Comparative Analysis of Sequence Polymorphism in Complete Organelle Genomes of the ‘Golden Tide’ Seaweed Sargassum horneri between Korean and Chinese Forms

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
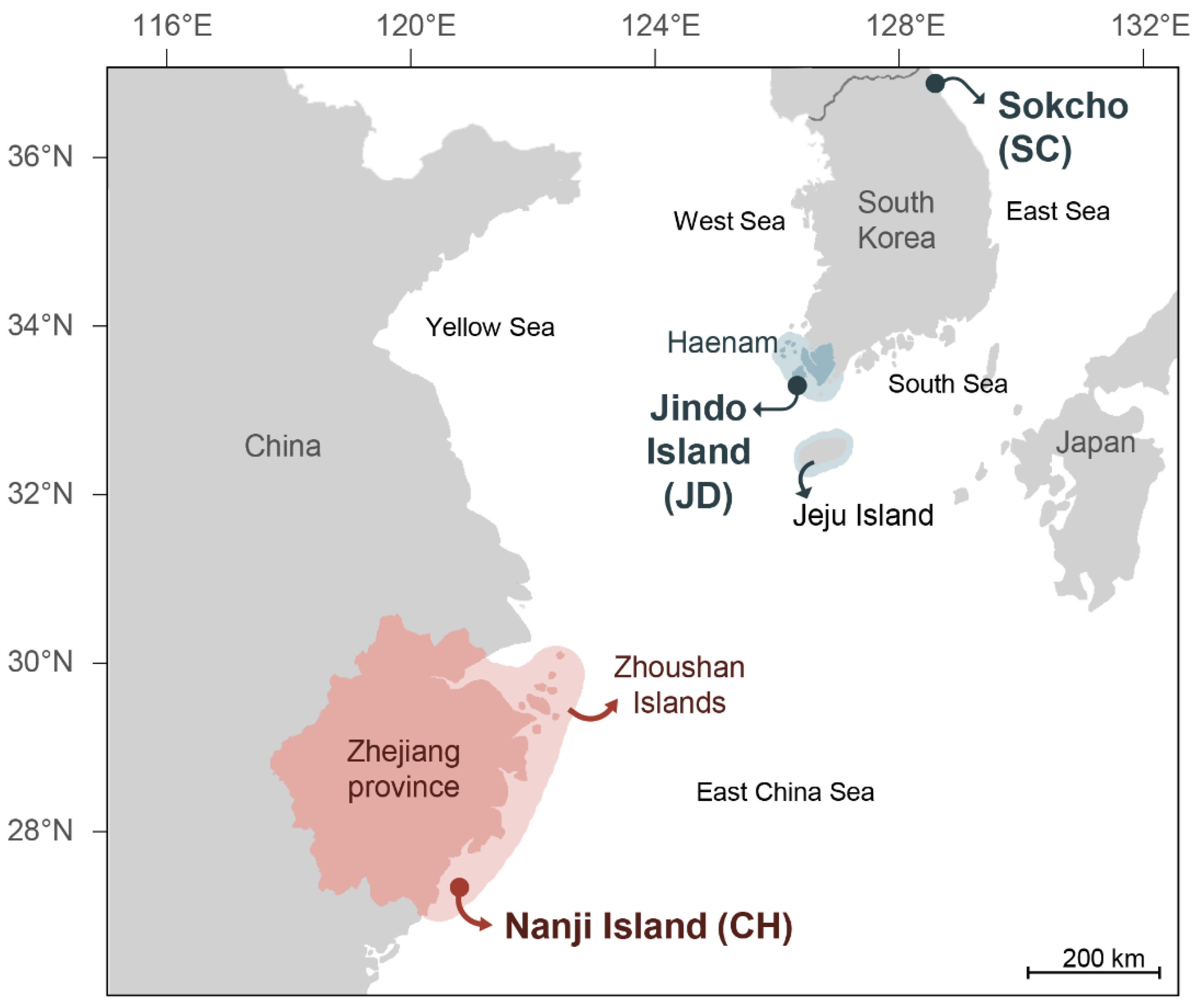
2.1. Sample Collection and DNA Extraction
2.2. Genome Sequencing, Assembly, and Mapping
2.3. Comparative Analyses of Nucleotide Diversity and Synonymous (Ks) and Non-Synonymous Substitution (Ka) Rates
3. Results
3.1. Mitochondrial and Chloroplast Genome Features of Korean S. horneri
3.2. Comparative Analysis of Genome Sequences between Korean and Chinese Individuals
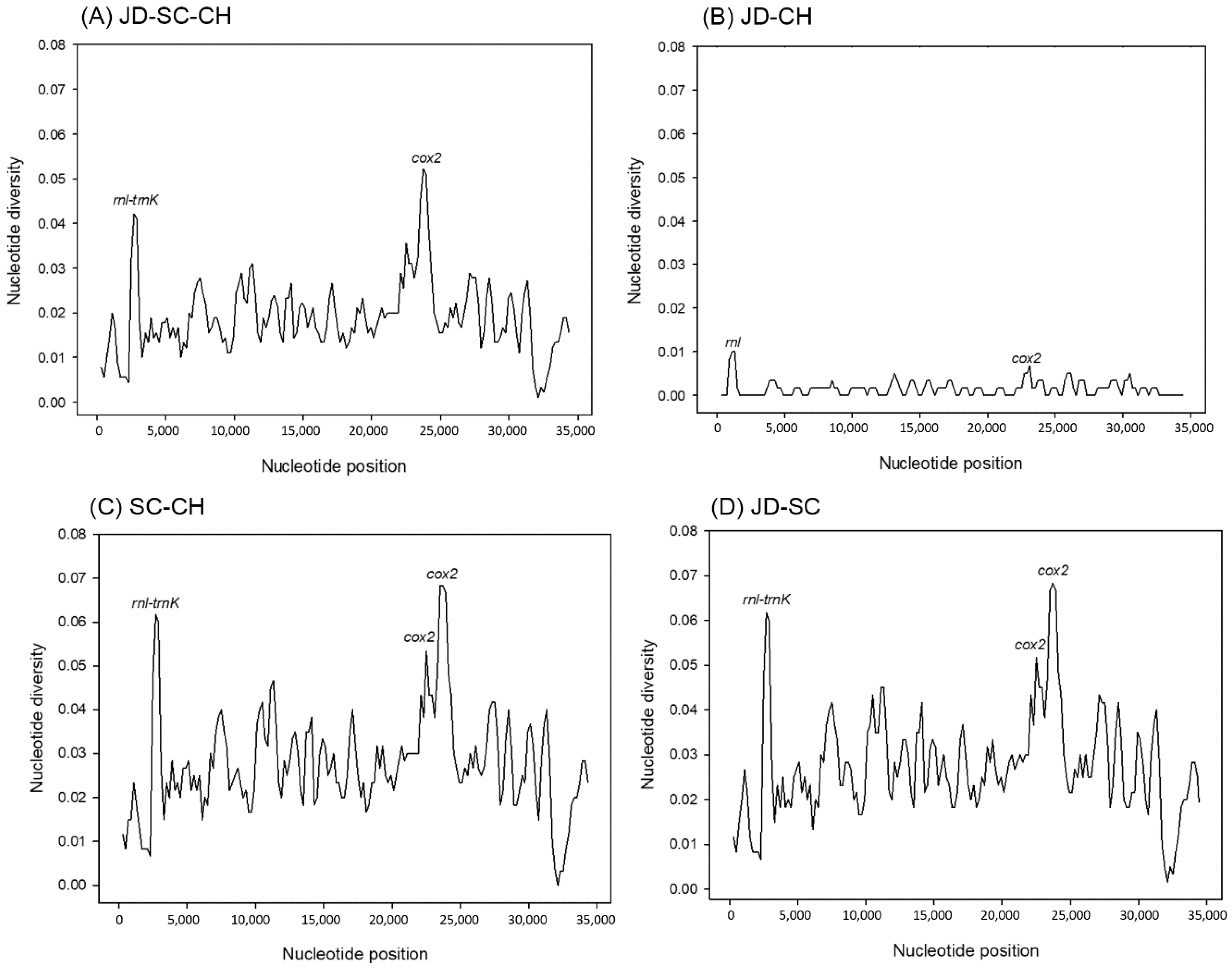
3.3. Nucleotide Diversity and Ka/Ks Ratio
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smetacek, V.; Zingone, A. Green and golden seaweed tides on the rise. Nature 2013, 504, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Hu, C.; Barnes, B.B.; Mitchum, G.; Lapointe, B.; Montoya, J.P. The great Atlantic Sargassum belt. Science 2019, 365, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, X.; Zhuang, M.; Wang, S.; Chen, L.; Shen, H.; He, P. An increase in new Sargassum (Phaeophyceae) blooms along the coast of the East China Sea and Yellow Sea. Phycologia 2019, 58, 374–381. [Google Scholar] [CrossRef]
- Byeon, S.Y.; Oh, H.-J.; Kim, S.; Yun, S.H.; Kang, J.H.; Park, S.R.; Lee, H.J. The origin and population genetic structure of the ‘golden tide’ seaweeds, Sargassum horneri, in Korean waters. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T. Studies on the distribution and drift of the floating seaweeds. Bull. Tohoku Reg. Fish. Res. Lab. 1963, 23, 141–186. [Google Scholar]
- Komatsu, T.; Tatsukawa, K.; Filippi, J.B.; Sagawa, T.; Matsunaga, D.; Mikami, A.; Ishida, K.; Ajisaka, T.; Tanaka, K.; Aoki, M. Distribution of drifting seaweeds in eastern East China Sea. J. Mar. Syst. 2007, 67, 245–252. [Google Scholar] [CrossRef]
- Komatsu, T.; Mizuno, S.; Natheer, A.; Kantachumpoo, A.; Tanaka, K.; Morimoto, A.; Hsiao, S.-T.; Rothäusler, E.A.; Shishidou, H.; Aoki, M. Unusual distribution of floating seaweeds in the East China Sea in the early spring of 2012. J. Appl. Phycol. 2014, 26, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Hwang, E.K.; Lee, S.J.; Ha, D.S.; Park, C.S. Sargassum golden tides in the Shinan-gun and Jeju Island, Korea. Korean J. Fish. Aquat. Sci. 2016, 49, 689–693. [Google Scholar]
- Choi, S.K.; Oh, H.-J.; Yun, S.-H.; Lee, H.J.; Lee, K.; Han, Y.S.; Kim, S.; Park, S.R. Population dynamics of the ‘golden tides’ seaweed, Sargassum horneri, on the southwestern coast of Korea: The extent and formation of golden tides. Sustainability 2020, 12, 2903. [Google Scholar] [CrossRef] [Green Version]
- Guiry, M.; Guiry, G. Sargassum horneri (Turner) C.Agardh 1820. Available online: http://www.algaebase.org (accessed on 1 September 2020).
- Yoshida, G.; Arima, S.; Terawaki, T. Growth and maturation of the ‘autumn-fruiting type’of Sargassum horneri (Fucales, Phaeophyta) and comparisons with the ‘spring-fruiting type’. Phycol. Res. 1998, 46, 183–189. [Google Scholar] [CrossRef]
- Dawes, C.J.; Mathieson, A.C. The Seaweeds of Florida, 1st ed.; University Press of Florida: Gainesville, FL, USA, 2008. [Google Scholar]
- Komatsu, T.; Ariyama, H.; Nakahara, H.; Sakamoto, W. Spatial and temporal distributions of water temperature in a Sargassum forest. J. Oceanogr. Soc. Jpn. 1982, 38, 63–72. [Google Scholar] [CrossRef]
- Thiel, M.; Gutow, L. The ecology of rafting in the marine environment. I. The floating substrata. Oceanogr. Mar. Biol. 2005, 42, 181–264. [Google Scholar]
- Hu, Z.M.; Uwai, S.; Yu, S.H.; Komatsu, T.; Ajisaka, T.; Duan, D.L. Phylogeographic heterogeneity of the brown macroalga Sargassum horneri (Fucaceae) in the northwestern Pacific in relation to late Pleistocene glaciation and tectonic configurations. Mol. Ecol. 2011, 20, 3894–3909. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Jin, Z.; Wang, Y.; Bi, Y.; Melton, J.T. Plastid genome of Dictyopteris divaricata (Dictyotales, Phaeophyceae): Understanding the evolution of plastid genomes in brown algae. Mar. Biotechnol. 2017, 19, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, X.; Che, Z. Mitochondrial genome sequences uncover evolutionary relationships of two Sargassum subgenera, Bactrophycus and Sargassum. J. Appl. Phycol. 2017, 29, 3261–3270. [Google Scholar] [CrossRef]
- Ševčíková, T.; Klimeš, V.; Zbránková, V.; Strnad, H.; Hroudová, M.; Vlček, Č.; Eliáš, M. A comparative analysis of mitochondrial genomes in eustigmatophyte algae. Genome Biol. Evol. 2016, 8, 705–722. [Google Scholar] [CrossRef] [Green Version]
- Keeling, P.J. The endosymbiotic origin, diversification and fate of plastids. Philos. Trans. R. Soc. B 2010, 365, 729–748. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Pan, J.; Zhang, Z.; Moejes, F.W. Organelle genomes of Sargassum confusum (Fucales, Phaeophyceae): mtDNA vs cpDNA. J. Appl. Phycol. 2018, 30, 2715–2722. [Google Scholar] [CrossRef]
- Liu, F.; Pang, S. Chloroplast genome of Sargassum horneri (Sargassaceae, Phaeophyceae): Comparative chloroplast genomics of brown algae. J. Appl. Phycol. 2016, 28, 1419–1426. [Google Scholar] [CrossRef]
- Liu, F.; Pang, S.; Li, X.; Li, J. Complete mitochondrial genome of the brown alga Sargassum horneri (Sargassaceae, Phaeophyceae): Genome organization and phylogenetic analyses. J. Appl. Phycol. 2015, 27, 469–478. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction; Humana Press, Springer: New York, NY, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogame, K.; Uwai, S.; Shimada, S.; Masuda, M. A study of sexual and asexual populations of Scytosiphon lomentaria (Scytosiphonaceae, Phaeophyceae) in Hokkaido, northern Japan, using molecular markers. Eur. J. Phycol. 2005, 40, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Uwai, S.; Kogame, K.; Yoshida, G.; Kawai, H.; Ajisaka, T. Geographical genetic structure and phylogeography of the Sargassum horneri/filicinum complex in Japan, based on the mitochondrial cox3 haplotype. Mar. Biol. 2009, 156, 901–911. [Google Scholar] [CrossRef]
- Doebeli, M.; Dieckmann, U. Speciation along environmental gradients. Nature 2003, 421, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Ballard, J.W.O.; Kreitman, M. Is mitochondrial DNA a strictly neutral marker? Trens. Ecol. Evol. 1995, 10, 485–488. [Google Scholar] [CrossRef]
- Liu, F.; Liu, X.; Wang, Y.; Jin, Z.; Moejes, F.W.; Sun, S. Insights on the Sargassum horneri golden tides in the Yellow Sea inferred from morphological and molecular data. Limnol. Oceanogr. 2018, 63, 1762–1773. [Google Scholar] [CrossRef]
- Voisin, M.; Engel, C.R.; Viard, F. Differential shuffling of native genetic diversity across introduced regions in a brown alga: Aquaculture vs. maritime traffic effects. Proc. Natl. Acad. Sci. USA 2005, 102, 5432–5437. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, Y.; Lei, X.; Zhang, Z. Natural selection of protein structural and functional properties: A single nucleotide polymorphism perspective. Genome. Biol. 2008, 9, R69. [Google Scholar] [CrossRef] [Green Version]
- Sieber, P.; Platzer, M.; Schuster, S. The definition of open reading frame revisited. Trends Genet. 2018, 34, 167–170. [Google Scholar] [CrossRef]
- Nevado, B.; Wong, E.L.; Osborne, O.G.; Filatov, D.A. Adaptive evolution is common in rapid evolutionary radiations. Curr. Biol. 2019, 29, 3081–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheller, H.V.; Jensen, P.E.; Haldrup, A.; Lunde, C.; Knoetzel, J. Role of subunits in eukaryotic Photosystem I. Biochim. Biophys. Acta 2001, 1507, 41–60. [Google Scholar] [CrossRef] [Green Version]
- Venkat, A.; Hahn, M.W.; Thornton, J.W. Multinucleotide mutations cause false inferences of lineage-specific positive selection. Nat. Ecol. Evol. 2018, 2, 1280–1288. [Google Scholar] [CrossRef]
- Schrider, D.R.; Hourmozdi, J.N.; Hahn, M.W. Pervasive multinucleotide mutational events in eukaryotes. Curr. Biol. 2011, 21, 1051–1054. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.; Koskella, B.; Schaack, S. Mutation pressure and the evolution of organelle genomic architecture. Science 2006, 311, 1727–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Le Cam, S.; Daguin-Thiébaut, C.; Bouchemousse, S.; Engelen, A.H.; Mieszkowska, N.; Viard, F. A genome-wide investigation of the worldwide invader Sargassum muticum shows high success albeit (almost) no genetic diversity. Evol. Appl. 2019, 13, 500–514. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | JD | SC | ||
|---|---|---|---|---|
| Molecular Features | Mt Genome | Cp Genome | Mt Genome | Cp Genome |
| GC% | 36.1 | 30.6 | 36.5 | 30.6 |
| rRNA | 3 | 6 | 3 | 6 |
| tRNA | 25 | 28 | 25 | 28 |
| Protein coding genes | 35 | 137 | 35 | 137 |
| Mitochondrial Genome | Chloroplast Genome | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Position | Locus | CH | JD | SC | Position | Locus | CH | JD | SC |
| 1192 | rnl | T | G | G | 1880 | rnl | A | T | T |
| 1295 | rnl | T | G | G | 1881 | rnl | T | A | A |
| 3842 | rps8 | T | C | C | 1911 | rnl | T | C | C |
| 4024 | rps8 | C | A | A | 4201 | rns | G | A | A |
| 4524 | rpl6 | A | G | G | 9208 | ycf47 | T | C | C |
| 5846 | rps4 | G | A | A | 12,192 | psbV | C | T | T |
| 8384 | trnL | - | C | C | 17,056 | dnaK | A | G | G |
| 10,071 | rpl14 | G | A | A | 20,737 | secA | G | A | A |
| 11,590 | rpl16, rps3 | G | A | A | 26,794 | chlB | C | T | T |
| 13,118 | rpl2 | C | T | T | 45,360 | acsF | A | C | C |
| 16,567 | nad2 | A | T | T | 46,202 | rpl35 | G | A | A |
| 17,023 | nad2 | A | T | T | 47,715 | ycf34-trnL IGS | T | - | - |
| 17,224 | nad2 | A | G | G | 67,316 | orf467 | A | C | C |
| 20,832 | cob | C | T | T | 67,665 | orf467 | T | C | C |
| 22,306 | cox2 | A | G | G | 75,251 | petL | T | C | C |
| 23,256 | cox2 | A | G | G | 75,509 | ycf4 | G | C | C |
| 24,936 | nad4 | C | T | T | 77,134 | psaI | A | T | T |
| 25,965 | nad4-nad5 IGS | C | A | A | 77,137 | psaI | G | A | A |
| 26,158 | nad5 | A | T | T | 77,142 | psaI | C | A | A |
| 29,327 | nad11 | C | T | T | 77,148 | psaI | A | T | T |
| 29,522 | nad3 | C | T | T | 77,151 | psaI | A | T | T |
| 30,348 | atp8 | G | A | A | 77,153 | psaI | C | A | A |
| 30,415 | trnS | C | T | T | 77,156 | psaI | G | C | C |
| 31,478 | rpl31 | T | - | - | 77,158 | psaI | T | A | A |
| 34,505* | nad7-trnP IGS (indel region) | - | A | C | 79,770 | rns | C | T | T |
| 34,506 | - | A | A | 82,090 | rnl | A | T | T | |
| 34,507 | - | T | T | 82,091 | rnl | T | A | A | |
| 34,508 | - | A | A | 89,686 | ycf33-ycf39 IGS | T | - | - | |
| 34,509 | - | T | T | 91,872 | sufB | A | G | G | |
| 34,510 | - | T | T | 92,695 | sufB | A | G | G | |
| 34,511 | - | T | T | 98,854 | psbD-trnN IGS (indel region) | A | - | - | |
| 34,512 | - | A | A | 98,855 | T | - | - | ||
| 34,513 | - | G | G | 98,856 | A | - | - | ||
| 34,514 | - | C | C | 98,857 | T | - | - | ||
| 34,515 | - | T | T | 98,858 | A | - | - | ||
| 34,516 | - | G | G | 98,859 | T | - | - | ||
| 34,517 | - | A | A | 10,0113 | dnaB | G | A | A | |
| 34,518 | - | A | A | 10,0384 | dnaB | A | G | G | |
| 10,3700 | petF | C | A | A | |||||
| 10,3801 | petF | T | C | C | |||||
| 10,3851 | petF | G | A | A | |||||
| 11,0124 | tufA | T | G | G | |||||
| 11,4149 | rps13 | A | G | G | |||||
| 11,7303 | rps8 | A | G | G | |||||
| 12,3529 | rpl3 | T | A | A | |||||
| Genomic Locus | JD-CH Ka/Ks | SC-CH Ka/Ks | JD-SC Ka/Ks |
|---|---|---|---|
| dnaB | 1.7333 | 0.1813 | 0.2180 |
| orf467 | 3.8889 | 0.1740 | 0.1662 |
| petF | 0.0000 | 1.1905 | 0.2925 |
| psaI | 0.6808 | 5.9401 | 0.0000 |
| secA | 3.6000 | 0.0372 | 0.0174 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byeon, S.Y.; Cheon, K.-S.; Kim, S.; Yun, S.-H.; Oh, H.-J.; Park, S.R.; Kim, T.-H.; Kim, J.K.; Lee, H.J. Comparative Analysis of Sequence Polymorphism in Complete Organelle Genomes of the ‘Golden Tide’ Seaweed Sargassum horneri between Korean and Chinese Forms. Sustainability 2020, 12, 7280. https://0-doi-org.brum.beds.ac.uk/10.3390/su12187280
Byeon SY, Cheon K-S, Kim S, Yun S-H, Oh H-J, Park SR, Kim T-H, Kim JK, Lee HJ. Comparative Analysis of Sequence Polymorphism in Complete Organelle Genomes of the ‘Golden Tide’ Seaweed Sargassum horneri between Korean and Chinese Forms. Sustainability. 2020; 12(18):7280. https://0-doi-org.brum.beds.ac.uk/10.3390/su12187280
Chicago/Turabian StyleByeon, Seo Yeon, Kyeong-Sik Cheon, Sangil Kim, Suk-Hyun Yun, Hyun-Ju Oh, Sang Rul Park, Tae-Hoon Kim, Jang Kyun Kim, and Hyuk Je Lee. 2020. "Comparative Analysis of Sequence Polymorphism in Complete Organelle Genomes of the ‘Golden Tide’ Seaweed Sargassum horneri between Korean and Chinese Forms" Sustainability 12, no. 18: 7280. https://0-doi-org.brum.beds.ac.uk/10.3390/su12187280