox partners and other proteins with allosteric regulatory effect [76,77,81,129], or substrate-substrate interaction—consisting in CYP activity inhibition due to substrate interference/competition mechanism, are also implicated in CYP function [25,100,140,141,142].
Transcriptional activation is described as the main process of induction of CYP genes and protein levels [143]. Yet, mRNA and protein stabilization, or inhibition of protein degradation pathways will also lead to altered levels of CYP activity [144]. Proteasome-mediated degradation, phosphorylation, and long non-coding RNA (lncRNAs)-related mechanisms, are among the non-canonical post-transcriptional regulation pathways of CYPs [145,146,147]. Induction of expression of CYP enzymes, and of DMEs in general, involves a complex expression-regulation network dependent on cell-membrane- and nuclear-receptors, promoter-regulation sequences of the cis-acting elements, and of trans-acting activators and repressors, which may be shared among the same enzyme family and between different DME families. Expression of genes encoding CYPs involved in xenobiotic metabolism (mainly of CYP families 1–4) is highly inducible and can be transcriptionally activated by xenobiotics through xenobiotic receptor-dependent mechanisms (Figure 5) [26,148]. Several receptors mediate the induction of these CYPs, such as: the aryl hydrocarbon receptor (AhR)—CYP1 genes; the pregnane nuclear receptor (PXR)—CYP2A6, 2B, 2C, and 3A genes; the constitutive androstane receptor (CAR)—CYP1A, 2A6, 2B, 2C8, 2C9 and 3A4 genes [5,144,148,149,150,151,152,153]. Typically, CYP1A1 and 1A2 are highly inducible by numerous xenobiotics that act as AhR ligands. Genes of these two CYP1A members are arranged in a head-to-head orientation, sharing a common bi-directional promoter with at least 13 AhR response elements [154,155]. Additionally, transactivation of both CYP1A promoters by CAR is also possible through a common cis-regulatory estrogen receptor element (ER8) in the 5′-flanking region [149]. Transcriptional regulation of CYP2A6 gene involves PXR and CAR activators via direct repeat 4 (DR4) elements [150]. In vivo and in vitro studies are indicative that transcriptional upregulation of CYP2A6 may also occurs through an estrogen receptor-dependent pathway [156,157]. CYP2B6 expression is majorly regulated by an orphan CAR (NR113) via a phenobarbital-responsive enhancer module (PBREM), while PXR (NR112) contributes to smaller fraction in CYP2B6 induction through a distal xenobiotics-responsive enhancer module (XREM) [151,152]. The CYP2C genes are variable in their relative inducibility, which is dependent on ligands of the PXR and CAR, glucocorticoid (GR), and vitamin D nuclear receptor (VDR) pathways [158,159]. CYP2C9 is the highest expressed member of this subfamily in the liver, requiring cross-talk between distal PXR and CAR sites and proximal hepatocyte nuclear factor 4α (HNF4α) binding sites in its promoter [160,161]. The proximal PXR responsive element (prPXRE), REM, and the constitutive liver enhancer module 4 (CLEM4) are cis-regulatory elements responsible for inducible transcriptional regulation of CYP3A genes via xenosensors PXR and CAR [153,160,162]. Peroxisome proliferator-activated receptor (PPAR) also contributes to inducible and constitutive regulation of CYP3A4, the major expressed member of the subfamily [163]. Constitutive transcriptional regulators of CYP3A genes include members of CCAAT/enhancer-binding proteins (C/EBP) and HNFs [161,164,165]. Although most of the studies on inducibility of expression of CYP4A subfamily genes have been performed in mouse and rat models, accumulated data are indicative that PPAR mediates induction of expression of members of this subfamily in humans [116]. Expression of the CYP4F2 gene seems to be transactivated by the sterol regulatory element-binding protein (SREBP) and AMP-activated protein kinase (AMPK) activators [118,166].
Inhibition of CYP enzymes impairs the biotransformation of clinically used drugs or environmental compounds, resulting in higher plasma concentrations of these xenobiotics, which may lead to toxicity (Figure 4) [130,144]. On the other end, if the compound is a prodrug, the efficacy of the therapeutic regimen could be decreased as concentrations of the metabolite (the active drug), may fall below effective levels. Inhibitors of CYP enzymes can be classified into three mechanistically distinct groups, namely agents that form (i) reversible complexes (competitive or noncompetitive), (ii) quasi-irreversible complexes with the heme-iron atom, (iii) irreversible complexes through covalent binding to particular residues of the CYP protein. The latter disrupt critical interactions with its redox partners, i.e., CPR or b5, or of the heme moiety, accelerating degradation and/or oxidative fragmentation of the prosthetic heme [81,140,141,142]. In competitive reversible inhibition two structurally different molecules transiently compete for the same CYP isoenzyme irrespective of whether these are substrates for the enzyme. In noncompetitive reversible inhibition, a molecule binds to a site other than the active site, causing allosteric modulation of CYP function [5,140].
Dietary factors such as phytochemicals affect CYP expression and activity which may be of importance in diet-drug interactions. Several studies evidenced the inhibitory properties of flavonoids by structural interference with CYP proteins [167]. Additionally, soy components seem to promiscuously modulate several nuclear receptors including AhR, PXR, PPAR and liver X receptor (LXR), altering drug pharmacokinetics and therapeutic efficacy [168] (Figure 5). Other factors have been described to be involved in the induction or inhibition of CYP enzymes, in particularly conditions implying underlying chronic inflammation, such as bacterial, parasitic or viral infections (HIV, hepatitis C), sepsis, rheumatoid arthritis, liver transplant, multiple myeloma, chronic liver disease and cancer [130,131]. Proinflammatory states with secretion of large amounts of cytokines seem to be implicated in divergent xenobiotic metabolism. Cytokines such as interleukins IL-1β, IL-2, IL-4, IL-6, IL-10, and IL-23, interferon gamma (IFNγ), transforming growth factor beta (TGFβ), tumor necrosis factor alpha (TNFα), but also factors involved in infection such as lipopolysaccharides (LPS), have been reported to directly or indirectly modulate CYP expression, as demonstrated in hepatic cell lines, animal models and humans (e.g., patients with cancer undergoing immunotherapy) [130,131,142,169,170,171,172,173,174]. These inflammation factors generally exert a down-regulation of CYP expression, although in some cases, the reverse was noted. The importance of a particular cytokine in CYP regulation will depend on many factors, including its concentration in the liver, particularly in the vicinity of the hepatocytes, time course of its production, modulation by other cytokines, and concentrations of natural antagonists (e.g., IL-1-ra) and facilitators (e.g., specific soluble receptors) [25,131,174]. Cross-talks between cytokine levels and xenobiotic nuclear receptors (i.e., PXR and CAR) have been reported to contribute to the modulation of the CYP activity. During inflammation, nuclear factor κB (NF-κB) represses both PXR and CYP expression through protein-protein interactions with the PXR pathway. Upregulation of protein kinase C (PKC) and cAMP-dependent protein kinase A (PKA) seem to be involved in the repression of CYP expression associated with liver inflammation [146,175]. Moreover, extra-hepatic conditions (e.g., infection, tumors) involving inflammation also reduce the capacity of hepatic drug metabolism due to downregulation of hepatic CYP expression, probably mediated via inflammatory cytokines released by remotely inflamed organs, reaching the liver via systemic circulation [176,177].
8. Final Remarks
This short review is intended to highlight main aspects of the central role of CYP enzymes in xenobiotic metabolism, of high significance in clinical pharmacology and toxicology. Although with over seven decades of research and major scientific breakthroughs, several questions and challenges still maintain regarding CYP-mediated metabolism. Current research is mainly focused on obtaining more detailed and precise knowledge regarding: (i) functional properties and differences of CYP isoenzymes; (ii) their interspecies functional variance; (iii) tissue distribution and cellular location; (iv) regulatory mechanisms of gene expression; (v) population genetic and epigenetic determinants and variability; (vi) physio-pathological and environmental factors influencing expression and activity; (vii) genotype–phenotype correlation; (viii) and overall clinical impact. The scientific literature cited in this short-review, and many more studies not referred, are evidence of the remarkable efforts and achievements in understanding CYPs as the central Phase I enzyme family responsible for the oxidative biotransformation of xenobiotics, integrated in a vast and complex physiological detoxification network.
Author Contributions
Bibliographic analysis, F.E. and M.K.; writing—original draft preparation, F.E. and M.K.; writing—review and editing, all authors; funding acquisition, M.K. and J.R. All authors have read and agreed to the published version of the manuscript.
Funding
The elaboration of this manuscript was in part funded by grant UID/BIM/0009/2020 of the Portuguese Fundação para a Ciência e a Tecnologia (FCT) for the Research Center for Toxicogenomics and Human Health (ToXomics).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Croom, E. Metabolism of Xenobiotics of Human Environments. In Progress in Molecular Biology and Translational Science; Ernest, H., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 112, pp. 31–88. ISBN 978-0-12-415813-9. [Google Scholar]
- Juchau, M.R.; Chen, H. Developmental Enzymology. In Handbook of Developmental Neurotoxicology; William, S., Jr., Louis, W.C., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; pp. 321–337. ISBN 978-0-12-648860-9. [Google Scholar]
- Evans, T.J. Chapter 2—Toxicokinetics and Toxicodynamics. In Small Animal Toxicology, 3rd ed.; Peterson, M.E., Talcott, P.A., Eds.; W.B. Saunders: Saint Louis, MO, USA, 2013; pp. 13–19. ISBN 978-1-4557-0717-1. [Google Scholar]
- Johnson, C.H.; Patterson, A.D.; Idle, J.R.; Gonzalez, F.J. Xenobiotic Metabolomics: Major Impact on the Metabolome. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Stanley, L.A. Drug Metabolism. In Pharmacognosy; Simone, B., Rupika, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 527–545. ISBN 978-0-12-802104-0. [Google Scholar]
- Rendic, S.; Guengerich, F.P. Survey of Human Oxidoreductases and Cytochrome P450 Enzymes Involved in the Metabolism of Xenobiotic and Natural Chemicals. Chem. Res. Toxicol. 2015, 28, 38–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.F.; Ioannides, C.; Parke, D.V. Cytochromes P450 and Species Differences in Xenobiotic Metabolism and Activation of Carcinogen. Environ. Health Perspect. 1998, 106, 633–641. [Google Scholar] [CrossRef]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Bachmann, K. Drug Metabolism. In Pharmacology; Miles, H., William, M., Kenneth, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 131–173. ISBN 978-0-12-369521-5. [Google Scholar]
- Tillement, J.-P.; Tremblay, D. Clinical Pharmacokinetic Criteria for Drug Research. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 11–30. ISBN 978-0-08-045044-5. [Google Scholar]
- Murray, M.; Zhou, F. Trafficking and Other Regulatory Mechanisms for Organic Anion Transporting Polypeptides and Organic Anion Transporters That Modulate Cellular Drug and Xenobiotic Influx and That Are Dysregulated in Disease. Br. J. Pharmacol. 2017, 174, 1908–1924. [Google Scholar] [CrossRef] [Green Version]
- Janov, P.; Iller, M. Phase II Drug Metabolism. In Topics on Drug Metabolism; Paxton, J., Ed.; InTech: London, UK, 2012; pp. 35–60. ISBN 978-953-51-0099-7. [Google Scholar]
- Guengerich, F.P. Cytochrome P450 and Chemical Toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Strolin Benedetti, M.; Tipton, K.F.; Whomsley, R. Amine Oxidases and Monooxygenases in the in Vivo Metabolism of Xenobiotic Amines in Humans: Has the Involvement of Amine Oxidases Been Neglected? Amine Oxidases Monooxygenases Amine Metab. Hum. Fundam. Clin. Pharmacol. 2007, 21, 467–480. [Google Scholar] [CrossRef]
- Gan, J.; Ma, S.; Zhang, D. Non-Cytochrome P450-Mediated Bioactivation and Its Toxicological Relevance. Drug Metab. Rev. 2016, 48, 473–501. [Google Scholar] [CrossRef] [PubMed]
- Furge, L.L.; Guengerich, F.P. Cytochrome P450 Enzymes in Drug Metabolism and Chemical Toxicology: An Introduction. Biochem. Mol. Biol. Educ. 2006, 34, 66–74. [Google Scholar] [CrossRef]
- Bernhardt, R. Cytochromes P450 as Versatile Biocatalysts. J. Biotechnol. 2006, 124, 128–145. [Google Scholar] [CrossRef]
- Grillo, M.P. Bioactivation by Phase-II-Enzyme-Catalyzed Conjugation of Xenobiotics. In Encyclopedia of Drug Metabolism and Interactions; Lyubimov, A.V., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; ISBN 978-0-470-92192-0. [Google Scholar] [CrossRef]
- Iyanagi, T. Molecular Mechanism of Phase I and Phase II Drug-Metabolizing Enzymes: Implications for Detoxification. In International Review of Cytology; Elsevier: Amsterdam, The Netherlands, 2007; Volume 260, pp. 35–112. ISBN 978-0-12-374114-1. [Google Scholar]
- Roy, U.; Barber, P.; Tse-Dinh, Y.-C.; Batrakova, E.V.; Mondal, D.; Nair, M. Role of MRP Transporters in Regulating Antimicrobial Drug Inefficacy and Oxidative Stress-Induced Pathogenesis during HIV-1 and TB Infections. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Döring, B.; Petzinger, E. Phase 0 and Phase III Transport in Various Organs: Combined Concept of Phases in Xenobiotic Transport and Metabolism. Drug Metab. Rev. 2014, 46, 261–282. [Google Scholar] [CrossRef]
- Petzinger, E.; Geyer, J. Drug Transporters in Pharmacokinetics. Naunyn. Schmiedebergs Arch. Pharmacol. 2006, 372, 465–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, K. Cytochrome P450 and Anticancer Drugs. Curr. Drug Metab. 2006, 7, 23–37. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Human Cytochrome P450 Enzymes. In Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz de Montellano, P.R., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 523–785. ISBN 978-3-319-12108-6. [Google Scholar]
- Frederiks, C.N.; Lam, S.W.; Guchelaar, H.J.; Boven, E. Genetic Polymorphisms and Paclitaxel- or Docetaxel-Induced Toxicities: A Systematic Review. Cancer Treat. Rev. 2015, 41, 935–950. [Google Scholar] [CrossRef]
- Stingl, J.C.; Brockmöller, J.; Viviani, R. Genetic Variability of Drug-Metabolizing Enzymes: The Dual Impact on Psychiatric Therapy and Regulation of Brain Function. Mol. Psychiatry 2013, 18, 273–287. [Google Scholar] [CrossRef]
- Annalora, A.J.; Marcus, C.B.; Iversen, P.L. Alternative Splicing in the Cytochrome P450 Superfamily Expands Protein Diversity to Augment Gene Function and Redirect Human Drug Metabolism. Drug Metab. Dispos. 2017, 45, 375–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebert, D.W.; Nelson, D.R.; Feyereisen, R. Evolution of the Cytochrome P450 Genes. Xenobiotica Fate Foreign Compd. Biol. Syst. 1989, 19, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Omura, T. Future Perception in P450 Research. J. Inorg. Biochem. 2018, 186, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Substrate Oxidation by Cytochrome P450 Enzymes. In Cytochrome P450; Ortiz de Montellano, P.R., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 111–176. ISBN 978-3-319-12107-9. [Google Scholar]
- Estabrook, R.W. A Passion for P450s (Rememberances of the Early History of Research on Cytochrome P450). Drug Metab. Dispos. Biol. Fate Chem. 2003, 31, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Mueller, G.C.; Miller, J.A. The Metabolism of 4-Dimethylaminoazobenzene by Rat Liver Homogenates. J. Biol. Chem. 1948, 176, 535–544. [Google Scholar] [CrossRef]
- Klingenberg, M. Pigments of Rat Liver Microsomes. Arch. Biochem. Biophys. 1958, 75, 376–386. [Google Scholar] [CrossRef]
- Garfinkel, D. Studies on Pig Liver Microsomes. I. Enzymic and Pigment Composition of Different Microsomal Fractions. Arch. Biochem. Biophys. 1958, 77, 493–509. [Google Scholar] [CrossRef]
- Omura, T.; Sato, R. A New Cytochrome in Liver Microsomes. J. Biol. Chem. 1962, 237, 1375–1376. [Google Scholar] [CrossRef]
- Conney, A.H. Pharmacological Implications of Microsomal Enzyme Induction. Pharmacol. Rev. 1967, 19, 317–366. [Google Scholar]
- Hildebrandt, A.; Remmer, H.; Estabrook, R.W. Cytochrome P-450 of Liver Microsomes—One Pigment or Many. Biochem. Biophys. Res. Commun. 1968, 30, 607–612. [Google Scholar] [CrossRef]
- Gillette, J.R.; Davis, D.C.; Sasame, H.A. Cytochrome P-450 and Its Role in Drug Metabolism. Annu. Rev. Pharmacol. 1972, 12, 57–84. [Google Scholar] [CrossRef]
- Wada, O.; Yano, Y. Adaptive Responses of the Liver to Foreign Compounds, with Special Reference to Microsomal Drug-Matabolizing Enzymes. Rev. Environ. Health 1974, 1, 261–282. [Google Scholar] [PubMed]
- Parke, D.V. Induction of the Drug-Metabolizing Enzymes. Basic Life Sci. 1975, 6, 207–271. [Google Scholar] [CrossRef] [PubMed]
- Estabrook, R.W.; Cooper, D.Y.; Rosenthal, O. The Light Reversible Carbon Monoxide Inhibition of the Steroid C21-Hydroxylase System of the Adrenal Cortex. Biochem. Z. 1963, 338, 741–755. [Google Scholar]
- Cooper, D.Y.; Estabrook, R.W.; Rosenthal, O. The Stoichiometry of C21 Hydroxylation of Steroids by Adrenocortical Microsomes. J. Biol. Chem. 1963, 238, 1320–1323. [Google Scholar] [CrossRef]
- Lu, A.Y.; Coon, M.J. Role of Hemoprotein P-450 in Fatty Acid Omega-Hydroxylation in a Soluble Enzyme System from Liver Microsomes. J. Biol. Chem. 1968, 243, 1331–1332. [Google Scholar] [CrossRef]
- Imai, Y.; Sato, R. A Gel-Electrophoretically Homogeneous Preparation of Cytochrome P-450 from Liver Microsomes of Phenobarbital-Pretreated Rabbits. Biochem. Biophys. Res. Commun. 1974, 60, 8–14. [Google Scholar] [CrossRef]
- Haugen, D.A.; van der Hoeven, T.A.; Coon, M.J. Purified Liver Microsomal Cytochrome P-450. Separation and Characterization of Multiple Forms. J. Biol. Chem. 1975, 250, 3567–3570. [Google Scholar] [CrossRef]
- Alvares, A.P.; Schilling, G.; Levin, W.; Kuntzman, R. Studies on the Induction of CO-Binding Pigments in Liver Microsomes by Phenobarbital and 3-Methylcholanthrene. Biochem. Biophys. Res. Commun. 1967, 29, 521–526. [Google Scholar] [CrossRef]
- Sladek, N.E.; Mannering, G.J. Induction of Drug Metabolism. II. Qualitative Differences in the Microsomal N-Demethylating Systems Stimulated by Polycyclic Hydrocarbons and by Phenobarbital. Mol. Pharmacol. 1969, 5, 186–199. [Google Scholar] [PubMed]
- Schenkman, J.B.; Remmer, H.; Estabrook, R.W. Spectral Studies of Drug Interaction with Hepatic Microsomal Cytochrome. Mol. Pharmacol. 1967, 3, 113–123. [Google Scholar] [PubMed]
- Katagiri, M.; Ganguli, B.N.; Gunsalus, I.C. A Soluble Cytochrome P-450 Functional in Methylene Hydroxylation. J. Biol. Chem. 1968, 243, 3543–3546. [Google Scholar] [CrossRef]
- Groves, J.T.; McClusky, G.A. Aliphatic Hydroxylation by Highly Purified Liver Microsomal Cytochrome P-450. Evidence for a Carbon Radical Intermediate. Biochem. Biophys. Res. Commun. 1978, 81, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Mizukami, Y.; Sogawa, K.; Suwa, Y.; Muramatsu, M.; Fujii-Kuriyama, Y. Gene Structure of a Phenobarbital-Inducible Cytochrome P-450 in Rat Liver. Proc. Natl. Acad. Sci. USA 1983, 80, 3958–3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebert, D.W.; Gonzalez, F.J. P450 Genes: Structure, Evolution, and Regulation. Annu. Rev. Biochem. 1987, 56, 945–993. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Ingelman-Sundberg, M.; Evans, W.E. Pharmacogenomics and Individualized Drug Therapy. Annu. Rev. Med. 2006, 57, 119–137. [Google Scholar] [CrossRef]
- Johansson, I.; Ingelman-Sundberg, M. Genetic Polymorphism and Toxicology—With Emphasis on Cytochrome P450. Toxicol. Sci. 2011, 120, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solus, J.F.; Arietta, B.J.; Harris, J.R.; Sexton, D.P.; Steward, J.Q.; McMunn, C.; Ihrie, P.; Mehall, J.M.; Edwards, T.L.; Dawson, E.P. Genetic Variation in Eleven Phase I Drug Metabolism Genes in an Ethnically Diverse Population. Pharmacogenomics 2004, 5, 895–931. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Feng, S. Role of Metabolic Enzymes P450 (CYP) on Activating Procarcinogen and Their Polymorphisms on the Risk of Cancers. Curr. Drug Metab. 2015, 16, 850–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; de Grassi, A.; Lee, C.; et al. Relative Impact of Nucleotide and Copy Number Variation on Gene Expression Phenotypes. Science 2007, 315, 848–853. [Google Scholar] [CrossRef] [Green Version]
- Ingelman-Sundberg, M. The Human Genome Project and Novel Aspects of Cytochrome P450 Research. Toxicol. Appl. Pharmacol. 2005, 207, 52–56. [Google Scholar] [CrossRef]
- Nelson, D.R. The Cytochrome P450 Homepage. Hum. Genomics 2009, 4, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Barnes, H.J.; Arlotto, M.P.; Waterman, M.R. Expression and Enzymatic Activity of Recombinant Cytochrome P450 17 Alpha-Hydroxylase in Escherichia Coli. Proc. Natl. Acad. Sci. USA 1991, 88, 5597–5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, J.R.; Coon, M.J.; Porter, T.D. Alcohol-Inducible Cytochrome P-450IIE1 Lacking the Hydrophobic NH2-Terminal Segment Retains Catalytic Activity and Is Membrane-Bound When Expressed in Escherichia Coli. J. Biol. Chem. 1991, 266, 7321–7324. [Google Scholar] [CrossRef]
- Li, Y.C.; Chiang, J.Y. The Expression of a Catalytically Active Cholesterol 7 Alpha-Hydroxylase Cytochrome P450 in Escherichia Coli. J. Biol. Chem. 1991, 266, 19186–19191. [Google Scholar] [CrossRef]
- Fisher, C.W.; Caudle, D.L.; Martin-Wixtrom, C.; Quattrochi, L.C.; Tukey, R.H.; Waterman, M.R.; Estabrook, R.W. High-Level Expression of Functional Human Cytochrome P450 1A2 in Escherichia Coli. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1992, 6, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Kranendonk, M.; Mesquita, P.; Laires, A.; Vermeulen, N.P.; Rueff, J. Expression of Human Cytochrome P450 1A2 in Escherichia Coli: A System for Biotransformation and Genotoxicity Studies of Chemical Carcinogens. Mutagenesis 1998, 13, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranendonk, M.; Carreira, F.; Theisen, P.; Laires, A.; Fisher, C.W.; Rueff, J.; Estabrook, R.W.; Vermeulen, N.P. Escherichia Coli MTC, a Human NADPH P450 Reductase Competent Mutagenicity Tester Strain for the Expression of Human Cytochrome P450 Isoforms 1A1, 1A2, 2A6, 3A4, or 3A5: Catalytic Activities and Mutagenicity Studies. Mutat. Res. 1999, 441, 73–83. [Google Scholar] [CrossRef]
- Poulos, T.L.; Finzel, B.C.; Gunsalus, I.C.; Wagner, G.C.; Kraut, J. The 2.6-A Crystal Structure of Pseudomonas Putida Cytochrome P-450. J. Biol. Chem. 1985, 260, 16122–16130. [Google Scholar] [CrossRef]
- Poulos, T.L.; Finzel, B.C.; Howard, A.J. High-Resolution Crystal Structure of Cytochrome P450cam. J. Mol. Biol. 1987, 195, 687–700. [Google Scholar] [CrossRef]
- Ravichandran, K.G.; Boddupalli, S.S.; Hasermann, C.A.; Peterson, J.A.; Deisenhofer, J. Crystal Structure of Hemoprotein Domain of P450BM-3, a Prototype for Microsomal P450’s. Science 1993, 261, 731–736. [Google Scholar] [CrossRef]
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Matak Vinković, D.; Jhoti, H. Crystal Structure of Human Cytochrome P450 2C9 with Bound Warfarin. Nature 2003, 424, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The Structure of Human Microsomal Cytochrome P450 3A4 Determined by X-Ray Crystallography to 2.05-A Resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef] [Green Version]
- Rowland, P.; Blaney, F.E.; Smyth, M.G.; Jones, J.J.; Leydon, V.R.; Oxbrow, A.K.; Lewis, C.J.; Tennant, M.G.; Modi, S.; Eggleston, D.S.; et al. Crystal Structure of Human Cytochrome P450 2D6. J. Biol. Chem. 2006, 281, 7614–7622. [Google Scholar] [CrossRef] [Green Version]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the Oxidation of Polycyclic Aromatic Hydrocarbons Exhibited by the Structure of Human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, M.P.; Palma, B.B.; Gilep, A.A.; Laires, A.; Oliveira, J.S.; Usanov, S.A.; Rueff, J.; Kranendonk, M. The Stimulatory Role of Human Cytochrome b5 in the Bioactivation Activities of Human CYP1A2, 2A6 and 2E1: A New Cell Expression System to Study Cytochrome P450-Mediated Biotransformation (a Corrigendum Report on Duarte et al. (2005) Mutagenesis 20, 93–100). Mutagenesis 2007, 22, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Palma, B.B.; Silva, E.; Sousa, M.; Urban, P.; Rueff, J.; Kranendonk, M. Functional Characterization of Eight Human CYP1A2 Variants: The Role of Cytochrome b5. Pharmacogenet. Genomics 2013, 23, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Nakamura, M.; Komatsu, T.; Ohyama, K.; Hatanaka, N.; Asahi, S.; Shimada, N.; Guengerich, F.P.; Shimada, T.; Nakajima, M.; et al. Roles of NADPH-P450 Reductase and Apo- and Holo-Cytochrome b5 on Xenobiotic Oxidations Catalyzed by 12 Recombinant Human Cytochrome P450s Expressed in Membranes of Escherichia Coli. Protein Expr. Purif. 2002, 24, 329–337. [Google Scholar] [CrossRef]
- Yamaori, S.; Yamazaki, H.; Suzuki, A.; Yamada, A.; Tani, H.; Kamidate, T.; Fujita, K.; Kamataki, T. Effects of Cytochrome b(5) on Drug Oxidation Activities of Human Cytochrome P450 (CYP) 3As: Similarity of CYP3A5 with CYP3A4 but Not CYP3A7. Biochem. Pharmacol. 2003, 66, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Storbeck, K.-H.; Swart, A.C.; Fox, C.L.; Swart, P. Cytochrome b5 Modulates Multiple Reactions in Steroidogenesis by Diverse Mechanisms. J. Steroid Biochem. Mol. Biol. 2015, 151, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Hlavica, P. Mechanistic Basis of Electron Transfer to Cytochromes P450 by Natural Redox Partners and Artificial Donor Constructs. Adv. Exp. Med. Biol. 2015, 851, 247–297. [Google Scholar] [CrossRef] [PubMed]
- Waskell, L.; Kim, J.-J.P. Electron Transfer Partners of Cytochrome P450. In Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz de Montellano, P.R., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 33–68. ISBN 978-3-319-12108-6. [Google Scholar]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human Cytochromes P450 in Health and Disease. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Adesnik, M.; Coon, M.J.; Estabrook, R.W.; Gonzalez, F.J.; Guengerich, F.P.; Gunsalus, I.C.; Johnson, E.F.; Kemper, B.; Levin, W. The P450 Gene Superfamily: Recommended Nomenclature. DNA 1987, 6, 1–11. [Google Scholar] [CrossRef]
- Maron, D.M.; Ames, B.N. Revised Methods for the Salmonella Mutagenicity Test. Mutat. Res. 1983, 113, 173–215. [Google Scholar] [CrossRef]
- Rueff, J.; Rodrigues, A.S.; Kranendonk, M. A Personally Guided Tour on Some of Our Data with the Ames Assay-A Tribute to Professor Bruce Ames. Mutat. Res. 2019, 846, 503094. [Google Scholar] [CrossRef]
- Hakura, A.; Shimada, H.; Nakajima, M.; Sui, H.; Kitamoto, S.; Suzuki, S.; Satoh, T. Salmonella/Human S9 Mutagenicity Test: A Collaborative Study with 58 Compounds. Mutagenesis 2005, 20, 217–228. [Google Scholar] [CrossRef]
- Baillie, T.A.; Rettie, A.E. Role of Biotransformation in Drug-Induced Toxicity: Influence of Intra- and Inter-Species Differences in Drug Metabolism. Drug Metab. Pharmacokinet. 2011, 26, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Helsby, N.; Palmer, B.D.; Tingle, M.; Baguley, B.C.; Kestell, P.; Ching, L.-M. Metabolism of Thalidomide in Liver Microsomes of Mice, Rabbits, and Humans. J. Pharmacol. Exp. Ther. 2004, 310, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Gordon, G.B.; Spielberg, S.P.; Blake, D.A.; Balasubramanian, V. Thalidomide Teratogenesis: Evidence for a Toxic Arene Oxide Metabolite. Proc. Natl. Acad. Sci. USA 1981, 78, 2545–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D.E.; Buhler, D.R. Purified Form of Cytochrome P-450 from Rainbow Trout with High Activity toward Conversion of Aflatoxin B1 to Aflatoxin B1-2,3-Epoxide. Cancer Res. 1983, 43, 4752–4756. [Google Scholar]
- Eaton, D.L.; Gallagher, E.P. Mechanisms of Aflatoxin Carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 135–172. [Google Scholar] [CrossRef]
- Nelson, D.R. Cytochrome P450 Nomenclature. Methods Mol. Biol. Clifton N. J. 1998, 107, 15–24. [Google Scholar] [CrossRef]
- Nelson, D.R. Progress in Tracing the Evolutionary Paths of Cytochrome P450. Biochim. Biophys. Acta 2011, 1814, 14–18. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Waterman, M.R.; Egli, M. Recent Structural Insights into Cytochrome P450 Function. Trends Pharmacol. Sci. 2016, 37, 625–640. [Google Scholar] [CrossRef] [Green Version]
- Omura, T. Forty Years of Cytochrome P450. Biochem. Biophys. Res. Commun. 1999, 266, 690–698. [Google Scholar] [CrossRef]
- Collins, S.L.; Patterson, A.D. The Gut Microbiome: An Orchestrator of Xenobiotic Metabolism. Acta Pharm. Sin. B 2020, 10, 19–32. [Google Scholar] [CrossRef]
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Mapping Human Microbiome Drug Metabolism by Gut Bacteria and Their Genes. Nature 2019, 570, 462–467. [Google Scholar] [CrossRef]
- Selwyn, F.P.; Cheng, S.L.; Klaassen, C.D.; Cui, J.Y. Regulation of Hepatic Drug-Metabolizing Enzymes in Germ-Free Mice by Conventionalization and Probiotics. Drug Metab. Dispos. Biol. Fate Chem. 2016, 44, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Aleksunes, L.M.; Klaassen, C.D. Coordinated Regulation of Hepatic Phase I and II Drug-Metabolizing Genes and Transporters Using AhR-, CAR-, PXR-, PPARα-, and Nrf2-Null Mice. Drug Metab. Dispos. Biol. Fate Chem. 2012, 40, 1366–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.F. On the Recognition of Mammalian Microsomal Cytochrome P450 Substrates and Their Characteristics: Towards the Prediction of Human P450 Substrate Specificity and Metabolism. Biochem. Pharmacol. 2000, 60, 293–306. [Google Scholar] [CrossRef]
- Urban, P.; Truan, G.; Pompon, D. Access Channels to the Buried Active Site Control Substrate Specificity in CYP1A P450 Enzymes. Biochim. Biophys. Acta 2015, 1850, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A. Interindividual Differences in Caffeine Metabolism and Factors Driving Caffeine Consumption. Pharmacol. Rev. 2018, 70, 384–411. [Google Scholar] [CrossRef] [Green Version]
- Song, M.-K.; Yoon, J.-S.; Song, M.; Choi, H.-S.; Shin, C.-Y.; Kim, Y.-J.; Ryu, W.-I.; Lee, H.-S.; Ryu, J.-C. Gene Expression Analysis Identifies DNA Damage-Related Markers of Benzo[a]Pyrene Exposure in HepG2 Human Hepatocytes. Toxicol. Environ. Health Sci. 2012, 4, 19–29. [Google Scholar] [CrossRef]
- Barnes, J.L.; Zubair, M.; John, K.; Poirier, M.C.; Martin, F.L. Carcinogens and DNA Damage. Biochem. Soc. Trans. 2018, 46, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Pathways of Acetaminophen Metabolism at the Therapeutic versus Toxic Doses. Pharmacogenet. Genomics 2015, 25, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Guan, X.; Dai, Z.; He, R.; Ding, X.; Yang, L.; Ge, G. Molecular Probes for Human Cytochrome P450 Enzymes: Recent Progress and Future Perspectives. Coord. Chem. Rev. 2021, 427, 213600. [Google Scholar] [CrossRef]
- Isin, E.M.; Guengerich, F.P. Complex Reactions Catalyzed by Cytochrome P450 Enzymes. Biochim. Biophys. Acta 2007, 1770, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Masters, B.S.S. The Journey from NADPH-Cytochrome P450 Oxidoreductase to Nitric Oxide Synthases. Biochem. Biophys. Res. Commun. 2005, 338, 507–519. [Google Scholar] [CrossRef]
- Hamdane, D.; Xia, C.; Im, S.-C.; Zhang, H.; Kim, J.-J.P.; Waskell, L. Structure and Function of an NADPH-Cytochrome P450 Oxidoreductase in an Open Conformation Capable of Reducing Cytochrome P450. J. Biol. Chem. 2009, 284, 11374–11384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aigrain, L.; Pompon, D.; Moréra, S.; Truan, G. Structure of the Open Conformation of a Functional Chimeric NADPH Cytochrome P450 Reductase. EMBO Rep. 2009, 10, 742–747. [Google Scholar] [CrossRef] [Green Version]
- Ellis, J.; Gutierrez, A.; Barsukov, I.L.; Huang, W.-C.; Grossmann, J.G.; Roberts, G.C.K. Domain Motion in Cytochrome P450 Reductase: Conformational Equilibria Revealed by NMR and Small-Angle x-Ray Scattering. J. Biol. Chem. 2009, 284, 36628–36637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campelo, D.; Lautier, T.; Urban, P.; Esteves, F.; Bozonnet, S.; Truan, G.; Kranendonk, M. The Hinge Segment of Human NADPH-Cytochrome P450 Reductase in Conformational Switching: The Critical Role of Ionic Strength. Front. Pharmacol. 2017, 8, 755. [Google Scholar] [CrossRef]
- Campelo, D.; Esteves, F.; Brito Palma, B.; Costa Gomes, B.; Rueff, J.; Lautier, T.; Urban, P.; Truan, G.; Kranendonk, M. Probing the Role of the Hinge Segment of Cytochrome P450 Oxidoreductase in the Interaction with Cytochrome P450. Int. J. Mol. Sci. 2018, 19, 3914. [Google Scholar] [CrossRef] [Green Version]
- Rendic, S.; Guengerich, F.P. Contributions of Human Enzymes in Carcinogen Metabolism. Chem. Res. Toxicol. 2012, 25, 1316–1383. [Google Scholar] [CrossRef]
- Jin, Y.; Zollinger, M.; Borell, H.; Zimmerlin, A.; Patten, C.J. CYP4F Enzymes Are Responsible for the Elimination of Fingolimod (FTY720), a Novel Treatment of Relapsing Multiple Sclerosis. Drug Metab. Dispos. Biol. Fate Chem. 2011, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Edson, K.Z.; Rettie, A.E. CYP4 Enzymes as Potential Drug Targets: Focus on Enzyme Multiplicity, Inducers and Inhibitors, and Therapeutic Modulation of 20-Hydroxyeicosatetraenoic Acid (20-HETE) Synthase and Fatty Acid ω-Hydroxylase Activities. Curr. Top. Med. Chem. 2013, 13, 1429–1440. [Google Scholar] [CrossRef]
- Seguin, R.P.; Herron, J.M.; Lopez, V.A.; Dempsey, J.L.; Xu, L. Metabolism of Benzalkonium Chlorides by Human Hepatic Cytochromes P450. Chem. Res. Toxicol. 2019, 32, 2466–2478. [Google Scholar] [CrossRef]
- Hsu, M.-H.; Savas, U.; Lasker, J.M.; Johnson, E.F. Genistein, Resveratrol, and 5-Aminoimidazole-4-Carboxamide-1-β-D-Ribofuranoside Induce Cytochrome P450 4F2 Expression through an AMP-Activated Protein Kinase-Dependent Pathway. J. Pharmacol. Exp. Ther. 2011, 337, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Wahlang, B.; Falkner, K.C.; Cave, M.C.; Prough, R.A. Role of Cytochrome P450 Monooxygenase in Carcinogen and Chemotherapeutic Drug Metabolism. Adv. Pharmacol. San Diego Calif 2015, 74, 1–33. [Google Scholar] [CrossRef]
- Stepanov, I.; Upadhyaya, P.; Carmella, S.G.; Feuer, R.; Jensen, J.; Hatsukami, D.K.; Hecht, S.S. Extensive Metabolic Activation of the Tobacco-Specific Carcinogen 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanone in Smokers. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1764–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guengerich, F.P. Principles of Covalent Binding of Reactive Metabolites and Examples of Activation of Bis-Electrophiles by Conjugation. Arch. Biochem. Biophys. 2005, 433, 369–378. [Google Scholar] [CrossRef]
- Mizutani, T. PM Frequencies of Major CYPs in Asians and Caucasians. Drug Metab. Rev. 2003, 35, 99–106. [Google Scholar] [CrossRef]
- Distlerath, L.M.; Reilly, P.E.; Martin, M.V.; Davis, G.G.; Wilkinson, G.R.; Guengerich, F.P. Purification and Characterization of the Human Liver Cytochromes P-450 Involved in Debrisoquine 4-Hydroxylation and Phenacetin O-Deethylation, Two Prototypes for Genetic Polymorphism in Oxidative Drug Metabolism. J. Biol. Chem. 1985, 260, 9057–9067. [Google Scholar] [CrossRef]
- Chun, Y.-J.; Kim, D. Cancer Activation and Polymorphisms of Human Cytochrome P450 1B1. Toxicol. Res. 2016, 32, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Ingelman-Sundberg, M.; Sim, S.C.; Gomez, A.; Rodriguez-Antona, C. Influence of Cytochrome P450 Polymorphisms on Drug Therapies: Pharmacogenetic, Pharmacoepigenetic and Clinical Aspects. Pharmacol. Ther. 2007, 116, 496–526. [Google Scholar] [CrossRef]
- Burkhard, F.Z.; Parween, S.; Udhane, S.S.; Flück, C.E.; Pandey, A.V. P450 Oxidoreductase Deficiency: Analysis of Mutations and Polymorphisms. J. Steroid Biochem. Mol. Biol. 2017, 165, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udhane, S.S.; Parween, S.; Kagawa, N.; Pandey, A.V. Altered CYP19A1 and CYP3A4 Activities Due to Mutations A115V, T142A, Q153R and P284L in the Human P450 Oxidoreductase. Front. Pharmacol. 2017, 8, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteves, F.; Campelo, D.; Gomes, B.C.; Urban, P.; Bozonnet, S.; Lautier, T.; Rueff, J.; Truan, G.; Kranendonk, M. The Role of the FMN-Domain of Human Cytochrome P450 Oxidoreductase in Its Promiscuous Interactions With Structurally Diverse Redox Partners. Front. Pharmacol. 2020, 11, 299. [Google Scholar] [CrossRef] [Green Version]
- Esteves, F.; Urban, P.; Rueff, J.; Truan, G.; Kranendonk, M. Interaction Modes of Microsomal Cytochrome P450s with Its Reductase and the Role of Substrate Binding. Int. J. Mol. Sci. 2020, 21, 6669. [Google Scholar] [CrossRef]
- Morgan, E. Impact of Infectious and Inflammatory Disease on Cytochrome P450–Mediated Drug Metabolism and Pharmacokinetics. Clin. Pharmacol. Ther. 2009, 85, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.T. Regulation of Drug-Metabolizing Enzymes and Drug Metabolism by Inflammatory Responses. In Drug Metabolism in Diseases; Wen, X., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 21–58. ISBN 978-0-12-802949-7. [Google Scholar]
- Beedanagari, S.R.; Taylor, R.T.; Bui, P.; Wang, F.; Nickerson, D.W.; Hankinson, O. Role of Epigenetic Mechanisms in Differential Regulation of the Dioxin-Inducible Human CYP1A1 and CYP1B1 Genes. Mol. Pharmacol. 2010, 78, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.-Z.; Gao, W.; Yu, A.-M. MicroRNAs Regulate CYP3A4 Expression via Direct and Indirect Targeting. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37, 2112–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannenberg, L.O.; Edenberg, H.J. Epigenetics of Gene Expression in Human Hepatoma Cells: Expression Profiling the Response to Inhibition of DNA Methylation and Histone Deacetylation. BMC Genomics 2006, 7, 181. [Google Scholar] [CrossRef] [Green Version]
- Anttila, S.; Hakkola, J.; Tuominen, P.; Elovaara, E.; Husgafvel-Pursiainen, K.; Karjalainen, A.; Hirvonen, A.; Nurminen, T. Methylation of Cytochrome P4501A1 Promoter in the Lung Is Associated with Tobacco Smoking. Cancer Res. 2003, 63, 8623–8628. [Google Scholar]
- Tsuchiya, Y.; Nakajima, M.; Takagi, S.; Taniya, T.; Yokoi, T. MicroRNA Regulates the Expression of Human Cytochrome P450 1B1. Cancer Res. 2006, 66, 9090–9098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohri, T.; Nakajima, M.; Fukami, T.; Takamiya, M.; Aoki, Y.; Yokoi, T. Human CYP2E1 Is Regulated by MiR-378. Biochem. Pharmacol. 2010, 79, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamba, V.; Ghodke, Y.; Guan, W.; Tracy, T.S. MicroRNA-34a Is Associated with Expression of Key Hepatic Transcription Factors and Cytochromes P450. Biochem. Biophys. Res. Commun. 2014, 445, 404–411. [Google Scholar] [CrossRef] [Green Version]
- Ingelman-Sundberg, M.; Gomez, A. The Past, Present and Future of Pharmacoepigenomics. Pharmacogenomics 2010, 11, 625–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, M.A.; Hollenberg, P.F. Inhibition of Cytochrome P450 Enzymes. In Cytochrome P450; Ortiz de Montellano, P.R., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 177–259. ISBN 978-3-319-12107-9. [Google Scholar]
- Gentry, K.A.; Anantharamaiah, G.M.; Ramamoorthy, A. Probing Protein-Protein and Protein-Substrate Interactions in the Dynamic Membrane-Associated Ternary Complex of Cytochromes P450, b5, and Reductase. Chem. Commun. Camb. Engl. 2019, 55, 13422–13425. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Jamali, F. The Effect of Infliximab on Hepatic Cytochrome P450 and Pharmacokinetics of Verapamil in Rats with Pre-Adjuvant Arthritis: A Drug-Disease and Drug-Drug Interaction. Basic Clin. Pharmacol. Toxicol. 2009, 105, 24–29. [Google Scholar] [CrossRef]
- Tompkins, L.M.; Wallace, A.D. Mechanisms of Cytochrome P450 Induction. J. Biochem. Mol. Toxicol. 2007, 21, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, O.; Turpeinen, M.; Hakkola, J.; Honkakoski, P.; Hukkanen, J.; Raunio, H. Inhibition and Induction of Human Cytochrome P450 Enzymes: Current Status. Arch. Toxicol. 2008, 82, 667–715. [Google Scholar] [CrossRef]
- Ishihara, Y.; Hamaguchi, A.; Sekine, M.; Hirakawa, A.; Shimamoto, N. Accumulation of Cytochrome P450 Induced by Proteasome Inhibition during Cardiac Ischemia. Arch. Biochem. Biophys. 2012, 527, 16–22. [Google Scholar] [CrossRef]
- Pondugula, S.R.; Dong, H.; Chen, T. Phosphorylation and Protein-Protein Interactions in PXR-Mediated CYP3A Repression. Expert Opin. Drug Metab. Toxicol. 2009, 5, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Mahpour, A.; Mullen, A.C. Our Emerging Understanding of the Roles of Long Non-Coding RNAs in Normal Liver Function, Disease, and Malignancy. JHEP Rep. Innov. Hepatol. 2021, 3, 100177. [Google Scholar] [CrossRef] [PubMed]
- Congiu, M.; Mashford, M.L.; Slavin, J.L.; Desmond, P.V. Coordinate Regulation of Metabolic Enzymes and Transporters by Nuclear Transcription Factors in Human Liver Disease. J. Gastroenterol. Hepatol. 2009, 24, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, K.; Yoda, N.; Toriyabe, T.; Yamazoe, Y. Constitutive Androstane Receptor Transcriptionally Activates Human CYP1A1 and CYP1A2 Genes through a Common Regulatory Element in the 5’-Flanking Region. Biochem. Pharmacol. 2010, 79, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Nakajima, M.; Higashi, E.; Yoshida, R.; Nagata, K.; Yamazoe, Y.; Yokoi, T. Induction of Human CYP2A6 Is Mediated by the Pregnane X Receptor with Peroxisome Proliferator-Activated Receptor-Gamma Coactivator 1alpha. J. Pharmacol. Exp. Ther. 2006, 319, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, T.; Kawamoto, T.; Zelko, I.; Honkakoski, P.; Negishi, M. The Repressed Nuclear Receptor CAR Responds to Phenobarbital in Activating the Human CYP2B6 Gene. J. Biol. Chem. 1999, 274, 6043–6046. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Faucette, S.; Sueyoshi, T.; Moore, R.; Ferguson, S.; Negishi, M.; LeCluyse, E.L. A Novel Distal Enhancer Module Regulated by Pregnane X Receptor/Constitutive Androstane Receptor Is Essential for the Maximal Induction of CYP2B6 Gene Expression. J. Biol. Chem. 2003, 278, 14146–14152. [Google Scholar] [CrossRef] [Green Version]
- Willson, T.M.; Kliewer, S.A. PXR, CAR and Drug Metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Ueda, R.; Iketaki, H.; Nagata, K.; Kimura, S.; Gonzalez, F.J.; Kusano, K.; Yoshimura, T.; Yamazoe, Y. A Common Regulatory Region Functions Bidirectionally in Transcriptional Activation of the Human CYP1A1 and CYP1A2 Genes. Mol. Pharmacol. 2006, 69, 1924–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorge-Nebert, L.F.; Jiang, Z.; Chakraborty, R.; Watson, J.; Jin, L.; McGarvey, S.T.; Deka, R.; Nebert, D.W. Analysis of Human CYP1A1 and CYP1A2 Genes and Their Shared Bidirectional Promoter in Eight World Populations. Hum. Mutat. 2010, 31, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Benowitz, N.L.; Lessov-Schlaggar, C.N.; Swan, G.E.; Jacob, P. Female Sex and Oral Contraceptive Use Accelerate Nicotine Metabolism. Clin. Pharmacol. Ther. 2006, 79, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Higashi, E.; Fukami, T.; Itoh, M.; Kyo, S.; Inoue, M.; Yokoi, T.; Nakajima, M. Human CYP2A6 Is Induced by Estrogen via Estrogen Receptor. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 1935–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Goldstein, J.A. The Transcriptional Regulation of the Human CYP2C Genes. Curr. Drug Metab. 2009, 10, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Helsby, N.A.; Burns, K.E. Molecular Mechanisms of Genetic Variation and Transcriptional Regulation of CYP2C19. Front. Genet. 2012, 3, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsuki, S.; Schaefer, O.; Kawakami, H.; Inoue, T.; Liehner, S.; Saito, A.; Ishiguro, N.; Kishimoto, W.; Ludwig-Schwellinger, E.; Ebner, T.; et al. Simultaneous Absolute Protein Quantification of Transporters, Cytochromes P450, and UDP-Glucuronosyltransferases as a Novel Approach for the Characterization of Individual Human Liver: Comparison with MRNA Levels and Activities. Drug Metab. Dispos. Biol. Fate Chem. 2012, 40, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Jover, R.; Moya, M.; Gómez-Lechón, M.J. Transcriptional Regulation of Cytochrome P450 Genes by the Nuclear Receptor Hepatocyte Nuclear Factor 4-Alpha. Curr. Drug Metab. 2009, 10, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K.; Saito, T.; Takahashi, Y.; Ozeki, T.; Kiyotani, K.; Fujieda, M.; Yamazaki, H.; Kunitoh, H.; Kamataki, T. Identification of a Novel Polymorphic Enhancer of the Human CYP3A4 Gene. Mol. Pharmacol. 2004, 65, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Schröder, A.; Wollnik, J.; Wrzodek, C.; Dräger, A.; Bonin, M.; Burk, O.; Thomas, M.; Thasler, W.E.; Zanger, U.M.; Zell, A. Inferring Statin-Induced Gene Regulatory Relationships in Primary Human Hepatocytes. Bioinforma. Oxf. Engl. 2011, 27, 2473–2477. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Jiménez, C.P.; Gómez-Lechón, M.J.; Castell, J.V.; Jover, R. Transcriptional Regulation of the Human Hepatic CYP3A4: Identification of a New Distal Enhancer Region Responsive to CCAAT/Enhancer-Binding Protein Beta Isoforms (Liver Activating Protein and Liver Inhibitory Protein). Mol. Pharmacol. 2005, 67, 2088–2101. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Antona, C.; Bort, R.; Jover, R.; Tindberg, N.; Ingelman-Sundberg, M.; Gómez-Lechón, M.J.; Castell, J.V. Transcriptional Regulation of Human CYP3A4 Basal Expression by CCAAT Enhancer-Binding Protein Alpha and Hepatocyte Nuclear Factor-3 Gamma. Mol. Pharmacol. 2003, 63, 1180–1189. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein Sensors for Membrane Sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Santes-Palacios, R.; Marroquín-Pérez, A.L.; Hernández-Ojeda, S.L.; Camacho-Carranza, R.; Govezensky, T.; Espinosa-Aguirre, J.J. Human CYP1A1 Inhibition by Flavonoids. Toxicol. Vitro Int. J. Publ. Assoc. BIBRA 2020, 62, 104681. [Google Scholar] [CrossRef]
- Ronis, M.J.J. Effects of Soy Containing Diet and Isoflavones on Cytochrome P450 Enzyme Expression and Activity. Drug Metab. Rev. 2016, 48, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Elkahwaji, J.; Robin, M.A.; Berson, A.; Tinel, M.; Lettéron, P.; Labbe, G.; Beaune, P.; Elias, D.; Rougier, P.; Escudier, B.; et al. Decrease in Hepatic Cytochrome P450 after Interleukin-2 Immunotherapy. Biochem. Pharmacol. 1999, 57, 951–954. [Google Scholar] [CrossRef]
- Abdel-Razzak, Z.; Garlatti, M.; Aggerbeck, M.; Barouki, R. Determination of Interleukin-4-Responsive Region in the Human Cytochrome P450 2E1 Gene Promoter. Biochem. Pharmacol. 2004, 68, 1371–1381. [Google Scholar] [CrossRef]
- Gorski, J.C.; Hall, S.D.; Becker, P.; Affrime, M.B.; Cutler, D.L.; Haehner-Daniels, B. In Vivo Effects of Interleukin-10 on Human Cytochrome P450 Activity. Clin. Pharmacol. Ther. 2000, 67, 32–43. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Ukairo, O.; Khetani, S.R.; McVay, M.; Kanchagar, C.; Seghezzi, W.; Ayanoglu, G.; Irrechukwu, O.; Evers, R. Establishment of a Hepatocyte-Kupffer Cell Coculture Model for Assessment of Proinflammatory Cytokine Effects on Metabolizing Enzymes and Drug Transporters. Drug Metab. Dispos. Biol. Fate Chem. 2015, 43, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Siewert, E.; Bort, R.; Kluge, R.; Heinrich, P.C.; Castell, J.; Jover, R. Hepatic Cytochrome P450 Down-Regulation during Aseptic Inflammation in the Mouse Is Interleukin 6 Dependent. Hepatol. Baltim. Md. 2000, 32, 49–55. [Google Scholar] [CrossRef]
- Shah, R.R.; Smith, R.L. Inflammation-Induced Phenoconversion of Polymorphic Drug Metabolizing Enzymes: Hypothesis with Implications for Personalized Medicine. Drug Metab. Dispos. 2015, 43, 400–410. [Google Scholar] [CrossRef] [Green Version]
- Charles, K.A.; Rivory, L.P.; Brown, S.L.; Liddle, C.; Clarke, S.J.; Robertson, G.R. Transcriptional Repression of Hepatic Cytochrome P450 3A4 Gene in the Presence of Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 7492–7497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, K.W.; Nicholson, T.E. Hepatic and Central Nervous System Cytochrome P450 Are Down-Regulated during Lipopolysaccharide-Evoked Localized Inflammation in Brain. J. Pharmacol. Exp. Ther. 2000, 294, 524–530. [Google Scholar] [PubMed]
- Abdulla, D.; Goralski, K.B.; Renton, K.W. The Regulation of Cytochrome P450 2E1 during LPS-Induced Inflammation in the Rat. Toxicol. Appl. Pharmacol. 2006, 216, 1–10. [Google Scholar] [CrossRef]
Figure 1.
Xenobiotic metabolism in the hepatocyte and the central role of CYPs in biotransformation. Besides hydroxylation, exemplified here, CYPs catalyze a variety of other biotransformation reactions (e.g., epoxidation, dealkylation, oxygenation, dehydrogenation, dehalogenation, among others). Non-CYP mediated metabolism may also occur in Phase I via flavin-containing monooxygenases (FMOs), NAD(P)H:quinone oxidoreductases (NQOs), amine oxidases, alcohol dehydrogenases, esterases and peroxidases. ABC: ATP binding cassette (e.g., multidrug resistance protein family—MRP); GA: glucuronic acid; GSH: glutathione; NTCP: sodium taurocholate cotransporting polypeptide; OAT: organic anion transporters; OATP: organic anion transporting polypeptides; OH: hydroxyl; PAPS: phosphoadenosine-phosphosulfate; SLC: solute carrier transporters; UDPGA: uridine diphosphate-glucuronic acid. Transferases: glutathione S-transferases (GST), methyltransferases, glycine N-acyltransferase (GLYAT), N-acetyltransferases (NAT), sulfotransferases (SULT), UDP-glucuronosyltransferases (UGT).
Figure 1.
Xenobiotic metabolism in the hepatocyte and the central role of CYPs in biotransformation. Besides hydroxylation, exemplified here, CYPs catalyze a variety of other biotransformation reactions (e.g., epoxidation, dealkylation, oxygenation, dehydrogenation, dehalogenation, among others). Non-CYP mediated metabolism may also occur in Phase I via flavin-containing monooxygenases (FMOs), NAD(P)H:quinone oxidoreductases (NQOs), amine oxidases, alcohol dehydrogenases, esterases and peroxidases. ABC: ATP binding cassette (e.g., multidrug resistance protein family—MRP); GA: glucuronic acid; GSH: glutathione; NTCP: sodium taurocholate cotransporting polypeptide; OAT: organic anion transporters; OATP: organic anion transporting polypeptides; OH: hydroxyl; PAPS: phosphoadenosine-phosphosulfate; SLC: solute carrier transporters; UDPGA: uridine diphosphate-glucuronic acid. Transferases: glutathione S-transferases (GST), methyltransferases, glycine N-acyltransferase (GLYAT), N-acetyltransferases (NAT), sulfotransferases (SULT), UDP-glucuronosyltransferases (UGT).

Figure 2.
Examples of reactions catalyzed by CYPs. (A) CYP-mediated bioactivation of thalidomide. (B) CYP-mediated hydroxylation of phenol. (C) CYP-mediated metabolism of caffeine, with multiple metabolites. (D) Bioactivation of benzo[a]pyrene mediated by CYP enzymes. (E) CYP-mediated metabolism of paracetamol and its potential toxicity. (F) CYP-mediated bioactivation of NNK. (G) Bioactivation of aflatoxin B1 by CYP enzymes. AFB1: Aflatoxin B1. APAP: acetaminophen; B[a]P: benzo[a]pyrene; BPDE: benzo[a]pyrene diol epoxide; DMX: dimethylxanthine; GST: glutathione S-transferases; NAPQI: N-acetyl-p-benzoquinone imine; NNAL: 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol; NNK: nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; PST: phenol sulfotransferase; SULT: sulfotransferases; TMX: trimethylxanthine; UGT: UDP-glucuronosyltransferases.
Figure 2.
Examples of reactions catalyzed by CYPs. (A) CYP-mediated bioactivation of thalidomide. (B) CYP-mediated hydroxylation of phenol. (C) CYP-mediated metabolism of caffeine, with multiple metabolites. (D) Bioactivation of benzo[a]pyrene mediated by CYP enzymes. (E) CYP-mediated metabolism of paracetamol and its potential toxicity. (F) CYP-mediated bioactivation of NNK. (G) Bioactivation of aflatoxin B1 by CYP enzymes. AFB1: Aflatoxin B1. APAP: acetaminophen; B[a]P: benzo[a]pyrene; BPDE: benzo[a]pyrene diol epoxide; DMX: dimethylxanthine; GST: glutathione S-transferases; NAPQI: N-acetyl-p-benzoquinone imine; NNAL: 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol; NNK: nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; PST: phenol sulfotransferase; SULT: sulfotransferases; TMX: trimethylxanthine; UGT: UDP-glucuronosyltransferases.



Figure 3.
Schematic representation of the nomenclature system of CYP genes (example of CYP3A4*1).

Figure 4.
Factors influencing inter- and intra-individual variability in CYP activity and expression, and their impact on xenobiotic metabolism (CYP circles: size indicate the importance of the different inter- and intra-individual levels of expression and function of CYP enzymes).
Figure 4.
Factors influencing inter- and intra-individual variability in CYP activity and expression, and their impact on xenobiotic metabolism (CYP circles: size indicate the importance of the different inter- and intra-individual levels of expression and function of CYP enzymes).

Figure 5.
Main mechanisms of CYP induction. AhR: aryl hydrocarbon receptor; CAR: constitutive androstane receptor; CLEM4: constitutive liver enhancer module 4; DR4: direct repeat 4; ER8: estrogen receptor 8; HNF4α: hepatocyte nuclear factor 4α; PBREM: phenobarbital-responsive enhancer module; PPAR: proliferator-activated receptor; prPXRE: proximal PXR responsive element; PXR: pregnane nuclear receptor; XNR: xenobiotic-nuclear receptors; XNR-RE: xenobiotic-nuclear receptors-responsive elements; XREM: xenobiotics-responsive enhancer module. (Note: other regulatory factors and elements not referred may be involved in CYP expression induction).
Figure 5.
Main mechanisms of CYP induction. AhR: aryl hydrocarbon receptor; CAR: constitutive androstane receptor; CLEM4: constitutive liver enhancer module 4; DR4: direct repeat 4; ER8: estrogen receptor 8; HNF4α: hepatocyte nuclear factor 4α; PBREM: phenobarbital-responsive enhancer module; PPAR: proliferator-activated receptor; prPXRE: proximal PXR responsive element; PXR: pregnane nuclear receptor; XNR: xenobiotic-nuclear receptors; XNR-RE: xenobiotic-nuclear receptors-responsive elements; XREM: xenobiotics-responsive enhancer module. (Note: other regulatory factors and elements not referred may be involved in CYP expression induction).


{kind=link}
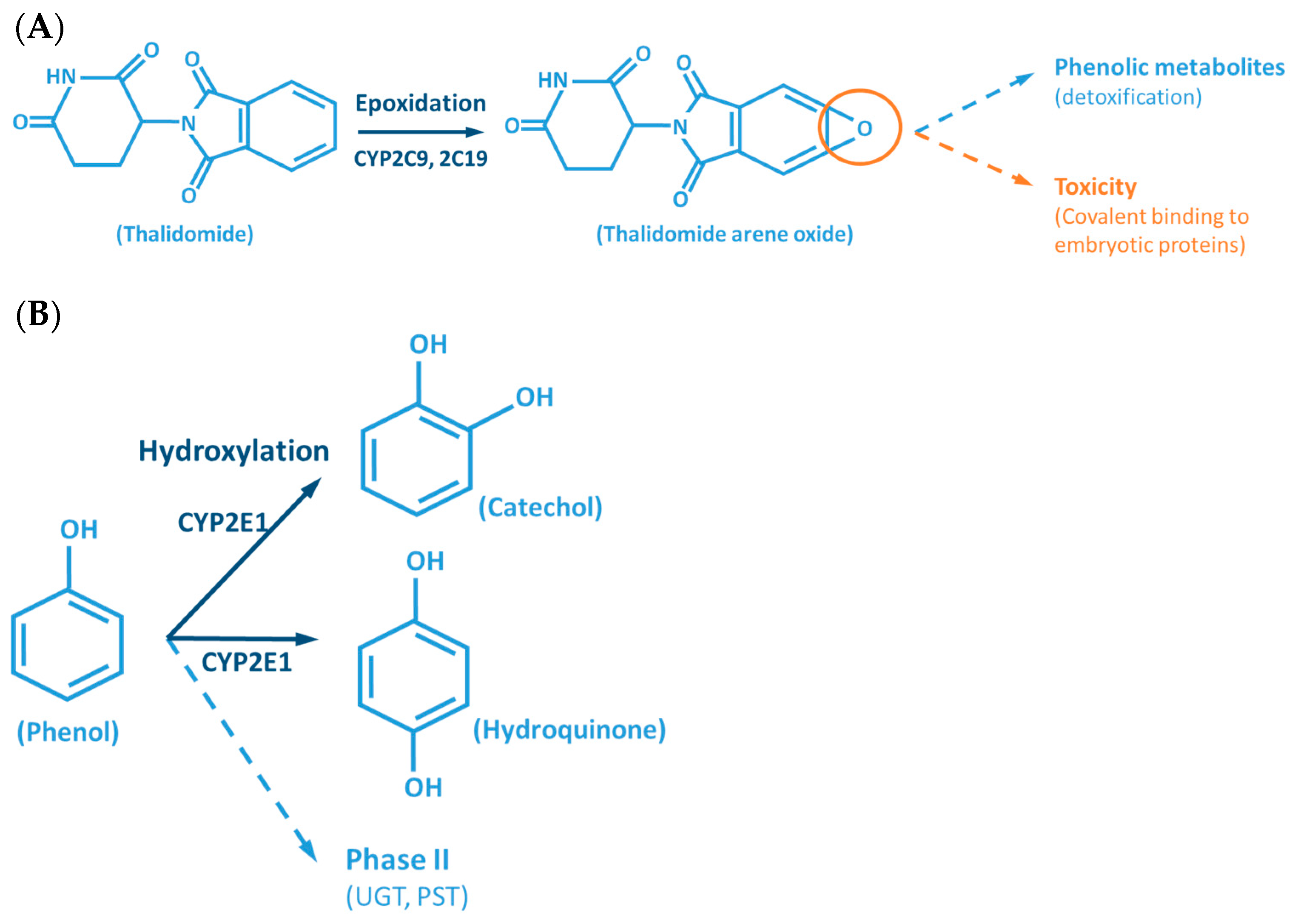{kind=link}
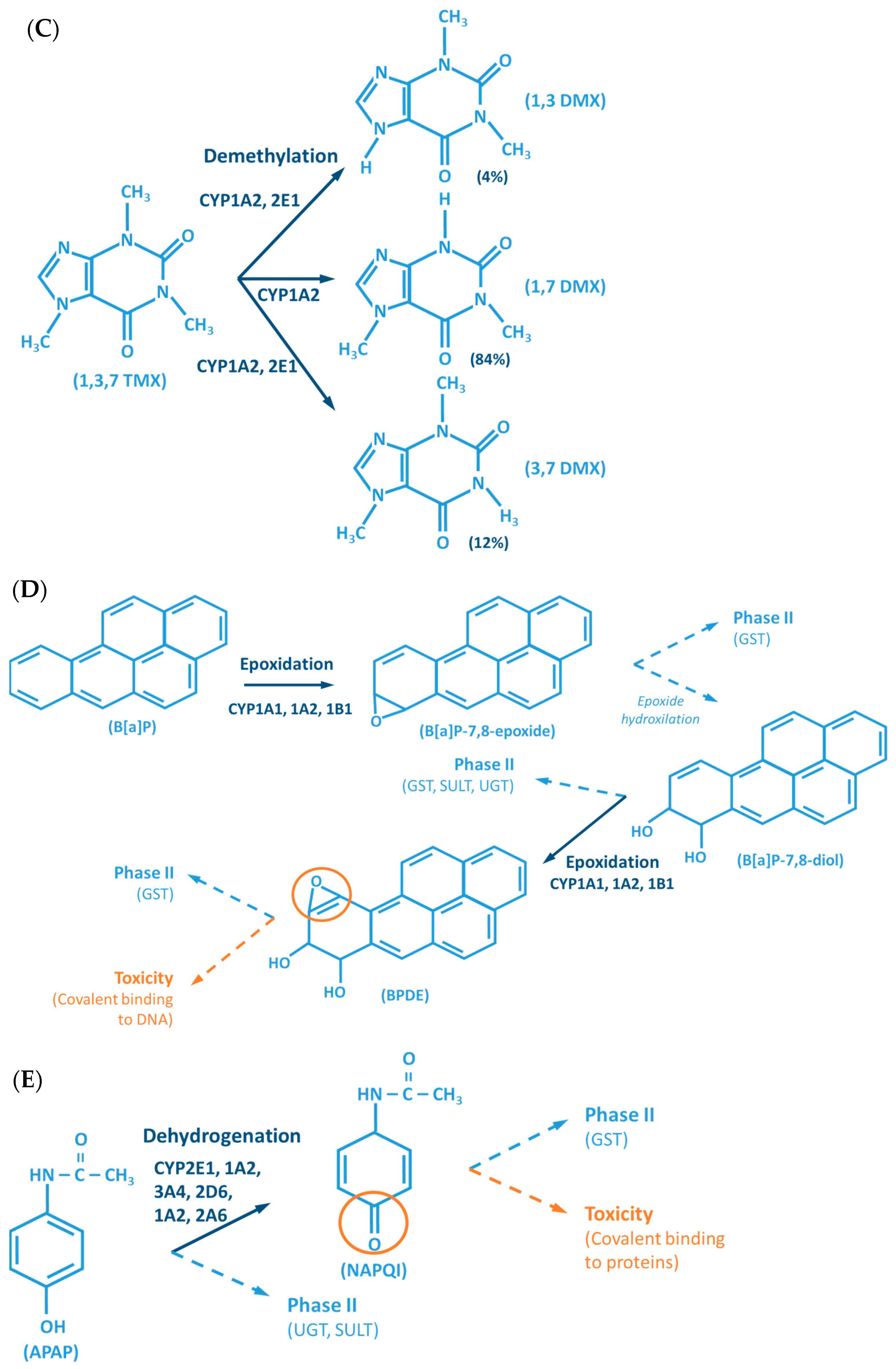{kind=link}
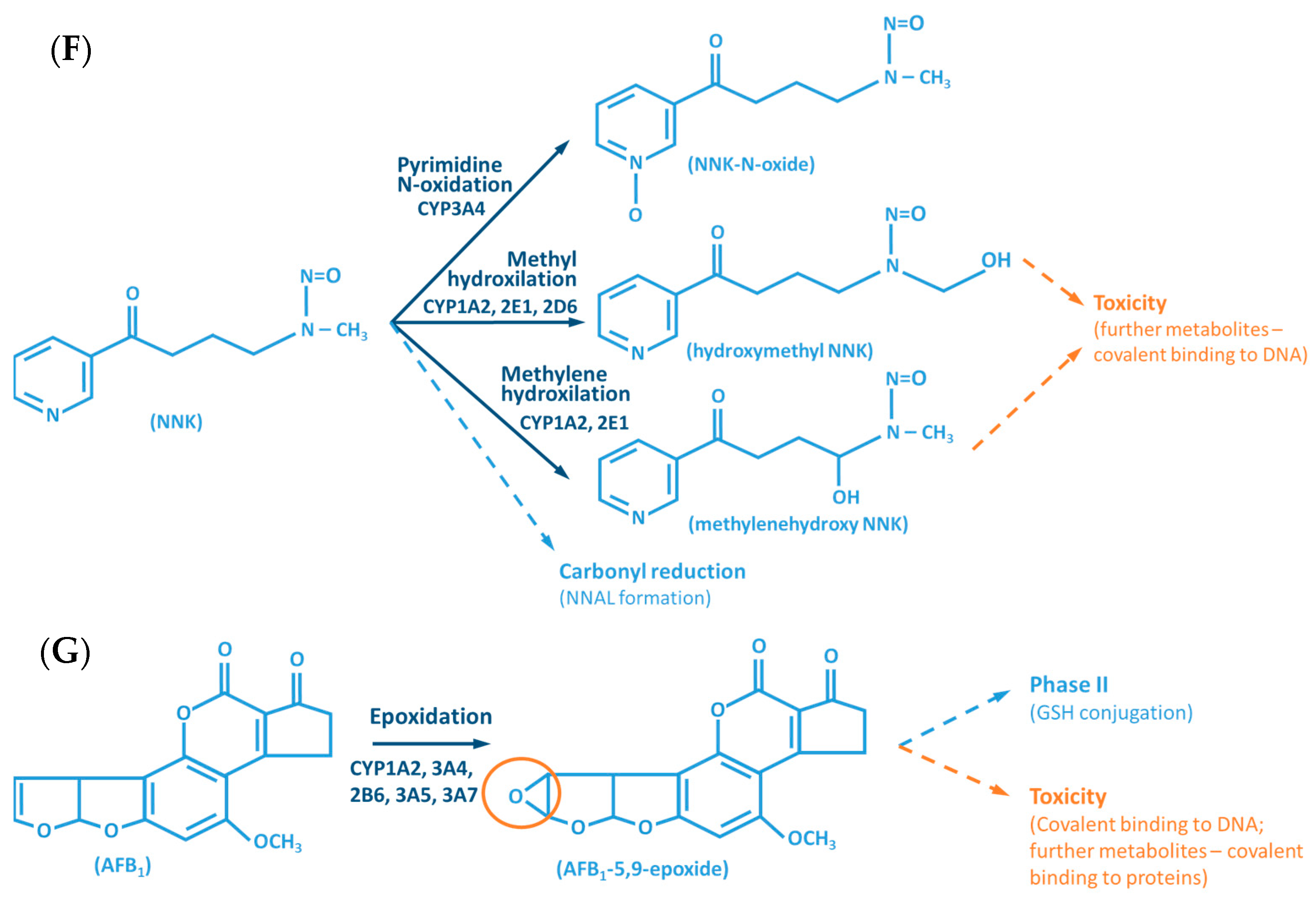{kind=link}
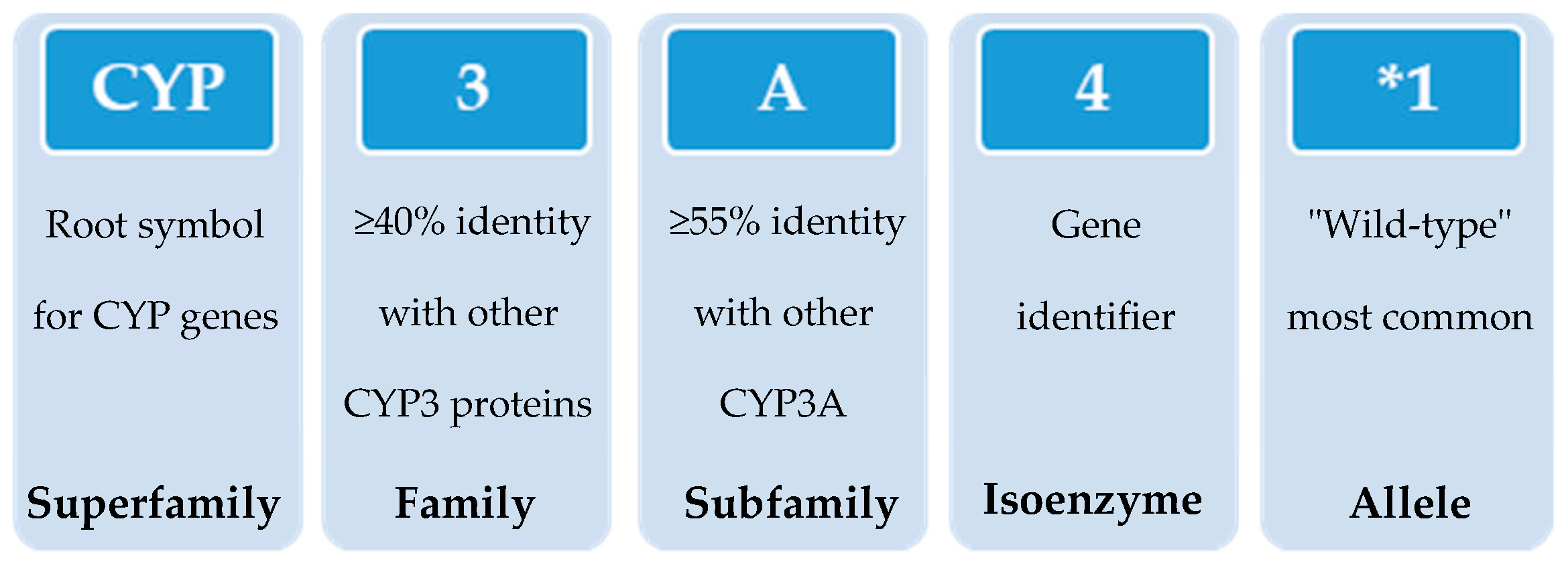{kind=link}
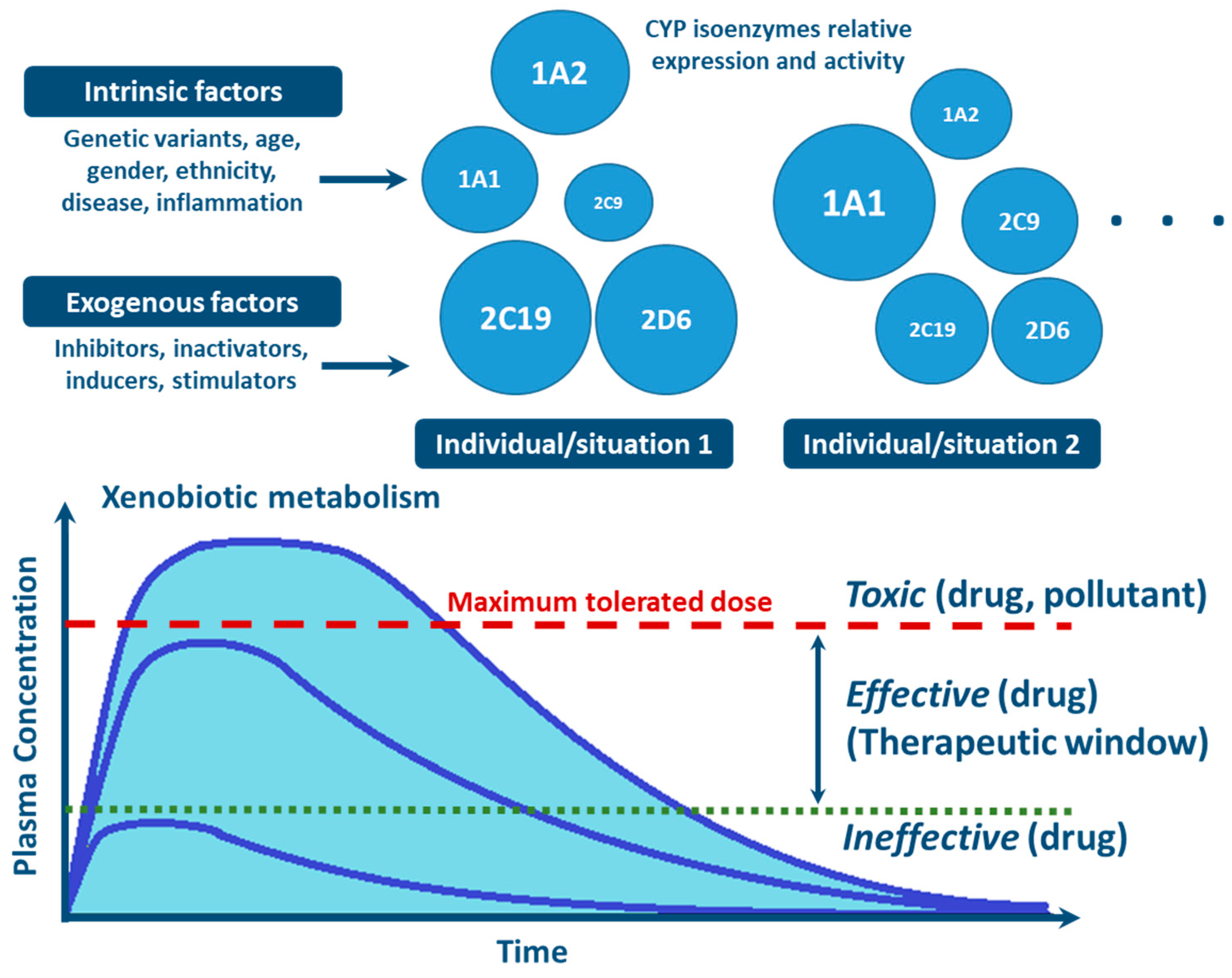{kind=link}
{kind=link}
Table 1.
Main classes of compounds metabolized by CYPs and major isoenzymes involved their biotransformation.
Table 1.
Main classes of compounds metabolized by CYPs and major isoenzymes involved their biotransformation.
| Classes of Compounds | CYP Isoenzymes |
|---|---|
| Sterols | 1B1, 7A1, 7B1, 8B1, 11A1, 11B1, 11B2, 17A1, 19A1, 21A2, 27A1, 39A1, 46A1, 51A1 |
| Xenobiotics | 1A1, 1A2, 2A6, 2A13, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2F1, 3A4, 3A5, 3A7 |
| Fatty acids | 2J2, 2U1, 4A11, 4B1, 4F11, 4F12, 4F22, 4V2, 4X1, 4Z1 |
| Eicosanoids | 4F2, 4F3, 4F8, 5A1, 8A1 |
| Vitamins | 2R1, 24A1, 26A1, 26B1, 26C1, 27B1, 27C1 |
| Unknown | 2A7, 2S1, 2W1, 4A22, 20A1 |
Microsomal CYPs (CPR as obligatory electron donor) in black and mitochondrial CYPs (adrenodoxin/adrenodoxin reductase as obligatory electron donor) in green.
Table 2.
Genetic variability and importance of the main CYP isoenzymes involved the metabolism of xenobiotics.
Table 2.
Genetic variability and importance of the main CYP isoenzymes involved the metabolism of xenobiotics.
| CYCYP Isoenzyme | Polymorphism Frequency | Functional Effects | Allelic Variants | Participation in the Metabolism of Xenobiotics |
|---|---|---|---|---|
| 1A1 | Relatively high | Rare | 13 |  |
| 1A2 | High | Rare | 21 | |
| 1B1 | Frequent missense mutations | Rare | 26 | |
| 2A6 | Higher in Orientals than in Caucasians | Significant | 45 | |
| 2B6 | High | Significant | 38 | |
| 2C8 | High | Significant | 14 | |
| 2C9 | Relatively rare in Caucasians | Very significant | 70 | |
| 2C19 | High | Very significant | 38 | |
| 2D6 | Very High | Highly significant | 145 | |
| 2E1 | Low | No significance | 7 | |
| 3A4 | Low | Low significance | 32 | |
| 3A5 | High | Significant | 9 | |
| Other (a) | - | - | - |
(a) Other CYP isoenzymes involved in smaller fraction of xenobiotic metabolism (2A13, 2C18, 2F1, 2J2, 3A7 and some members of the CYP4 family). Due to substrate overlapping specificity, CYP1B1 (typical sterols metabolizer) and 2J2 (typical fatty acids metabolizer) were also included in the CYP xenobiotic metabolizer analysis.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Esteves, F.; Rueff, J.; Kranendonk, M. The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family. J. Xenobiot. 2021, 11, 94-114. https://0-doi-org.brum.beds.ac.uk/10.3390/jox11030007
AMA Style
Esteves F, Rueff J, Kranendonk M. The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family. Journal of Xenobiotics. 2021; 11(3):94-114. https://0-doi-org.brum.beds.ac.uk/10.3390/jox11030007
Chicago/Turabian StyleEsteves, Francisco, José Rueff, and Michel Kranendonk. 2021. "The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family" Journal of Xenobiotics 11, no. 3: 94-114. https://0-doi-org.brum.beds.ac.uk/10.3390/jox11030007