Genetic Diversity and Population Divergence of a Rare, Endemic Grass (Elymus breviaristatus) in the Southeastern Qinghai-Tibetan Plateau

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
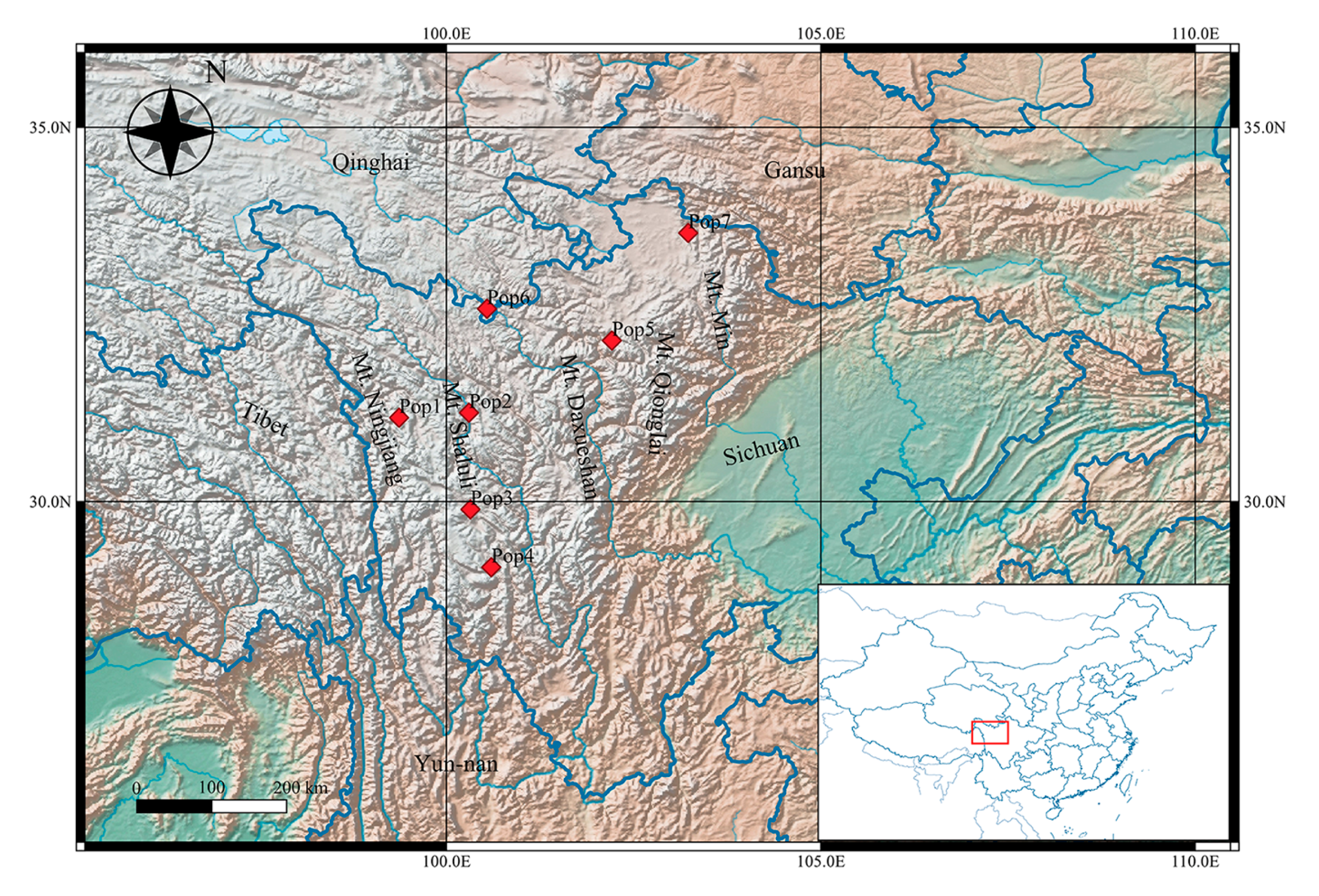
2.1. Plant Sampling
2.2. DNA Extraction and PCR Amplification
2.3. Data Statistic and Analysis
3. Results
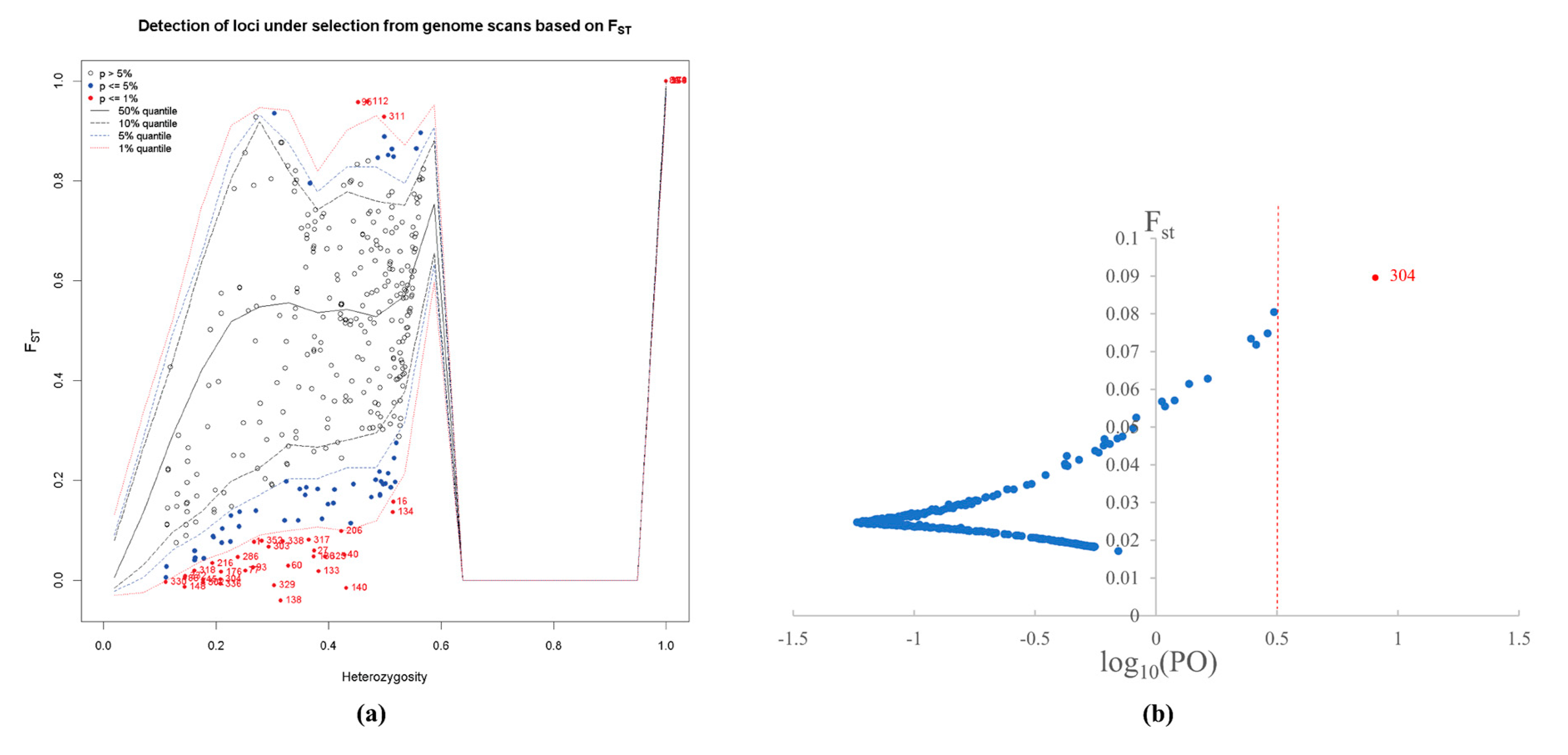
3.1. SRAP-PCR Amplification and Outlier Analysis
3.2. Intra-Population Variation
3.3. Genetic Differentiation among Populations
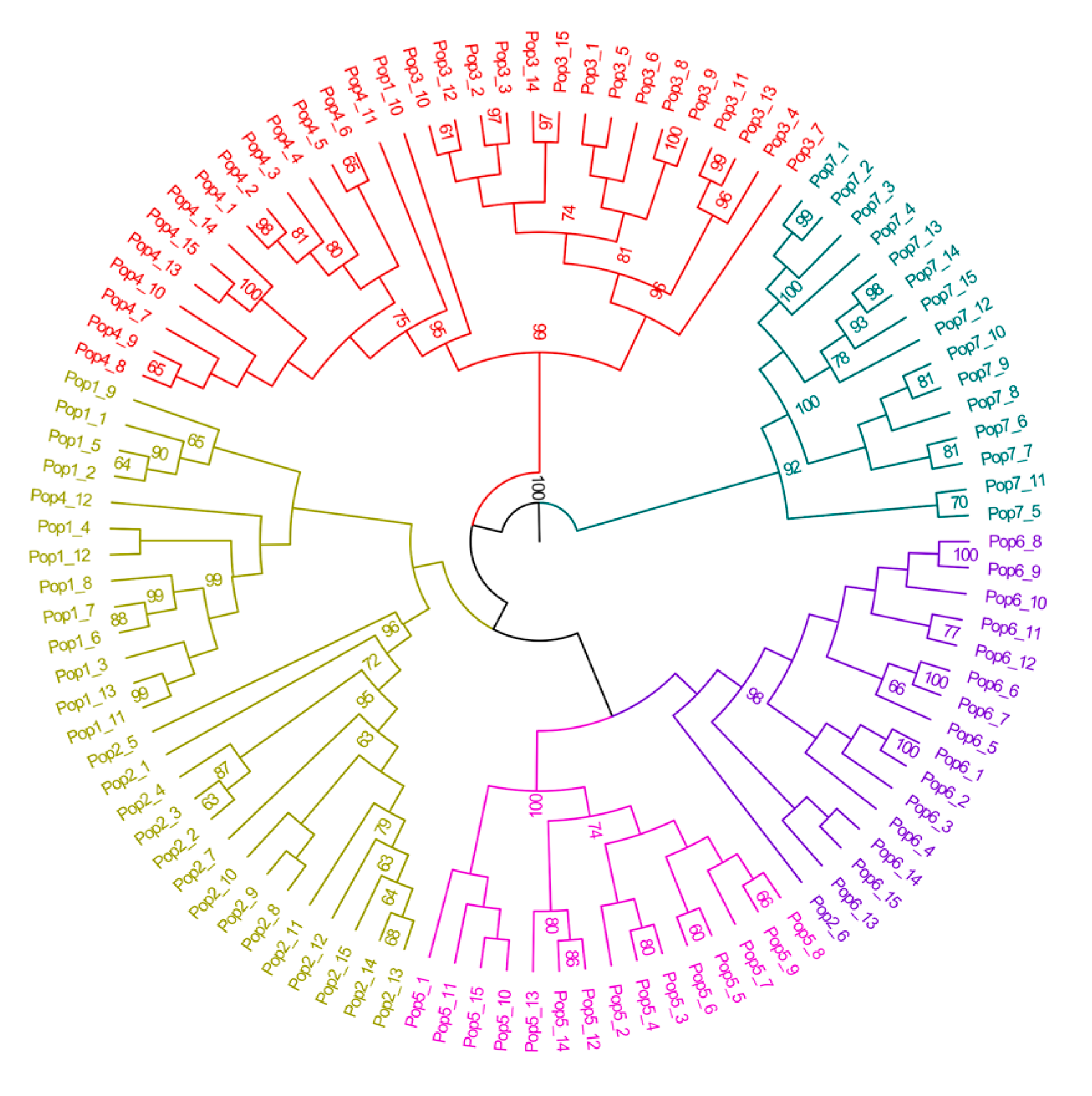
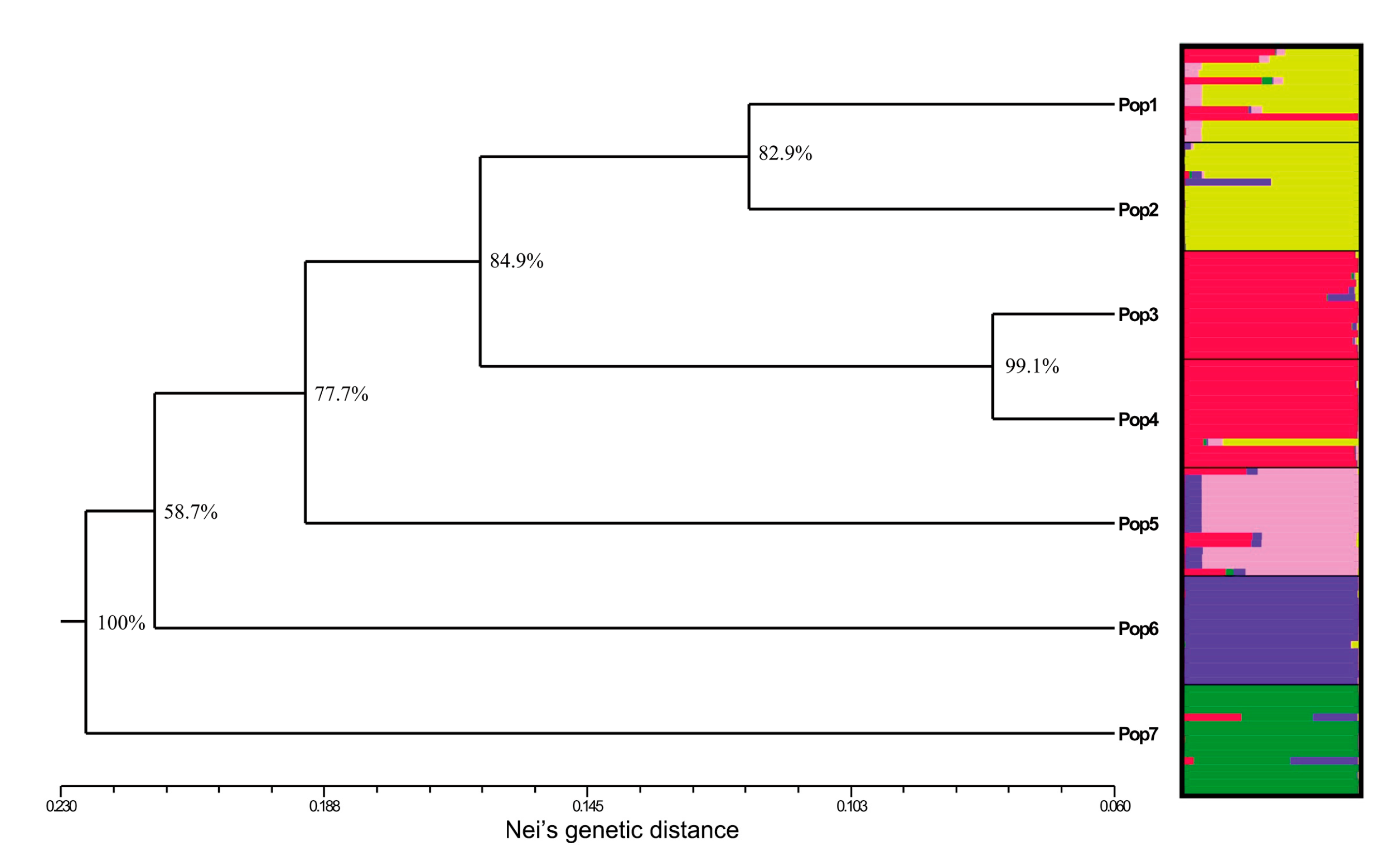
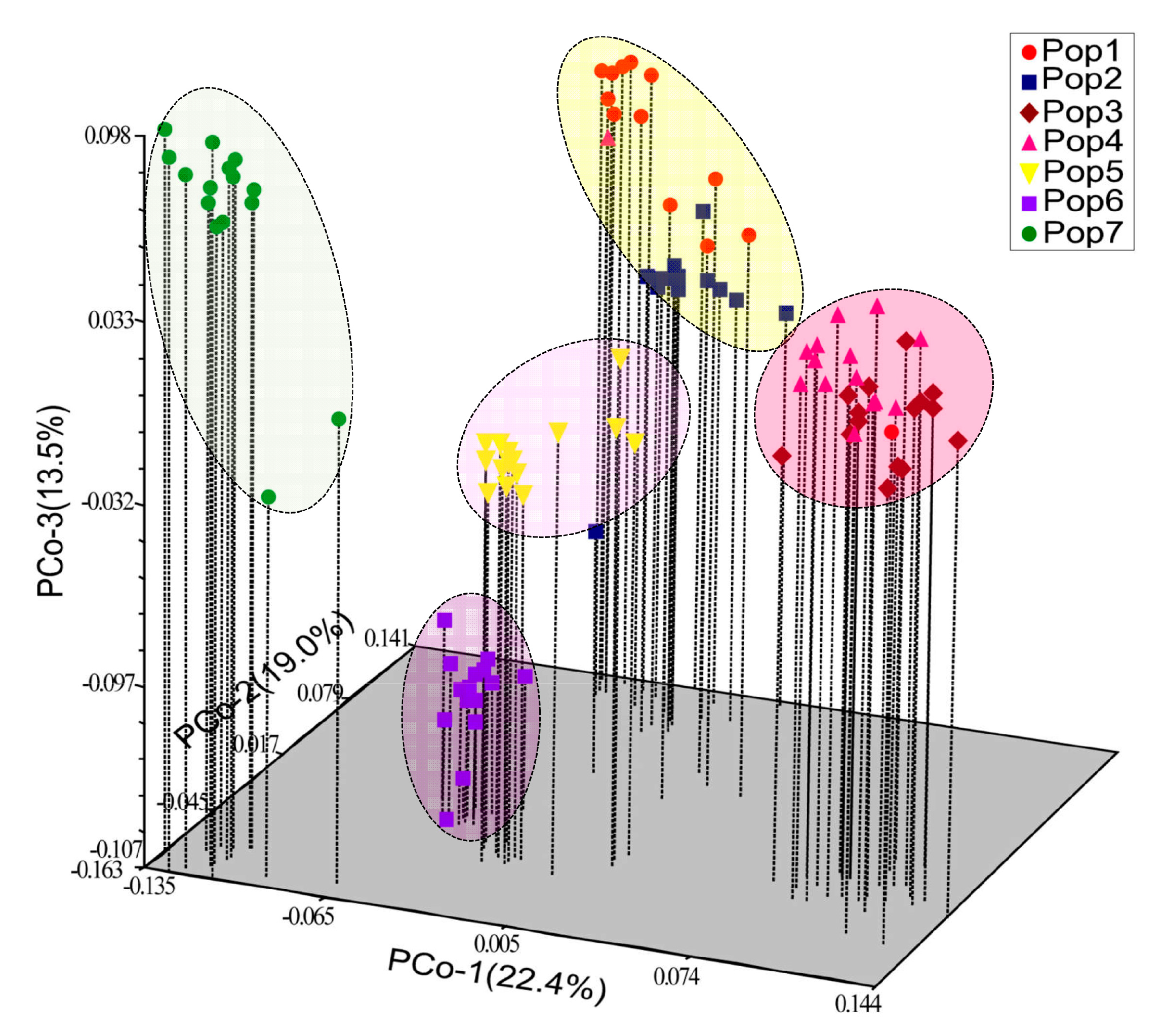
3.4. Inter-Population Structure
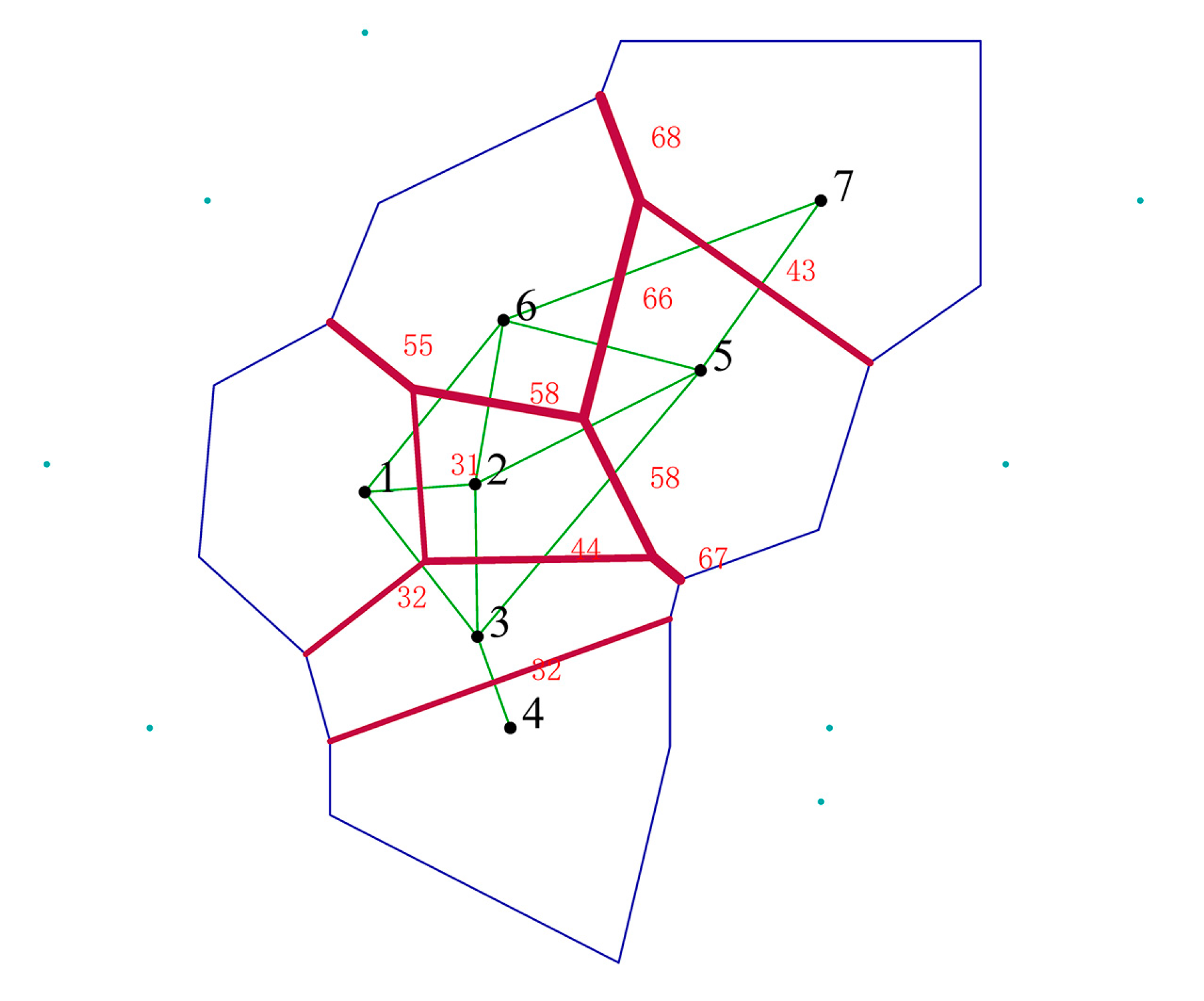
3.5. The Identification of Genetic Barriers
3.6. Correlation of Genetic Diversity with Geographic Distances and Eco-Geographic Factors
4. Discussion
4.1. Genetic Diversity at Species and Population Levels
4.2. Population Genetic Structure
4.3. Contribution of Geographic Barriers and Bioclimatic Factors to Genetic Structure
4.4. Suggestion of Conservation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tang, N.; Mo, G.; van Tuyl, J.M.; Arens, P.; Liu, J.; Tang, D. Genetic diversity and structure of Lilium pumilum DC. in southeast of Qinghai-Tibet plateau. Plant Syst. Evol. 2014, 300, 1453–1464. [Google Scholar] [CrossRef]
- Bajpai, P.K.; Warghat, A.R.; Sharma, R.K.; Yadav, A.; Thakur, A.K.; Srivastava, R.B.; Stobdan, T. Structure and genetic diversity of natural populations of morus alba in the trans-himalayan ladakh region. Biochem. Genet. 2014, 52, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; He, S.L.; Li, D.Z.; Yi, T.S. Nuclear genetic variation of Rosa odorata var. gigantea (Rosaceae): Population structure and conservation implications. Tree Genet. Genomes 2016, 12, 65. [Google Scholar] [CrossRef]
- Van der Meer, S.; Jacquemyn, H. Genetic diversity and spatial genetic structure of the grassland perennial saxifraga granulata along two river systems. PLoS ONE 2015, 10, e130463. [Google Scholar] [CrossRef] [PubMed]
- Gentili, R.; Abeli, T.; Parolo, G.; Ciappetta, S.; Montagnani, C.; Müller, J.V.; Rossi, G.; Citterio, S. Genetic structure of Leucojum aestivum L. in the Po Valley (N-Italy) drives conservation management actions. Conserv. Genet. 2018, 19, 827–838. [Google Scholar] [CrossRef]
- Jian, H.; Li, S.; Guo, J.; Li, S.; Wang, Q.; Yan, H.; Qiu, X.; Zhang, Y.; Cai, Z.; Volis, S.; et al. High genetic diversity and differentiation of an extremely narrowly distributed and critically endangered decaploid rose (Rosa praelucens): Implications for its conservation. Conserv. Genet. 2018, 19, 761–776. [Google Scholar] [CrossRef]
- Liu, Y.P.; Ren, Z.M.; Harris, A.J.; Peterson, P.M.; Wen, J.; Su, X. Phylogeography of Orinus (Poaceae), a dominant grass genus on the Qinghai-Tibet Plateau. Bot. J. Linn. Soc. 2018, 186, 202–223. [Google Scholar] [CrossRef]
- Fu-Sheng, Y.; Ai-Li, Q.; Yu-Fei, L.; Xiao-Quan, W. Great genetic differentiation among populations of Meconopsis integrifolia and its implication for plant speciation in the Qinghai-Tibetan Plateau. PLoS ONE 2012, 7, e37196. [Google Scholar] [CrossRef]
- Love, A. Conspectus of the Triticeae. Fedd. Rep. 1984, 95, 425–521. [Google Scholar] [CrossRef]
- Hodge, C.D.; Wang, H.; Sun, G. Phylogenetic analysis of the maternal genome of tetraploid StStYY Elymus (Triticeae: Poaceae) species and the monogenomic Triticeae based on rps16 sequence data. Plant Sci. 2010, 178, 463–468. [Google Scholar] [CrossRef]
- Zhou, Y. Phylogenetic relationships among ten Elymus species based on random amplified polymorphic DNA. Acta Phytotaxon. Sin. 1999, 37, 425–432. [Google Scholar]
- Zhang, Y.B.; Ke-Ping, M.A. Geographic distribution characteristics of the national key protected wild plants in China. Chin. J. Appl. Ecol. 2008, 19, 1670–1675. [Google Scholar]
- Xiao-yan, G.; Zhi-hui, G.; Xin-quan, Z.; Kai, Z.; Chao-jie, Z.; Kai-xin, F.; Xin, L.; Xiao, M. Phenotypic variations in seven ex-situ conservation populations of Elymus breviaristatus. Acta Prataculturae Sin. 2015, 24, 141–152. [Google Scholar]
- Yan, X.B.; Guo, Y.X.; Zhou, H.; Lu, B.R.; Wang, K. Genetic patterns of ten Elymus species from the Tibetan and Inner Mongolian plateaus of China. Grass Forage Sci. 2010, 61, 398–404. [Google Scholar] [CrossRef]
- Kai-Xin, F.; Liu, X.; Zhang, X.Q.; Zhang, J.B.; Gou, W.L.; Zhang, C.L.; Sun, M.; Ma, X. Evaluation of drought resistance of six Elymus breviaristatus accessions from the northwest plateau of Sichuan province at germination stage. Chin. J. Grassl. 2017, 39, 41–47. [Google Scholar]
- Sun, M.D. Breeding and domesticating tongde Elymus breviaristatus cv.tongde. Qinghai Prataculture 2008, 17, 2–7. [Google Scholar]
- Yang, C.R.; Baum, B.R.; Chen, W.H.; Zhang, H.Q.; Liu, X.Y.; Fan, X.; Sha, L.N.; Kang, H.Y.; Wang, Y.; Zhou, Y.H. Genomic constitution and taxonomy of the Chinese hexaploids Elymus cylindricus and E. breviaristatus (Poaceae: Triticeae). Bot. J. Linn. Soc. 2016, 182, 650–657. [Google Scholar] [CrossRef]
- Gu, X.Y.; Guo, Z.H.; Ma, X.; Bai, S.Q.; Zhang, X.Q.; Zhang, C.B.; Chen, S.Y.; Peng, Y.; Yan, Y.H.; Huang, L.K.; et al. Population genetic variability and structure of Elymus breviaristatus (Poaceae: Triticeae) endemic to Qinghai-Tibetan Plateau inferred from SSR markers. Biochem. Syst. Ecol. 2015, 58, 247–256. [Google Scholar] [CrossRef]
- Robarts, D.W.H.; Wolfe, A.D. Sequence-related amplified polymorphism (SRAP) markers: A potential resource for studies in plant molecular biology. Appl. Plant Sci. 2014, 2, 15–18. [Google Scholar] [CrossRef]
- Li, B.; Wang, J.; Liu, Z.; Zhuang, X.; Huang, J. Genetic diversity and ex situ conservation of Loropetalum subcordatum, an endangered species endemic to China. BMC Genet. 2018, 19, 12. [Google Scholar] [CrossRef]
- Chen, Z.H.; Miao, J.M.; Zhong, J.C.; Ma, X.; Chen, S.Y.; Zhang, X.Q. Genetic diversity of wild Elymus nutans germplasm detected by SRAP markers. Acta Prataculturae Sin. 2009, 18, 192–200. [Google Scholar] [CrossRef]
- Gu, X.; Guo, Z.; Zhang, X.; Zhou, Y.; Bai, S.; Zhang, C.; Jiang, Z.; Liu, X.; Zhou, C.; Ma, X. Genetic diversity of Elymus sibiricus germplasm resources revealed by SRAP markers. Acta Prataculturae Sin. 2014, 23, 205–216. [Google Scholar]
- Lynch, M. Analysis of population genetic structure with RAPD markers. Mol. Ecol. 1994, 3, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Isolation by Distance. Genetics 1943, 28, 114–138. [Google Scholar] [PubMed]
- Wang, T. AFLP Genome scan to detect genetic structure and candidate loci under selection for local adaptation of the invasive weed mikania micrantha. PLoS ONE 2012, 7, e41310. [Google Scholar] [CrossRef]
- Matthieu, F.; Oscar, G. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef]
- Excoffier, L. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.J.; Donnelly, P.J. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.S.; Regnaut, S.J.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Dice, L.R. Measures of the amount of ecologic association between species. Ecology 1945, 26, 297–302. [Google Scholar] [CrossRef]
- Gaudett, M.; Salomon, B.; Sun, G. Molecular variation and population structure in Elymus trachycaulus and comparison with its morphologically similar E. alaskanus. Plant Syst. Evol. 2005, 250, 81–91. [Google Scholar] [CrossRef]
- Zhang, X.; Salomon, B.; von Bothmer, R. Application of random amplified polymorphic DNA markers to evaluate intraspecific genetic variation in the Elymus alaskanus complex (Poaceae). Genet. Resour. Crop Evol. 2002, 49, 399–409. [Google Scholar] [CrossRef]
- Díaz, O.; Sun, G.; Salomon, B.; von Bothmer, R. Levels and distribution of allozyme and RAPD variation in populations of Elymus fibrosus (Schrenk) Tzvel. (Poaceae). Genet. Resour. Crop Evol. 2000, 47, 11–24. [Google Scholar] [CrossRef]
- Sun, G.L.; Díaz, O.; Salomon, B.; Bothmer, R.V. Genetic diversity and structure in a natural Elymus caninus population from Denmark based on microsatellite and isozyme analyses. Plant Syst. Evol. 2001, 227, 235–244. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, X.; Zhou, Y.; Bai, S.; Liu, W. Assessing genetic diversity of Elymus sibiricus (Poaceae: Triticeae) populations from Qinghai-Tibet Plateau by ISSR markers. Biochem. Syst. Ecol. 2008, 36, 514–522. [Google Scholar] [CrossRef]
- Ellis, J.R.; Pashley, C.H.; Burkejm, J.M.; McCauley, D.E. High genetic diversity in a rare and endangered sunflower as compared to a common congener. Mol. Ecol. 2010, 15, 2345–2355. [Google Scholar] [CrossRef]
- Gitzendanner, M.A.; Soltis, P.S. Patterns of genetic variation in rare and widespread plant congeners. Am. J. Bot. 2000, 87, 783–792. [Google Scholar] [CrossRef]
- Turchetto, C.; Segatto, A.L.; Mäder, G.; Rodrigues, D.M.; Bonatto, S.L.; Freitas, L.B. High levels of genetic diversity and population structure in an endemic and rare species: Implications for conservation. AoB Plants 2016, 1, 1–17. [Google Scholar] [CrossRef]
- Cole, C.T. Genetic variation in rare and common plants. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 213–237. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.; Guo, Q.; Ren, G.; Wu, Y. Genetic variation within and between populations of a desert poplar (Populus euphratica) revealed by SSR markers. Ann. For. Sci. 2011, 68, 1143–1149. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Shen, B.; Gong, H. Genetic diversity of Simao pine in China revealed by SRAP markers. Peer J. 2019, 7, e6529. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.B.; Guo, Y.X.; Liu, F.Y.; Zhao, C.; Liu, Q.L.; Lu, B.R. Population structure affected by excess gene flow in self-pollinating Elymus nutans and E. burchan-buddae (Triticeae: Poaceae). Popul. Ecol. 2010, 52, 233–241. [Google Scholar] [CrossRef]
- Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 2004, 13, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Li, J.; Zheng, D.; Friedman, C.; Wang, H. Analysis of genetic population structure and diversity in Mallotus oblongifolius using ISSR and SRAP markers. Peer J. 2019, 7, e7173. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Linhart, Y.B.; Mitton, J.B. Relationships between life history characteristics and electrophoretically detectable genetic variation in plants. Annu. Rev. Ecol. Syst. 1979, 10, 173–200. [Google Scholar] [CrossRef]
- Nevo, E. Genetic diversity in wild cereals: Regional and local studies and their bearing on conservation ex situ and in situ. Genet. Resour. Crop Evol. 1998, 45, 355–370. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Conservation Genetics of Endemic Plant Species. In Conservation Genetics: Case Histories from Nature; Chapman & Hall: New York, NY, USA, 1996; pp. 281–304. [Google Scholar]
- Wilson, B.L.; Kitzmiller, J.; Rolle, W.; Hipkins, V.D. Isozyme variation and its environmental correlates in Elymus glaucus from the California Floristic Province. Can. J. Bot. 2001, 79, 139–153. [Google Scholar] [CrossRef]
- Hu, Z.Z. Contrast experiment of Elymus breviaristatus, Elymus sibiricas and Elymus nutans. Qinghai Pratacult. 2012, 21, 20–23. [Google Scholar]
- Yang, Z.; Wang, L.; Zhao, T. High genetic variability and complex population structure of the native Chinese hazelnut. Braz. J. Bot. 2018, 41, 687–697. [Google Scholar] [CrossRef]
- Chen, S.; Ma, X.; Zhang, X.; Chen, Z. Genetic variation and geographical divergence in Elymus nutans Griseb. (Poaceae: Triticeae) from West China. Biochem. Syst. Ecol. 2009, 37, 716–722. [Google Scholar] [CrossRef]
- Brzosko, E.; Ratkiewicz, M.; Wróblewska, A. Allozyme differentiation and genetic structure of the Lady’s slipper (Cypripedium calceolus) island populations in north-east Poland. Bot. J. Linn. Soc. 2015, 138, 433–440. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pop No. | Collection Site | Latitude | Longitude | Habit and Its Area | Sample Size | Population Size | Altitude (m) | Annual Temperature (°C) | Annual Precipitation (mm) |
|---|---|---|---|---|---|---|---|---|---|
| Pop1 | Anzi Town in Baiyu County of Garze region | N 31°7′16″ | E 99°22′4″ | Hillside; about 320 m2 | 13 | 47 | 3890(H) | 1.3(L) | 596(L) |
| Pop2 | Anle Town in Xinlong County of Garze region | N 31°11′16″ | E 100°18′5″ | river valley; about 180 m2 | 15 | 31 | 3170(L) | 5.8(H) | 638(L) |
| Pop3 | Benge Town in Litang County of Garze region | N 29°53′48″ | E 100°19′16″ | shrub grassland; about 176 m2 | 15 | 28 | 3920(H) | 3.7(M) | 710(H) |
| Pop4 | Labo Town in Litang County of Garze region | N 29°7′25″ | E 100°35′58″ | river valley; about 210 m2 | 15 | 19 | 3450(M) | 6.6(H) | 693(M) |
| Pop5 | Dazang Town in Maerkang County of Aba region | N 32°9′6″ | E 102°12′38″ | shrub grassland; about 136 m2 | 15 | 23 | 3110(L) | 6.7(H) | 758(H) |
| Pop6 | Nianlong Town in Seda County of Garze region | N 32°34′36″ | E 100°32′31″ | shrub grassland; about 142 m2 | 15 | 21 | 3680(M) | 0.8(L) | 662(M) |
| Pop7 | Baxi Town in Ruoergai County of Aba region | N33°35′21″ | E 103°13′46″ | shrub grassland; about 158 m2 | 15 | 18 | 3130(L) | 3.4(M) | 654(M) |
| Population | Sample Size | No. of PB | PPB (%) | Ao | Ae | He | Ho | HB | Hj | LD (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Pop1 | 13 | 289 | 62.96 | 1.6296 ± 0.4834 | 1.3939 ± 0.3789 | 0.2283 ± 0.2026 | 0.3070 ± 0.2667 | 0.2571 ± 0.0058 | 0.2532 ± 0.0095 | 1.8824 |
| Pop2 | 15 | 215 | 46.84 | 1.4684 ± 0.4995 | 1.2646 ± 0.3593 | 0.1544 ± 0.1954 | 0.1968 ± 0.2378 | 0.1962 ± 0.0070 | 0.1770 ± 0.0092 | 0.526 |
| Pop3 | 15 | 267 | 58.17 | 1.5817 ± 0.4938 | 1.3582 ± 0.3857 | 0.2061 ± 0.2055 | 0.2721 ± 0.2627 | 0.2407 ± 0.0062 | 0.2286 ± 0.0096 | 0.6967 |
| Pop4 | 15 | 315 | 68.63 | 1.6863 ± 0.4645 | 1.4359 ± 0.3875 | 0.2494 ± 0.2035 | 0.3198 ± 0.2576 | 0.2698 ± 0.0059 | 0.2715 ± 0.0096 | 0.812 |
| Pop5 | 15 | 204 | 44.44 | 1.4444 ± 0.4974 | 1.2708 ± 0.3668 | 0.1566 ± 0.1981 | 0.2060 ± 0.2528 | 0.2029 ± 0.0067 | 0.1796 ± 0.0093 | 0.9181 |
| Pop6 | 15 | 189 | 41.18 | 1.4118 ± 0.4927 | 1.2594 ± 0.3645 | 0.1491 ± 0.1986 | 0.1790 ± 0.2380 | 0.1932 ± 0.0073 | 0.1718 ± 0.0093 | 0.4614 |
| Pop7 | 15 | 220 | 47.93 | 1.4793 ± 0.5001 | 1.2668 ± 0.3515 | 0.1577 ± 0.1929 | 0.2036 ± 0.2389 | 0.1968 ± 0.0066 | 0.1788 ± 0.0091 | 1.2134 |
| Means | 14.7 | 242.7 | 52.88 | 1.5288 ± 0.1038 | 1.3214 ± 0.0734 | 0.1859 ± 0.0413 | 0.2406 ± 0.0577 | 0.2224 ± 0.0050 | 0.2086 ± 0.0094 | 0.93 |
| Species | 103 | 424 | 92.37 | 1.9237 ± 0.2657 | 1.5723 ± 0.3306 | 0.3314 ± 0.1580 | 0.4490 ± 0.2238 | 0.3608 ± 0.0031 | 0.3812 ± 0.0092 | 20.3691 |
| Nei’s Gene Diversity | Shannon’s Information Index | Lynch & Milligan (AFLPsurv) | Bayesian Method | ||||
|---|---|---|---|---|---|---|---|
| HT | 0.3314 | Hsp | 0.4490 | Ht | 0.3812 | HT-B | 0.3608 |
| HS | 0.1859 | Hpop | 0.2406 | Hw | 0.2486 | HS-B | 0.2224 |
| HS/HT | 0.5607 | Hpop/Hsp | 0.5359 | Hw/Ht | 0.6522 | HS-B/HT-B | 0.6164 |
| GST | 0.4393 | (Hsp-Hpop)/Hsp | 0.4641 | Fst | 0.3478 | GST-B | 0.3836 |
| Nm | 0.6381 | – | – | – | – | – | – |
| Different Pops | Source of Variation | df | Sum of Squares | Variance Components | Percentage of Variation (%) |
|---|---|---|---|---|---|
| Total | Among Pops | 6 | 3402.308 | 567.0514 | 48.90% |
| Within Pops | 96 | 3610.41 | 37.60844 | 51.10% | |
| Total | 102 | 7012.718 | |||
| Regions | Among Regions | 1 | 795.915 | 6.276007 | 8.15% |
| Among populations within regions | 5 | 2608.055 | 32.97922 | 42.82% | |
| Within populations | 96 | 3625.467 | 37.76528 | 49.03% | |
| Total | 102 | 7029.437 | |||
| Altitude | Among groups | 2 | 964.4179 | 482.2089 | 0.00% |
| Among populations within groups | 4 | 2438.802 | 609.7004 | 50.78% | |
| Within populations | 96 | 3613.344 | 37.639 | 49.22% | |
| Total | 102 | 7016.563 | |||
| AMT | Among groups | 2 | 959.8829 | 479.9415 | 0.00% |
| Among populations within groups | 4 | 2443.337 | 610.8341 | 50.83% | |
| Within populations | 96 | 3613.344 | 37.639 | 49.17% | |
| Total | 102 | 7016.563 | |||
| AMP | Among groups | 2 | 1191.462 | 595.7312 | 1.83% |
| Among populations within groups | 4 | 2211.757 | 552.9393 | 47.29% | |
| Within populations | 96 | 3613.344 | 37.639 | 50.89% | |
| Total | 102 | 7016.563 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Q.; Liu, Q.; Xiong, Y.; Xiong, Y.; Dong, Z.; Yang, J.; Liu, W.; Ma, X.; Bai, S. Genetic Diversity and Population Divergence of a Rare, Endemic Grass (Elymus breviaristatus) in the Southeastern Qinghai-Tibetan Plateau. Sustainability 2019, 11, 5863. https://0-doi-org.brum.beds.ac.uk/10.3390/su11205863
Yu Q, Liu Q, Xiong Y, Xiong Y, Dong Z, Yang J, Liu W, Ma X, Bai S. Genetic Diversity and Population Divergence of a Rare, Endemic Grass (Elymus breviaristatus) in the Southeastern Qinghai-Tibetan Plateau. Sustainability. 2019; 11(20):5863. https://0-doi-org.brum.beds.ac.uk/10.3390/su11205863
Chicago/Turabian StyleYu, Qingqing, Qian Liu, Yi Xiong, Yanli Xiong, Zhixiao Dong, Jian Yang, Wei Liu, Xiao Ma, and Shiqie Bai. 2019. "Genetic Diversity and Population Divergence of a Rare, Endemic Grass (Elymus breviaristatus) in the Southeastern Qinghai-Tibetan Plateau" Sustainability 11, no. 20: 5863. https://0-doi-org.brum.beds.ac.uk/10.3390/su11205863