Delphinidin Ameliorates Hepatic Triglyceride Accumulation in Human HepG2 Cells, but Not in Diet-Induced Obese Mice

Abstract
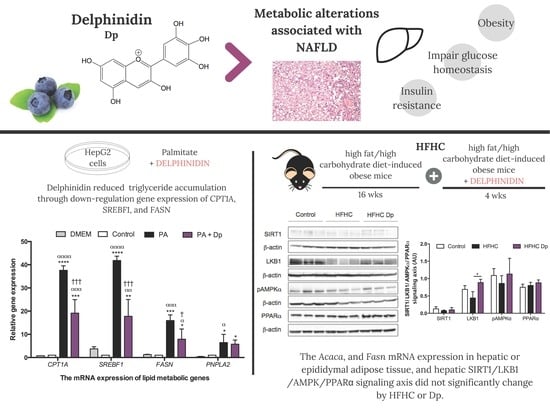
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
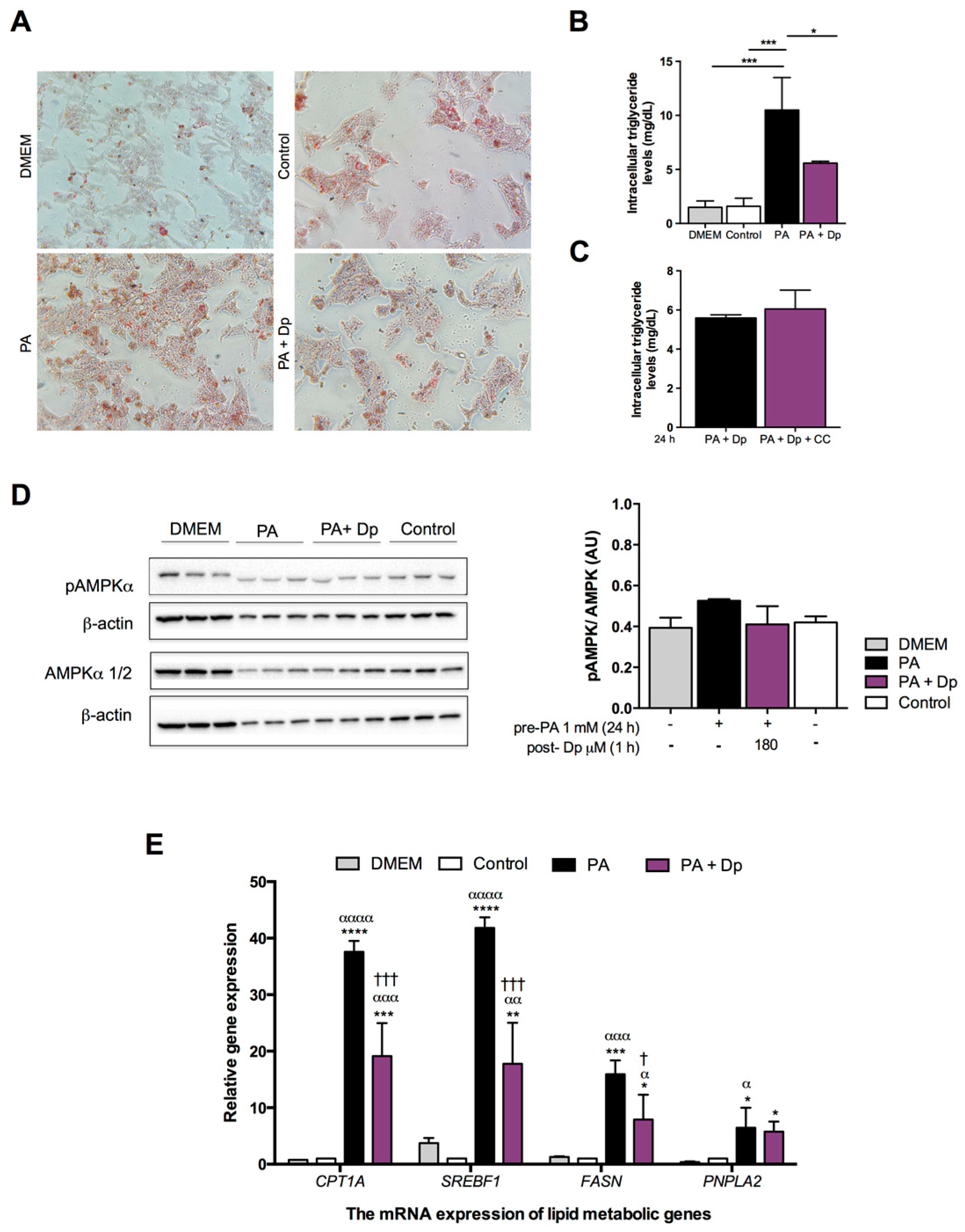
2.3. Preparation of PA and Intracellular Lipid Accumulation
2.4. Dp Treatment on the Fatty Liver Cell Model
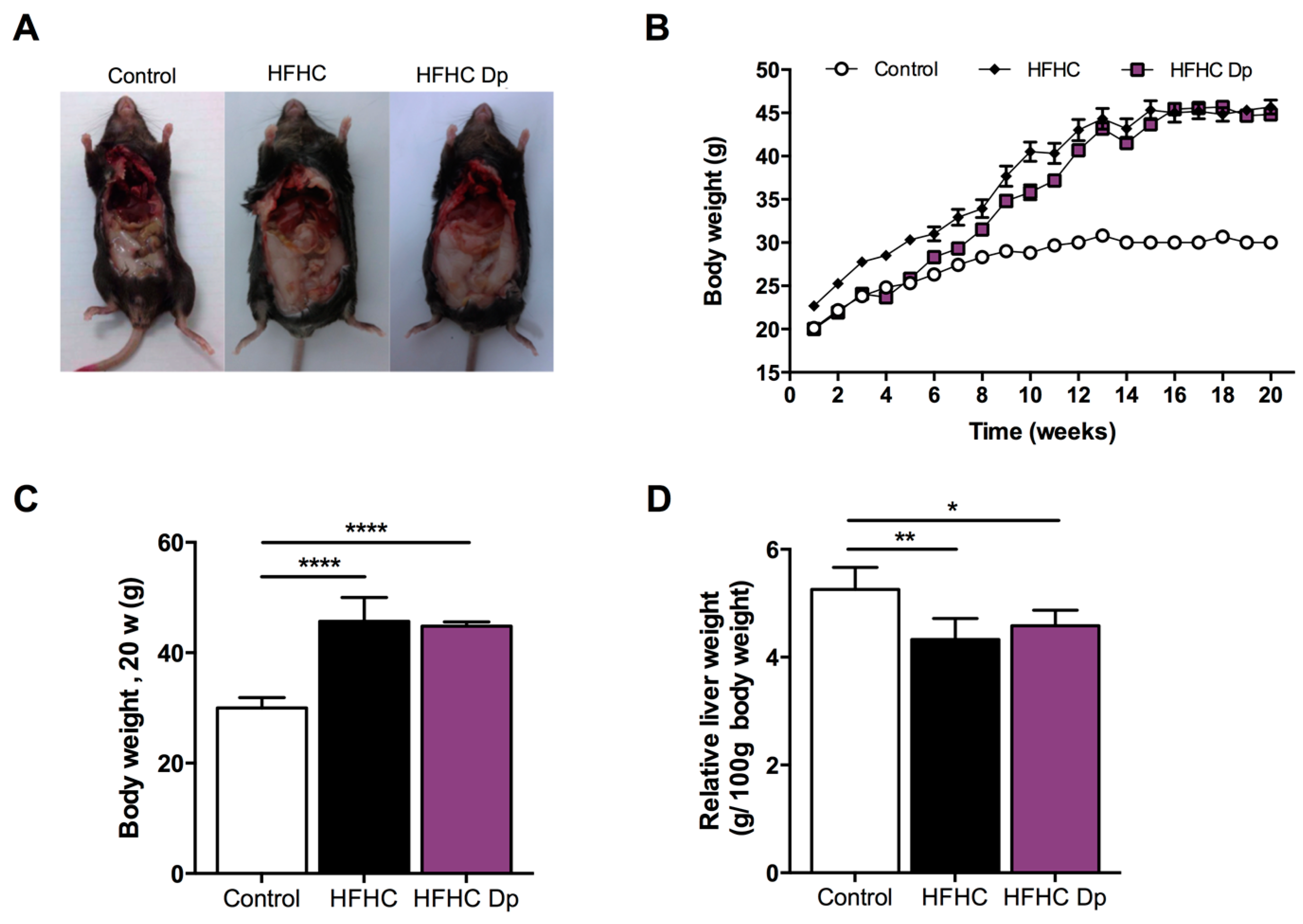
2.5. Animal Experiments
2.6. Diets, Food Intake, and Energy Intake
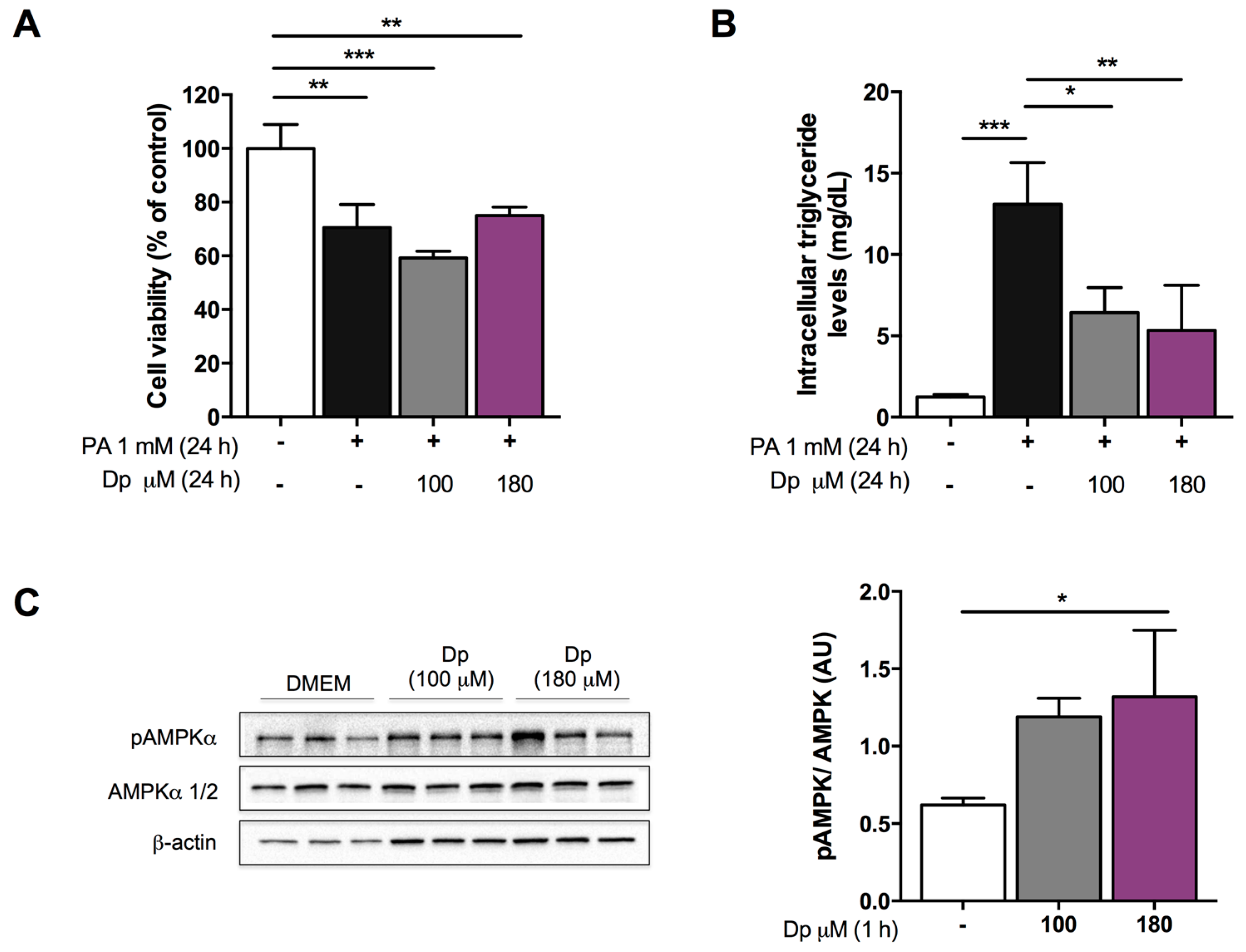
2.7. Cell Viability Assay
2.8. Determination of Intracellular Lipid Accumulation Levels
2.9. Weight, Fasting Glucose, and Insulin Sensitivity
2.10. Liver Histology Analysis
2.11. Western Blotting Analysis
2.12. Quantitative Real-Time PCR and Gene Expression Analysis
2.13. Statistical Analysis
3. Results
3.1. Dp Dose Selection in HepG2 Cells
3.2. Effects of Dp on Lipid Metabolism of the Fatty Liver Cell Model
3.3. Effects of Dp on the Body Weight, Liver Weight, Energy Intake, and Food Intake of Experimental Animals
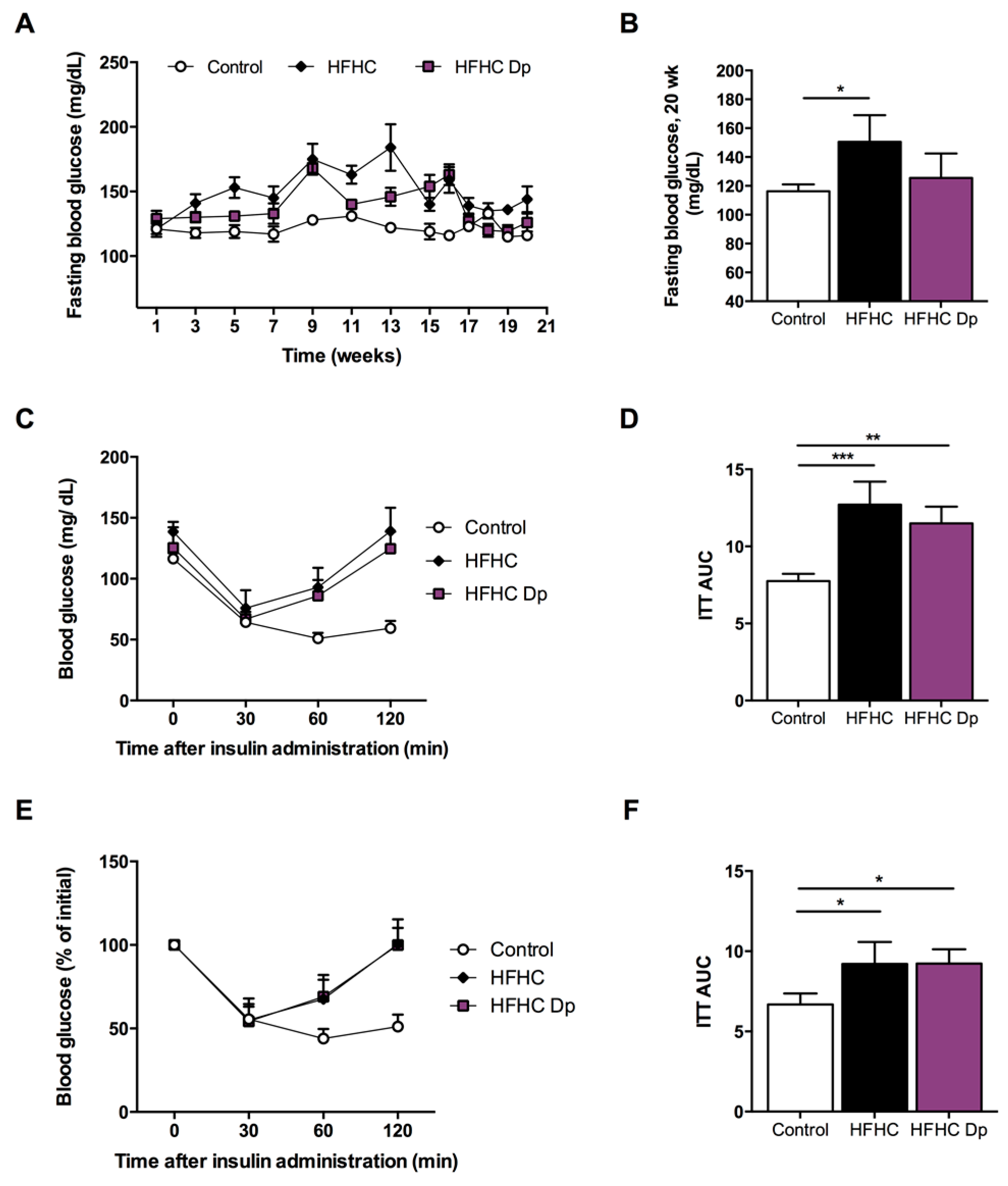
3.4. Effects of Dp on Glucose Homeostasis and Insulin Sensitivity of Experimental Animals
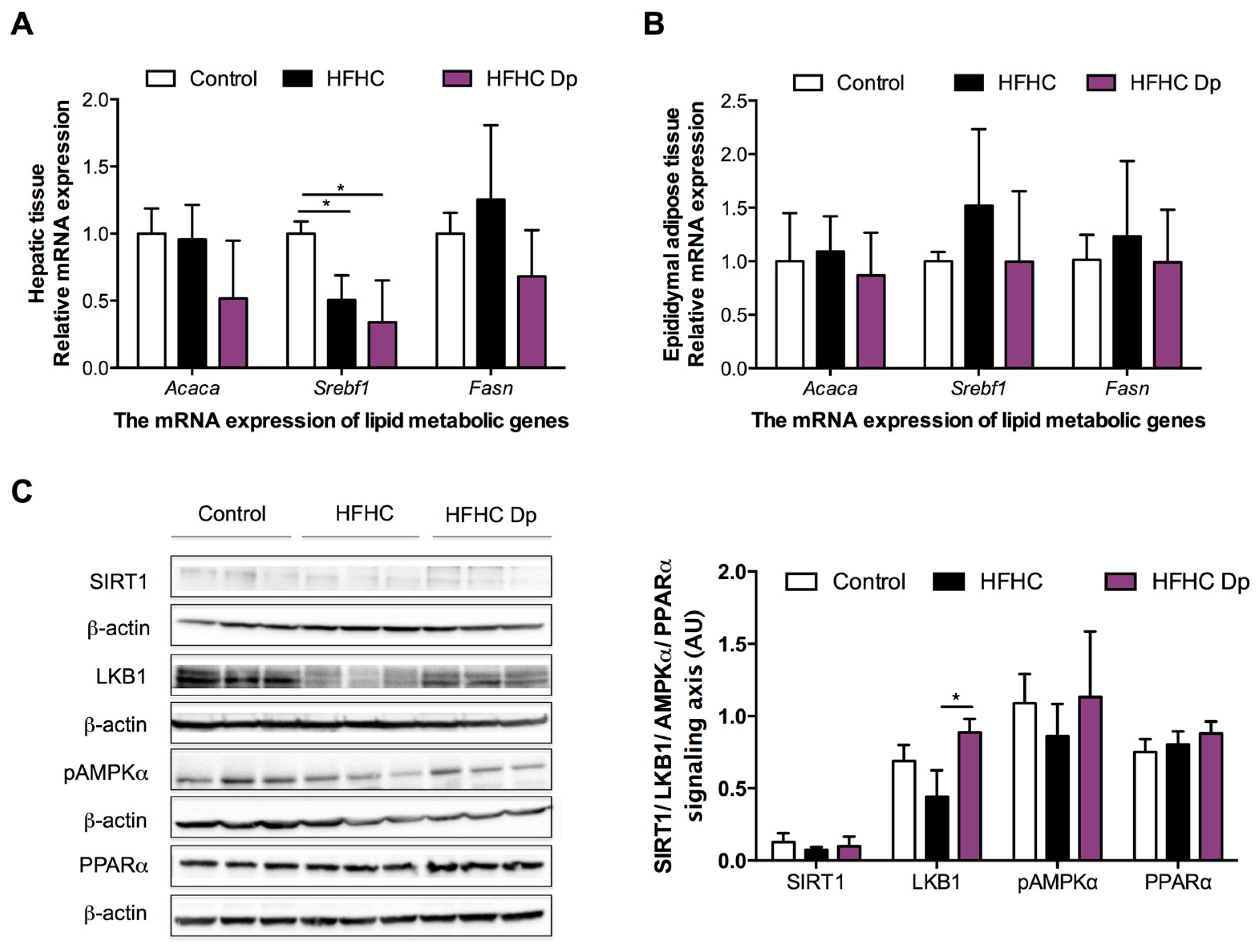
3.5. Effects of Dp on Lipid Metabolic Gene Expression of Experimental Animals
3.6. Effects of Dp on the SIRT1/LKB1/AMPK and PPARα Signaling Axis of Experimental Animals
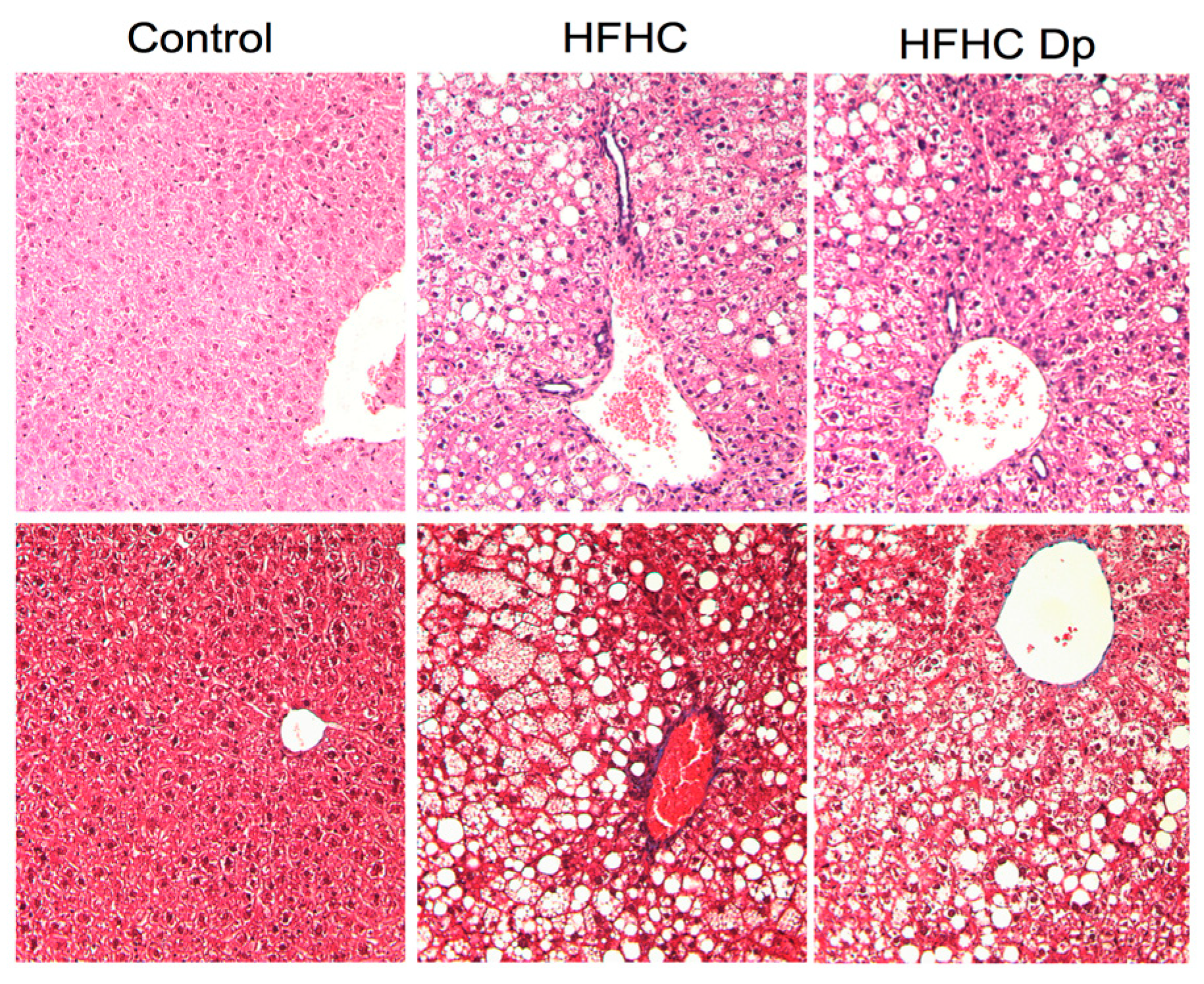
3.7. Effect of Dp on Liver Histology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC1 | acetyl-CoA carboxylase 1 |
| AMPK | AMP-activated protein kinase |
| ATGL | adipose triglyceride lipase |
| AUC | area under the curve |
| Bw | body weight |
| CC | compound C |
| cDNA | complementary DNA |
| CPT1A | carnitine palmitoyltransferase 1A |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | dimethyl sulfoxide |
| dNTP | deoxynucleotide triphosphate |
| Dp | delphinidin |
| DTT | dithiothreitol |
| FAS | fatty acid synthase |
| FBS | fetal bovine serum |
| H&E | hematoxylin and eosin |
| HCD | high-complex-carbohydrate/high-calorie diet |
| HFD | high-fat diet |
| HFHC | high-fat and high-carbohydrate diet |
| HH | high-fat/high-sucrose diet |
| HL | high-fat/low-sucrose diet |
| ITT | insulin tolerance test |
| LH | low-fat/high-sucrose diet |
| LKB1 | liver kinase B1 |
| LL | low-fat/low-sucrose diet |
| M-MLV | Moloney Murine Leukemia Virus |
| mRNA | messenger RNA |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NAFLD | non-alcoholic fatty liver disease |
| NAS | NASH activity score |
| NASH | non-alcoholic steatohepatitis |
| NEFA-free BSA | non-esterified fatty-acid-free bovine serum albumin |
| ORO | Oil Red O staining |
| PA | sodium palmitate |
| PBS | phosphate-buffered saline |
| PVDF | polyvinylidene difluoride |
| SEM | standard error of the mean |
| PPARα | peroxisome proliferator-activated receptor alpha |
| SIRT1 | sirtuin 1 |
| SREBP1c | sterol regulatory element binding protein-1c |
| SS | simple steatosis |
References
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, G.; Nobili, V.; Tiribelli, C. Epidemiology of fatty liver: An update. World J. Gastroenterol. 2014, 20, 9050–9054. [Google Scholar] [CrossRef] [PubMed]
- Calzadilla Bertot, L.; Adams, L.A. The natural course of non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, S.; Abeygunasekera, S.; Farrell, G.C.; Holmes-Walker, J.; Hui, J.M.; Fung, C.; Karim, R.; Lin, R.; Samarasinghe, D.; Liddle, C.; et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 2002, 35, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaja, A.J. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J. Gastroenterol. 2014, 20, 2515–2532. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, G.; Rastogi, A.; Trehanpati, N.; Sen, B.; Khosla, R.; Sarin, S.K. From cirrhosis to hepatocellular carcinoma: New molecular insights on inflammation and cellular senescence. Liver Cancer 2013, 2, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Chitturi, S.; Wong, G.L.; Yu, J.; Chan, H.L.; Farrell, G.C. Pathogenesis and novel treatment options for non-alcoholic steatohepatitis. Lancet Gastroenterol. Hepatol. 2016, 1, 56–67. [Google Scholar] [CrossRef]
- Suárez, M.; Boqué, N.; Del Bas, J.M.; Mayneris-Perxachs, J.; Arola, L.; Caimari, A. Mediterranean Diet and Multi-Ingredient-Based Interventions for the Management of Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 1052. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alonso, M.; Rimbach, G.; Sasai, M.; Nakahara, M.; Matsugo, S.; Uchida, Y.; Rivas-Gonzalo, J.C.; De Pascual-Teresa, S. Electron spin resonance spectroscopy studies on the free radical scavenging activity of wine anthocyanins and pyranoanthocyanins. Mol. Nutr. Food Res. 2005, 49, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.J.; Howard, L.R.; Prior, R.L.; Clark, J.R. Flavonoid glycosides and antioxidant capacity of various blackberry, blueberry and red grape genotypes determined by high-performance liquid chromatography/mass spectrometry. J. Sci. Food. Agric. 2004, 84, 1771–1782. [Google Scholar] [CrossRef]
- Kolehmainen, M.; Mykkänen, O.; Kirjavainen, P.V.; Leppänen, T.; Moilanen, E.; Adriaens, M.; Laaksonen, D.E.; Hallikainen, M.; Puupponen-Pimiä, R.; Pulkkinen, L.; et al. Bilberries reduce low-grade inflammation in individuals with features of metabolic syndrome. Mol. Nutr. Food Res. 2012, 56, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Pojer, E.; Mattivi, F.; Johnson, D.; Stockley, C.S. The case for anthocyanin consumption to promote human health: A review. Compr. Rev. Food Sci. Food Saf. 2013, 12, 483–508. [Google Scholar] [CrossRef]
- Kähkönen, M.P.; Heinonen, M. Antioxidant activity of anthocyanins and their aglycons. J. Agric. Food Chem. 2003, 51, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Slimestad, R.; Solheim, H. Anthocyanins from black currants (Ribes nigrum L.). J. Agric. Food Chem. 2002, 50, 3228–3231. [Google Scholar] [CrossRef] [PubMed]
- Takeoka, G.R.; Dao, L.T.; Full, G.H.; Wong, R.Y.; Harden, L.A.; Edwards, R.H.; Berrios, J.D. Characterization of black bean (Phaseolus vulgaris L.) anthocyanins. J. Agric. Food Chem. 1997, 45, 3395–3400. [Google Scholar] [CrossRef]
- Overall, J.; Bonney, S.A.; Wilson, M.; Beermann, A.; Grace, M.H.; Esposito, D.; Lila, M.A.; Komarnytsky, S. Metabolic effects of berries with structurally diverse anthocyanins. Int. J. Mol. Sci. 2017, 18, 422. [Google Scholar] [CrossRef] [PubMed]
- Sindi, H.A.; Marshall, L.J.; Morgan, M.R. Comparative chemical and biochemical analysis of extracts of Hibiscus sabdariffa. Food Chem. 2014, 164, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ling, W.; Yang, Y.; Chen, Y.; Tian, Z.; Du, Z.; Chen, J.; Xie, Y.; Liu, Z.; Yang, L. Role of Purified Anthocyanins in Improving Cardiometabolic Risk Factors in Chinese Men and Women with Prediabetes or Early Untreated Diabetes-A Randomized Controlled Trial. Nutrients 2017, 9, 1104. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xia, M.; Zou, T.; Ling, W.; Zhong, R.; Zhang, W. Cyanidin 3-glucoside attenuates obesity-associated insulin resistance and hepatic steatosis in high-fat diet-fed and db/db mice via the transcription factor FoxO1. J. Nutr. Biochem. 2012, 23, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.W.; Chang, C.L.; Kao, E.S.; Lin, J.H. Effect of Hibiscus sabdariffa extract on high fat diet-induced obesity and liver damage in hamsters. Food Nutr. Res. 2015, 59. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, G.; Zhong, R.; Wang, Y.; Wang, D.; Xia, M. Cyanidin-3-O-β-glucoside regulates fatty acid metabolism via an AMP-activated protein kinase-dependent signaling pathway in human HepG2 cells. Lipids Health Dis. 2012, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Qi, X.; Liu, Y.; Guo, J.; Zhu, R.; Chen, W.; Zheng, X.; Yu, T. Dietary supplementation with purified mulberry (Morus australis Poir) anthocyanins suppresses body weight gain in high-fat diet fed C57BL/6 mice. Food Chem. 2013, 141, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Horio, F.; Uchida, K.; Aoki, H.; Osawa, T. Dietary cyanidin 3-O-beta-d-glucoside-rich purple corn color prevents obesity and ameliorates hyperglycemia in mice. J. Nutr. 2003, 133, 2125–2130. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.P.; Choi, J.H.; Han, E.H.; Kim, H.G.; Wee, J.H.; Jung, K.O.; Jung, K.H.; Kwon, K.I.; Jeong, T.C.; Chung, Y.C.; et al. Purple sweet potato anthocyanins attenuate hepatic lipid accumulation through activating adenosine monophosphate-activated protein kinase in human HepG2 cells and obese mice. Nutr. Res. 2011, 31, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.J.; Hsu, M.J.; Huang, H.P.; Chung, D.J.; Chang, Y.C.; Wang, C.J. Mulberry anthocyanins inhibit oleic acid induced lipid accumulation by reduction of lipogenesis and promotion of hepatic lipid clearance. J. Agric. Food Chem. 2013, 61, 6069–6076. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.; Prentki, M. AMP kinase and malonyl-CoA: Targets for therapy of the metabolic syndrome. Nat. Rev. Drug Discov. 2004, 3, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Kim, J.Y.; Jun, H.J.; Kim, S.J.; Lee, J.H.; Hoang, M.H.; Kim, H.S.; Chang, H.I.; Hwang, K.Y.; Um, S.J.; et al. Cyanidin is an agonistic ligand for peroxisome proliferator-activated receptor-alpha reducing hepatic lipid. Biochim. Biophys. Acta 2013, 1831, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, P.; English, T.; Karki, S.; Qiang, L.; Tao, R.; Kim, J.; Luo, Z.; Farmer, S.R.; Kandror, K.V. SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J. Lipid Res. 2011, 52, 1693–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, K.T.; Mashek, M.T.; Bu, S.Y.; Greenberg, A.S.; Mashek, D.G. ATGL is a major hepatic lipase that regulates TAG turnover and fatty acid signaling and partitioning. Hepatology 2011, 53, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, L.; Wang, D.; Huo, Y.; Ji, B. Anthocyanin-rich extracts from blackberry, wild blueberry, strawberry, and chokeberry: Antioxidant activity and inhibitory effect on oleic acid-induced hepatic steatosis in vitro. J. Sci. Food Agric. 2016, 96, 2494–2503. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Kern, M.; Fridrich, D.; Reichert, J.; Skrbek, S.; Nussher, A.; Hofem, S.; Vatter, S.; Pahlke, G.; Rüfer, C.; Marko, D. Limited stability in cell culture medium and hydrogen peroxide formation affect the growth inhibitory properties of delphinidin and its degradation product gallic acid. Mol. Nutr. Food Res. 2007, 51, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Woo, S.L.; Guo, X.; Li, H.; Zheng, J.; Botchlett, R.; Liu, M.; Pei, Y.; Xu, H.; Cai, Y.; et al. Berberine ameliorates hepatic steatosis and suppresses liver and mice with diet-induced obesity. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Daveri, E.; Cremonini, E.; Mastaloudis, A.; Hester, S.N.; Wood, S.M.; Waterhouse, A.L.; Anderson, M.; Fraga, C.G.; Oteiza, P.I. Cyanidin and delphinidin modulate inflammation and altered redox signaling improving insulin resistance in high fat-fed mice. Redox Biol. 2018, 18, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wu, T.; Xu, D.; Chu, Q.; Lin, D.; Zheng, X. Dietary sweet cherry anthocyanins attenuates diet-induced hepatic steatosis by improving hepatic lipid metabolism in mice. Nutrition 2016, 32, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Jois, T.; Howard, V.; Youngs, K.; Cowley, M.A.; Sleeman, M.W. Dietary macronutrient composition directs ChREBP isoform expression and glucose metabolism in mice. PLoS ONE 2016, 12, E0168797. [Google Scholar] [CrossRef] [PubMed]
- Black, B.L.; Croom, J.; Eisen, E.J.; Petro, A.E.; Edwards, C.L.; Surwit, R.S. Differential effects of fat and sucrose on body composition in A/J and C57BL/6 mice. Metabolism 1998, 47, 1354–1359. [Google Scholar] [CrossRef]
- Lomba, A.; Martínez, J.A.; García-Díaz, D.F.; Paternain, L.; Marti, A.; Campión, J.; Milagro, F.I. Weight gain induced by an isocaloric pair-fed high fat diet: A nutriepigenetic study on FASN and NDUFB6 gene promoters. Mol. Genet. Metab. 2010, 101, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaya, T.; Tanaka, N.; Suzuki, T.; Sano, K.; Horiuchi, A.; Komatsu, M.; Nakajima, T.; Nishizawa, T.; Joshita, S.; Umemura, T.; et al. Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J. Hepatol. 2010, 53, 724–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, C.R.; Ertel, R.L.; Bauwens, J.D.; Schmuck, E.G.; Mulligan, J.D.; Saupe, K.W. A high-fat diet decreases AMPK activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats. J. Physiol. Biochem. 2013, 69, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.G.; She, H.; Cheng, J.H.; French, S.W.; Koop, D.R.; Xiong, S.; Tsukamoto, H. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology 2005, 42, 905–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudaba, N.; Marion, A.; Huet, C.; Pierre, R.; Viollet, B.; Foretz, M. AMPK Re-Activation Suppresses Hepatic Steatosis but its Downregulation Does Not Promote Fatty Liver Development. EBioMedicine 2018, 28, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Rojo, L.E.; Ribnicky, D.; Logendra, S.; Poulev, A.; Rojas-Silva, P.; Kuhn, P.; Dorn, R.; Grace, M.H.; Lila, M.A.; Raskin, I. In Vitro and in Vivo Anti-Diabetic Effects of Anthocyanins from Maqui Berry (Aristotelia chilensis). Food Chem. 2012, 131, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, R.A.; Morrison, M.C.; Sheedfar, F.; Mulder, P.; Schreurs, M.; Hommelberg, P.P.; Hofker, M.H.; Schalkwijk, C.; Kleemann, R.; Tietge, U.J.; et al. Effects of Anthocyanin and Flavanol Compounds on Lipid Metabolism and Adipose Tissue Associated Systemic Inflammation in Diet-Induced Obesity. Med. Inflamm. 2016, 2016, 20421072016. [Google Scholar] [CrossRef] [PubMed]
- Grace, M.H.; Ribnicky, D.M.; Kuhn, P.; Poulev, A.; Logendra, S.; Yousef, G.G.; Raskin, I.; Lila, M.A. Hypoglycemic activity of a novel anthocyanin-rich formulation from lowbush blueberry, Vaccinium angustifolium Aiton. Phytomedicine 2009, 16, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Beecher, G.R.; Holden, J.M.; Haytowitz, D.B.; Gebhardt, S.E.; Prior, R.L. Concentrations of anthocyanins in common foods in the United States and estimation of normal consumption. J. Agric. Food Chem. 2006, 54, 4069–4075. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Nonaka, H.; Komatsu, S.; Goto, M.; Morozumi, M.; Yamada, S.; Lin, I.C.; Yamashita, S.; Tachibana, H. Delphinidin Prevents Muscle Atrophy and Upregulates miR-23a Expression. J. Agric. Food Chem. 2016, 65, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, S.; Suzuki, K.; Muramatsu, M.; Nomura, A.; Inoue, F.; Into, T.; Yoshiko, Y.; Niida, S. Delphinidin, one of the major anthocyanidins, prevents bone loss through the inhibition of excessive osteoclastogenesis in osteoporosis model mice. PLoS ONE 2014, 9, e97177. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, I.; Faria, A.; Calhau, C.; de Freitas, V.; Mateus, N. Bioavailability of anthocyanins and derivatives. J. Funct. Foods 2014, 7, 54–66. [Google Scholar] [CrossRef]
- Matsumoto, H.; Inaba, H.; Kishi, M.; Tominaga, S.; Hirayama, M.; Tsuda, T. Orally administered delphinidin 3-rutinoside and cyanidin 3-rutinoside are directly absorbed in rats and humans and appear in the blood as the intact forms. J. Agric. Food Chem. 2001, 49, 1546–1551. [Google Scholar] [CrossRef] [PubMed]
- Titta, L.; Trinei, M.; Stendardo, M.; Berniakovich, I.; Petroni, K.; Tonelli, C.; Riso, P.; Porrini, M.; Minucci, S.; Pelicci, P.G.; et al. Blood orange juice inhibits fat accumulation in mice. Int. J. Obes. 2010, 34, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Wan, T.; Wang, S.; Ye, M.; Qiu, Y.; Jiang, R.; Pang, N.; Huang, Y.; Zhou, Y.; Jiang, X.; et al. Cyanidin-3-O-β-glucoside regulates the activation and the secretion of adipokines from brown adipose tissue and alleviates diet induced fatty liver. Biomed. Pharmacother. 2018, 105, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Gupta, M.K.; Wang, G.X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 128, 1199. [Google Scholar] [CrossRef] [PubMed]
- Kanuri, G.; Bergheim, I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int. J. Mol. Sci. 2013, 14, 11963–11980. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | HFHC | HFHC-Dp | |
|---|---|---|---|
| Prior to Dp treatment | |||
| Energy intake (kcal/day) | 11.15 ± 0.08 a | 11.7 ± 0.59 a | 11.27 ± 0.16 a |
| Food intake (g/day) | 3.66 ± 0.03 a | 2.42 ± 0.13 b | 2.33 ± 0.04 b |
| Energy intake from fat (kcal/day) | 3.21 ± 0.02 a | 5. 02 ± 0.27 b | 4.83 ± 0.09 b |
| Energy intake from fat (% kcal) | 28.8 | 45 | 45 |
| High-carbohydrate drinking water intake (mL/day) | 0.00 ± 0.00 a | 3.27 ± 0.12 b | 3.22 ± 0.15 b |
| Energy intake from high-carbohydrate drinking water (kcal/day) | 0.00 ± 0.00 a | 0.55 ± 0.02 b | 0.54 ± 0.03 b |
| During Dp treatment | |||
| Energy intake (kcal/day) | 10.74 ± 0.08 a | 11.21 ± 1.28 a | 11.19 ± 1.36 a |
| Food intake (g/day) | 3.53 ± 0.03 a | 2.33 ± 0.28 b | 2.33 ± 0.29 b |
| Energy intake from fat (kcal/day) | 3.09 ± 0.02 a | 4.81 ± 0.57 b | 4.82 ± 0.60 b |
| Energy intake from fat (% kcal) | 28.8 | 45 | 45 |
| High-carbohydrate drinking water intake (mL/day) | 0.00 ± 0.00 a | 3.06 ± 0.14 b | 2.93 ± 0.20 b |
| Energy intake from high-carbohydrate drinking water (kcal/day) | 0.00 ± 0.00 a | 0.51 ± 0.02 b | 0.49 ± 0.03 b |
| Grade | Control (n = 6) | HFHC (n = 6) | HFHC-Dp (n = 6) | |
|---|---|---|---|---|
| Steatosis | 0/1/2/3 | 6/0/0/0 a | 0/0/0/6 b | 0/0/1/5 b |
| Lobular inflammation | 0/1/2/3 | 6/0/0/0 a | 0/0/2/4 b | 0/0/0/6 b |
| Ballooning | 0/1/2 | 6/0/0/0 a | 0/3/3 b | 0/3/3 b |
| NAS score * | 0 ± 0.00 a | 7.17 ± 0.98 b | 7.33 ± 0.82 b | |
| Portal inflammation | 0/1/2/3/4 | 6/0/0/0 a | 1/5/0/0 a | 1/5/0/0 a |
| Fibrosis | 0/1/2/3/4 | 6/0/0/0 a | 5/1/0/0 a | 6/0/0/0 a |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parra-Vargas, M.; Sandoval-Rodriguez, A.; Rodriguez-Echevarria, R.; Dominguez-Rosales, J.A.; Santos-Garcia, A.; Armendariz-Borunda, J. Delphinidin Ameliorates Hepatic Triglyceride Accumulation in Human HepG2 Cells, but Not in Diet-Induced Obese Mice. Nutrients 2018, 10, 1060. https://0-doi-org.brum.beds.ac.uk/10.3390/nu10081060
Parra-Vargas M, Sandoval-Rodriguez A, Rodriguez-Echevarria R, Dominguez-Rosales JA, Santos-Garcia A, Armendariz-Borunda J. Delphinidin Ameliorates Hepatic Triglyceride Accumulation in Human HepG2 Cells, but Not in Diet-Induced Obese Mice. Nutrients. 2018; 10(8):1060. https://0-doi-org.brum.beds.ac.uk/10.3390/nu10081060
Chicago/Turabian StyleParra-Vargas, Marcela, Ana Sandoval-Rodriguez, Roberto Rodriguez-Echevarria, Jose Alfredo Dominguez-Rosales, Arturo Santos-Garcia, and Juan Armendariz-Borunda. 2018. "Delphinidin Ameliorates Hepatic Triglyceride Accumulation in Human HepG2 Cells, but Not in Diet-Induced Obese Mice" Nutrients 10, no. 8: 1060. https://0-doi-org.brum.beds.ac.uk/10.3390/nu10081060