DNA Methylation Signatures of Breastfeeding in Buccal Cells Collected in Mid-Childhood

 ,
,  , , , , and
, , , , and
Abstract
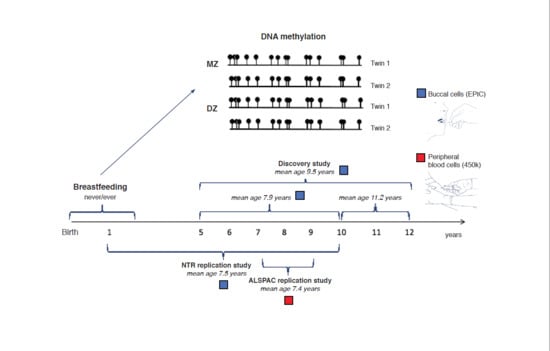
:
1. Introduction
2. Materials and Methods
2.1. Overview
2.2. Subjects and Samples
2.2.1. Discovery Study
2.2.2. NTR Replication Study
2.2.3. ALSPAC Replication Study
2.3. Phenotype Data
ALSPAC Replication Study
2.4. DNA Sample Collection
2.4.1. NTR (Discovery and Replication Study)
2.4.2. ALSPAC Replication Study
2.5. DNA-Methylation Measurements
2.5.1. NTR Discovery Study
2.5.2. NTR Replication Study
2.5.3. ALSPAC Replication Study
2.6. Cellular Proportions
2.6.1. NTR Discovery and Replication Study
2.6.2. ALSPAC Replication Study
2.7. Data Analyses
2.7.1. Associations between Breastfeeding and Pre- and Perinatal Factors
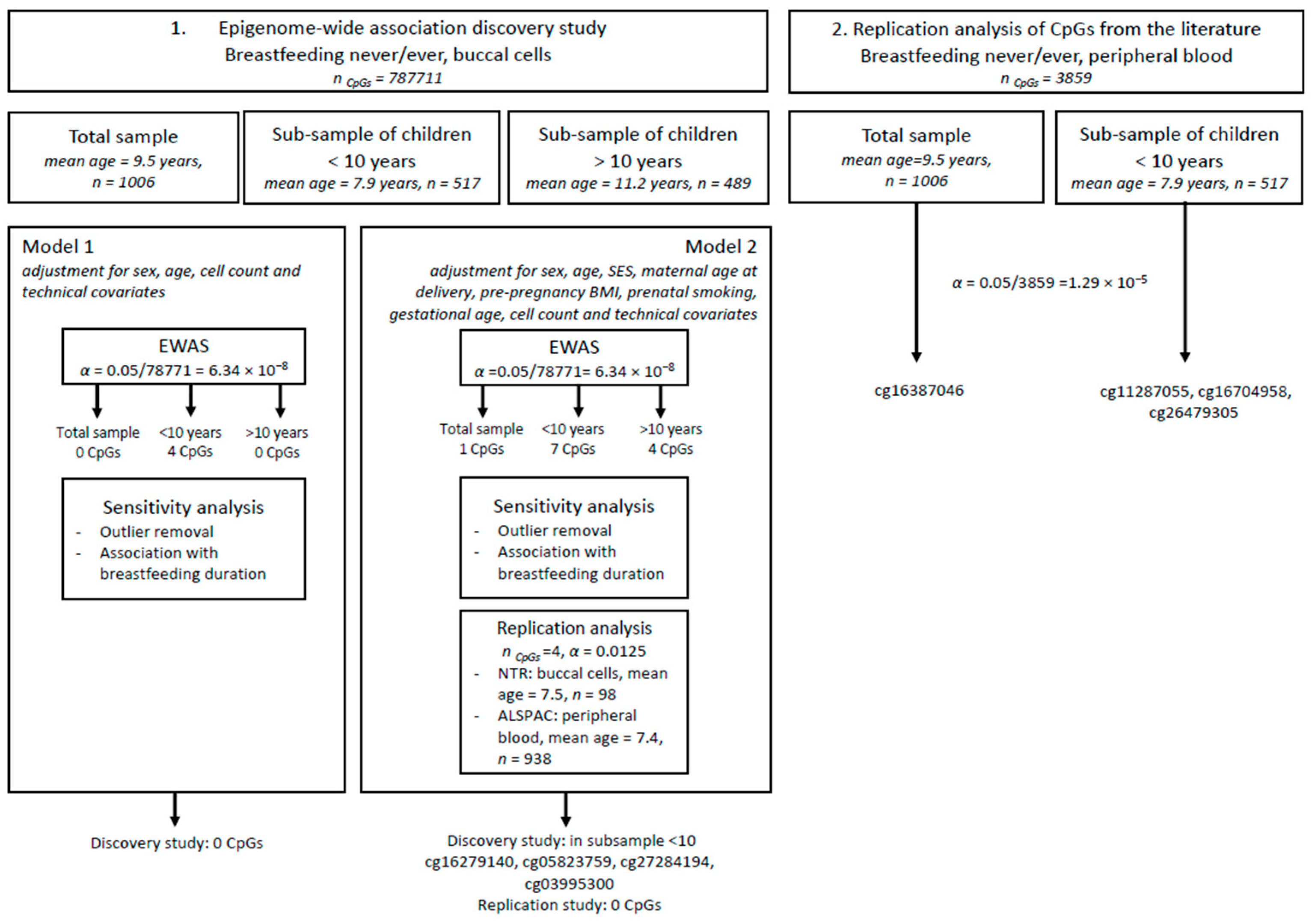
2.7.2. EWAS
Discovery Study
Twin Correlations
Replication
2.8. Methylation Data Annotation
2.9. Overlap with Previous Findings
3. Results
3.1. Descriptive Statistics
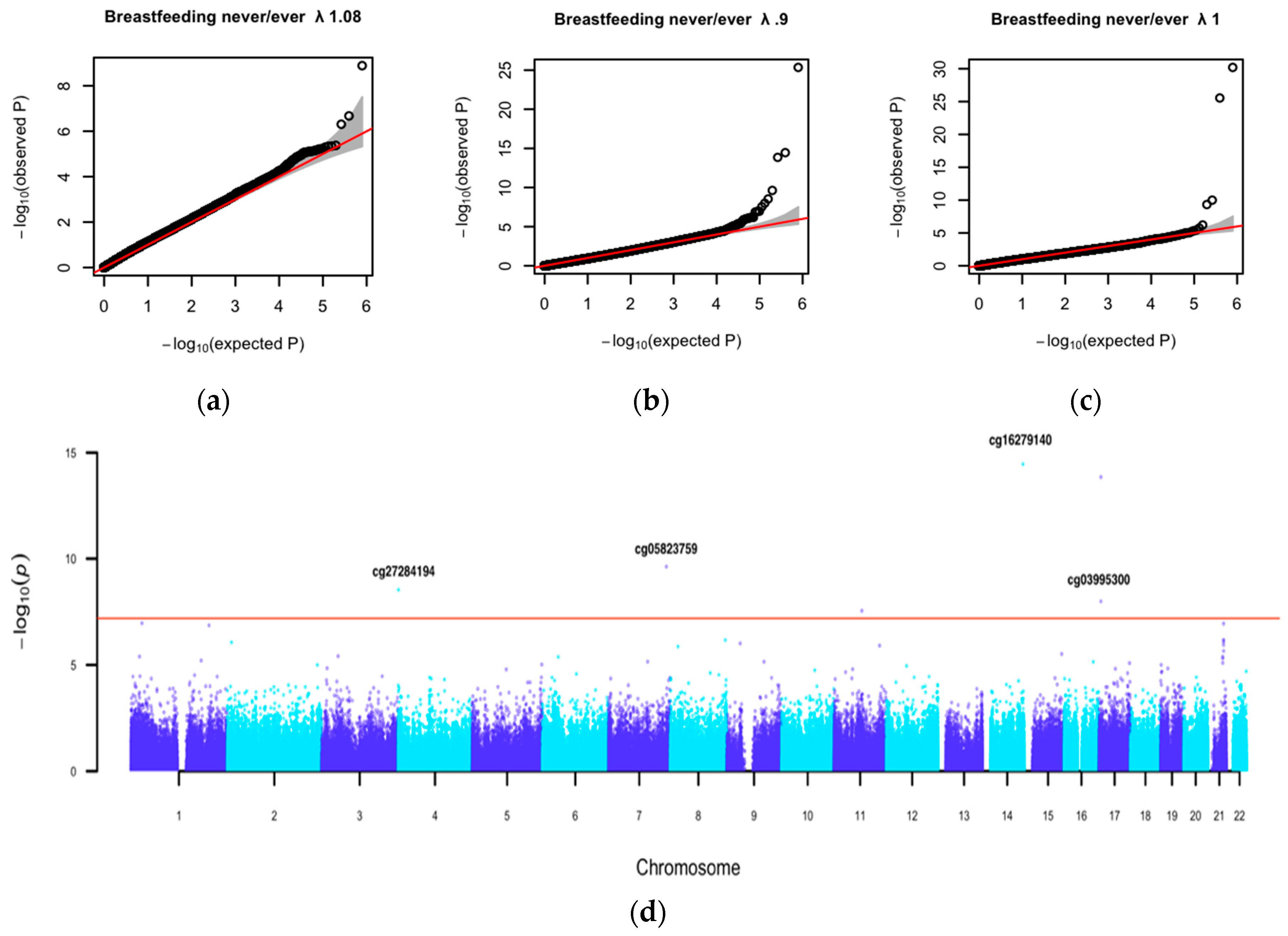
3.2. Association Analysis Findings
3.3. Replication Analysis of Our Findings in Other Samples
3.4. Replication Analysis of Findings from Previous EWAS
3.5. Methylation Data Annotation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CpG | cytosine-phosphate-guanine |
| EWAS | epigenome-wide association study |
| NTR | Netherlands Twin Register |
| ALSPAC | Avon Longitudinal Study of Parents and Children |
| SES | socio-economic status |
| GA | gestational age |
| BMI | body mass index |
| GEE | generalized estimating equations |
| mQTL | methylation quantitative trait locus |
| SNP | single nucleotide polymorphism |
| MZ | monozygotic |
| DZ | dizygotic |
References
- Godfrey, K.M.; Lillycrop, K.A.; Burdge, G.C.; Gluckman, P.D.; Hanson, M.A. Epigenetic Mechanisms and the Mismatch Concept of the Developmental Origins of Health and Disease. Pediatr. Res. 2007, 61, 5R–10R. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Low, F.M. The role of developmental plasticity and epigenetics in human health. Birth Defects Res. Part C Embryo Today Rev. 2011, 93, 12–18. [Google Scholar] [CrossRef]
- Verduci, E.; Banderali, G.; Barberi, S.; Radaelli, G.; Lops, A.; Betti, F.; Riva, E.; Giovannini, M. Epigenetic Effects of Human Breast Milk. Nutrients 2014, 6, 1711–1724. [Google Scholar] [CrossRef]
- Langley-Evans, S.C. Nutrition in Early Life and the Programming of Adult Disease: A Review. J. Hum. Nutr. Diet. 2015, 28, 1–14. [Google Scholar] [CrossRef]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.-C.; Boileau, P.; Le Bouc, Y.; Deal, C.L.; Lillycrop, K.; et al. Child Health, Developmental Plasticity, and Epigenetic Programming. Endocr. Rev. 2011, 32, 159–224. [Google Scholar] [CrossRef]
- Raghuraman, S.; Donkin, I.; Versteyhe, S.; Barrès, R.; Simar, D. The Emerging Role of Epigenetics in Inflammation and Immunometabolism. Trends Endocrinol. Metab. 2016, 27, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Paparo, L.; Di Costanzo, M.; Di Scala, C.; Cosenza, L.; Leone, L.; Nocerino, R.; Canani, R.B. The Influence of Early Life Nutrition on Epigenetic Regulatory Mechanisms of the Immune System. Nutrients 2014, 6, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Canani, R.B.; Di Costanzo, M.; Leone, L.; Bedogni, G.; Brambilla, P.; Cianfarani, S.; Nobili, V.; Pietrobelli, A.; Agostoni, C. Epigenetic mechanisms elicited by nutrition in early life. Nutr. Res. Rev. 2011, 24, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, P.D.; Buss, C.; Entringer, S.; Swanson, J.M. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Semin. Reprod. Med. 2009, 27, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A.; Beedle, A.S. Early Life Events and Their Consequences for Later Disease: A Life History and Evolutionary Perspective. Am. J. Hum. Biol. 2007, 19, 1–19. [Google Scholar] [CrossRef]
- Bateson, P.; Barker, D.; Clutton-Brock, T.; Deb, D.; D’Udine, B.; Foley, R.A.; Gluckman, P.; Godfrey, K.; Kirkwood, T.; Lahr, M.M.; et al. Developmental Plasticity and Human Health. Nature 2004, 430, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-W.; Friso, S. Epigenetics: A New Bridge between Nutrition and Health. Adv. Nutr. 2010, 1, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Peter, C.J.; Fischer, L.K.; Kundakovic, M.; Garg, P.; Jakovcevski, M.; Dincer, A.; Amaral, A.C.; Ginns, E.I.; Galdzicka, M.; Bryce, C.P.; et al. DNA Methylation Signatures of Early Childhood Malnutrition Associated With Impairments in Attention and Cognition. Boil. Psychiatry 2016, 80, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A.; Michels, K.B. Epigenetic Epidemiology of the Developmental Origins Hypothesis. Annu. Rev. Nutr. 2007, 27, 363–388. [Google Scholar] [CrossRef]
- Cutfield, W.S.; Hofman, P.L.; Mitchell, M.; Morison, I.M. Could Epigenetics Play a Role in the Developmental Origins of Health and Disease? Pediatr. Res. 2007, 61, 68R–75R. [Google Scholar] [CrossRef]
- Tammen, S.A.; Friso, S.; Choi, S.-W. Epigenetics: The Link between Nature and Nurture. Mol. Asp. Med. 2013, 34, 753–764. [Google Scholar] [CrossRef]
- Faa, G.; Manchia, M.; Pintus, R.; Gerosa, C.; Marcialis, M.A.; Fanos, V. Fetal programming of neuropsychiatric disorders. Birth Defects Res. Part C Embryo Today Rev. 2016, 108, 207–223. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Müller, F.; et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 5592. [Google Scholar] [CrossRef]
- Barker, D.J. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef]
- Ruiz, L.; García-Carral, C.; Rodriguez, J.M. Unfolding the Human Milk Microbiome Landscape in the Omics Era. Front. Microbiol. 2019, 10, 1378. [Google Scholar] [CrossRef] [PubMed]
- Fanos, V.; Reali, A.; Marcialis, M.A.; Bardanzellu, F. What You Have to Know about Human Milk Oligosaccharides. J. Pediatr. Neonatal Individ. Med. 2018, 7, e070137. [Google Scholar]
- Hassiotou, F.; Beltran, A.; Chetwynd, E.; Stuebe, A.M.; Twigger, A.-J.; Metzger, P.; Trengove, N.; Lai, C.T.; Filgueira, L.; Blancafort, P.; et al. Breastmilk Is a Novel Source of Stem Cells with Multilineage Differentiation Potential. Stem Cells 2012, 30, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Bardanzellu, F.; Fanos, V.; Reali, A. “Omics” in Human Colostrum and Mature Milk: Looking to Old Data with New Eyes. Nutrients 2017, 9, 843. [Google Scholar] [CrossRef] [PubMed]
- Dessì, A.; Briana, D.; Corbu, S.; Gavrili, S.; Marincola, F.C.; Georgantzi, S.; Pintus, R.; Fanos, V.; Malamitsi-Puchner, A. Metabolomics of Breast Milk: The Importance of Phenotypes. Metabolites 2018, 8, 79. [Google Scholar] [CrossRef]
- Forsum, E.; Lonnerdal, B. Variation in the contents of nutrients of breast milk during one feeding. Nutrients Rep. Int. 1979, 19, 815–820. [Google Scholar]
- Victora, C.G.; Bahl, R.; Barros, A.J.D.; França, G.V.A.; Horton, S.; Krasevec, J.; Murch, S.; Sankar, M.J.; Walker, N.; Rollins, N.C.; et al. Breastfeeding in the 21st Century: Epidemiology, Mechanisms, and Lifelong Effect. Lancet 2016, 387, 475–490. [Google Scholar] [CrossRef]
- McInerny, T.K. Breastfeeding, Early Brain Development, and Epigenetics—Getting Children off to Their Best Start. Breastfeed. Med. 2014, 9, 333–334. [Google Scholar] [CrossRef]
- Ip, S.; Chung, M.; Raman, G.; Chew, P.; Magula, N.; Devine, D.; Trikalinos, T.; Lau, J. Breastfeeding and maternal and infant health outcomes in developed countries. Évid. Rep. Assess. 2007, 153, 1–186. [Google Scholar]
- Wisnieski, L.; Kerver, J.; Holzman, C.; Todem, D.; Margerison-Zilko, C. Breastfeeding and Risk of Metabolic Syndrome in Children and Adolescents: A Systematic Review. J. Hum. Lact. 2018, 34, 515–525. [Google Scholar] [CrossRef]
- Chirico, G.; Marzollo, R.; Cortinovis, S.; Fonte, C.; Gasparoni, A. Antiinfective Properties of Human Milk. J. Nutr. 2008, 138, 1801S–1806S. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.R.; Ling, P.-R.; Blackburn, G.L. Review of Infant Feeding: Key Features of Breast Milk and Infant Formula. Nutrients 2016, 8, 279. [Google Scholar] [CrossRef] [PubMed]
- Else-Quest, N.M.; Hyde, J.S.; Clark, R. Breastfeeding, Bonding, and the Mother-Infant Relationship. Merrill-Palmer Quarterly; Wayne State University Press: Detroit, MI, USA, 2003; pp. 495–517. [Google Scholar] [CrossRef]
- WHO. Protecting, Promoting, and Supporting Breastfeeding in Facilities Providing Maternity and Newborn Services: The Revised Baby-Friendly Hospital Initiative 2018; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Michaelsen, K.F.; Lauritzen, L.; Mortensen, E.L. Effects of Breast-Feeding on Cognitive Function. In Breast-Feeding: Early Influences on Later Health; Springer: Dordrecht, The Netherlands, 2009; Volume 639, pp. 199–215. [Google Scholar] [CrossRef]
- Kramer, M.S.; Aboud, F.; Mironova, E.; Vanilovich, I.; Platt, R.W.; Matush, L.; Igumnov, S.; Fombonne, E.; Bogdanovich, N.; Ducruet, T.; et al. Breastfeeding and child cognitive development: New evidence from a large randomized trial. Arch. Gen. Psychiatry 2008, 65, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Horta, B.L.; De Mola, C.L.; Victora, C.G. Breastfeeding and intelligence: A systematic review and meta-analysis. Acta Paediatr. 2015, 104, 14–19. [Google Scholar] [CrossRef]
- Blesa, M.; Sullivan, G.; Anblagan, D.; Telford, E.J.; Quigley, A.J.; Sparrow, S.A.; Serag, A.; Semple, S.I.; Bastin, M.E.; Boardman, J.P. Early breast milk exposure modifies brain connectivity in preterm infants. NeuroImage 2019, 184, 431–439. [Google Scholar] [CrossRef]
- Bartels, M.; Van Beijsterveldt, C.E.M.; Boomsma, D.I. Breastfeeding, Maternal Education and Cognitive Function: A Prospective Study in Twins. Behav. Genet. 2009, 39, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Harder, T.; Bergmann, R.; Kallischnigg, G.; Plagemann, A. Duration of Breastfeeding and Risk of Overweight: A Meta-Analysis. Am. J. Epidemiol. 2005, 162, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Burke, V.; Beilin, L.J.; Simmer, K.; Oddy, W.H.; Blake, K.V.; Doherty, D.; Kendall, G.E.; Newnham, J.P.; Landau, L.I.; Stanley, F.J. Breastfeeding and Overweight: Longitudinal Analysis in an Australian Birth Cohort. J. Pediatr. 2005, 147, 56–61. [Google Scholar] [CrossRef]
- Gillman, M.W.; Rifas-Shiman, S.L.; Camargo, J.C.A.; Berkey, C.S.; Frazier, A.L.; Rockett, H.R.H.; Field, A.E.; Colditz, G.A. Risk of Overweight Among Adolescents Who Were Breastfed as Infants. JAMA 2001, 285, 2461. [Google Scholar] [CrossRef] [Green Version]
- Owen, C.G.; Whincup, P.H.; Odoki, K.; Gilg, J.A.; Cook, D.G. Infant feeding and blood cholesterol: A study in adolescents and a systematic review. Pediatrics 2002, 110, 597–608. [Google Scholar] [CrossRef]
- Shoji, H.; Shimizu, T. Effect of Human Breast Milk on Biological Metabolism in Infants. Pediatr. Int. 2019, 61, 6–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klement, E.; Cohen, R.V.; Boxman, J.; Joseph, A.; Reif, S. Breastfeeding and risk of inflammatory bowel disease: A systematic review with meta-analysis. Am. J. Clin. Nutr. 2004, 80, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amitay, E.L.; Keinan-Boker, L. Breastfeeding and childhood leukemia incidence: A meta-analysis and systematic review. JAMA Pediatr. 2015, 169, e151025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horta, B.L.; Victora, C.G.; World Health Organization. Short-Term Effects of Breastfeeding: A Systematic Review on the Benefits of Breastfeeding on Diarrhoea and Pneumonia Mortality; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Horta, B.L.; De Mola, C.L.; Victora, C.G. Long-term consequences of breastfeeding on cholesterol, obesity, systolic blood pressure and type 2 diabetes: A systematic review and meta-analysis. Acta Paediatr. 2015, 104, 30–37. [Google Scholar] [CrossRef]
- Lodge, C.; Tan, D.; Lau, M.; Dai, X.; Tham, R.; Lowe, A.; Bowatte, G.; Allen, K.; Dharmage, S.; Lodge, C. Breastfeeding and asthma and allergies: A systematic review and meta-analysis. Acta Paediatr. 2015, 104, 38–53. [Google Scholar] [CrossRef]
- Fall, C.H.; Borja, J.B.; Osmond, C.; Richter, L.; Bhargava, S.K.; Martorell, R.; Stein, A.D.; Barros, F.C.; Victora, C.G.; COHORTS group. Infant-Feeding Patterns and Cardiovascular Risk Factors in Young Adulthood: Data from Five Cohorts in Low- and Middle-Income Countries. Int. J. Epidemiol. 2011, 40, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, F.P.; Loret de Mola, C.; Davies, N.M.; Victora, C.G.; Relton, C.L. Breastfeeding Effects on DNA Methylation in the Offspring: A Systematic Literature Review. PLoS ONE 2017, 12, e0173070. [Google Scholar]
- Obermann-Borst, S.A.; Eilers, P.H.; Tobi, E.W.; De Jong, F.H.; Slagboom, P.E.; Heijmans, B.T.; Steegers-Theunissen, R.P.; Slagboom, P. Duration of breastfeeding and gender are associated with methylation of the LEPTIN gene in very young children. Pediatr. Res. 2013, 74, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Naumova, O.Y.; Odintsova, V.V.; Arincina, I.A.; Rychkov, S.Y.; Muhamedrahimov, R.J.; Shneider, Y.V.; Grosheva, A.N.; Zhukova, O.V.; Grigorenko, E.L. A Study of the Association between Breastfeeding and DNA Methylation in Peripheral Blood Cells of Infants. Russ. J. Genet. 2019, 55, 749–755. [Google Scholar] [CrossRef]
- Rossnerova, A.; Tulupova, E.; Tabashidze, N.; Schmuczerova, J.; Dostal, M.; Jr, P.R.; Gmuender, H.; Sram, R.J. Factors affecting the 27K DNA methylation pattern in asthmatic and healthy children from locations with various environments. Mutat. Res. Mol. Mech. Mutagen. 2013, 741, 18–26. [Google Scholar] [CrossRef]
- Sherwood, W.B.; Bion, V.; Lockett, G.A.; Ziyab, A.H.; Soto-Ramírez, N.; Mukherjee, N.; Kurukulaaratchy, R.J.; Ewart, S.; Zhang, H.; Arshad, S.H.; et al. Duration of breastfeeding is associated with leptin (LEP) DNA methylation profiles and BMI in 10-year-old children. Clin. Epigenet. 2019, 11, 128. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.-J.; et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küpers, L.K.; Monnereau, C.; Sharp, G.C.; Yousefi, P.; Salas, L.A.; Ghantous, A.; Page, C.M.; Reese, S.E.; Wilcox, A.J.; Czamara, D.; et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat. Commun. 2019, 10, 1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Ramírez, N.; Arshad, S.H.; Holloway, J.W.; Zhang, H.; Schauberger, E.; Ewart, S.; Patil, V.; Karmaus, W. The interaction of genetic variants and DNA methylation of the interleukin-4 receptor gene increase the risk of asthma at age 18 years. Clin. Epigenet. 2013, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Tao, M.-H.; Marian, C.; Shields, P.G.; Potischman, N.; Nie, J.; Krishnan, S.S.; Berry, D.L.; Kallakury, B.V.; Ambrosone, C.; Edge, S.B.; et al. Exposures in early life: Associations with DNA promoter methylation in breast tumors. J. Dev. Orig. Health Dis. 2013, 4, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, F.P.; Smith, G.D.; Simpkin, A.J.; Victora, C.G.; Relton, C.L.; Caramaschi, D. Association between Breastfeeding and DNA Methylation over the Life Course: Findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). bioRxiv 2019, 800722. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.K.; Kilaru, V.; Klengel, T.; Mercer, K.B.; Bradley, B.; Conneely, K.N.; Ressler, K.J.; Binder, E.B. DNA Extracted from Saliva for Methylation Studies of Psychiatric Traits: Evidence Tissue Specificity and Relatedness to Brain. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2015, 168, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Papavassiliou, P.; York, T.P.; Gursoy, N.; Hill, G.; Nicely, L.V.; Sundaram, U.; McClain, A.; Aggen, S.H.; Eaves, L.; Riley, B.; et al. The phenotype of persons having mosaicism for trisomy 21/Down syndrome reflects the percentage of trisomic cells present in different tissues. Am. J. Med. Genet. Part A 2009, 149, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Van Dongen, J.; Ehli, E.A.; Jansen, R.; Van Beijsterveldt, C.E.M.; Willemsen, G.; Hottenga, J.J.; Kallsen, N.A.; Peyton, S.A.; Breeze, C.E.; Kluft, C.; et al. Genome-wide analysis of DNA methylation in buccal cells: A study of monozygotic twins and mQTLs. Epigenet. Chromatin 2018, 11, 54. [Google Scholar] [CrossRef]
- Van Beijsterveldt, C.E.M.; Groen-Blokhuis, M.; Hottenga, J.J.; Franić, S.; Hudziak, J.J.; Lamb, D.; Huppertz, C.; de Zeeuw, E.; Nivard, M.; Schutte, N.; et al. The Young Netherlands Twin Register (YNTR): Longitudinal Twin and Family Studies in Over 70,000 Children. Twin Res. Hum. Genet. 2013, 16, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Boomsma, D.I.; Fanos Cagliari, V.; Mussap Genoa, M.; Del Vecchio Bari, A.; Sun Shanghai, B.; Faa Cagliari, G.; Giordano Philadelphia, A. Aggression in Children: Unravelling the Interplay of Genes and Environment through (Epi)Genetics and Metabolomics. J. Pediatr. Neonatal Individ. Med. 2015, 4. [Google Scholar] [CrossRef]
- Bartels, M.; Hendriks, A.; Mauri, M.; Krapohl, E.; Whipp, A.; Bolhuis, K.; Conde, L.C.; Luningham, J.; Ip, H.F.; Hagenbeek, F.; et al. Childhood aggression and the co-occurrence of behavioural and emotional problems: Results across ages 3-16 years from multiple raters in six cohorts in the EU-ACTION project. Eur. Child Adolesc. Psychiatry 2018, 27, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagenbeek, F.A.; Roetman, P.J.; Pool, R.; Kluft, C.; Harms, A.C.; van Dongen, J.; Colins, O.F.; Talens, S.; Van Beijsterveldt, C.E.M.; de Zeeuw, E.L.; et al. Urinary Amine and Organic Acid Metabolites Evaluated as Markers for Childhood Aggression: The ACTION Biomarker Study. Frontiers in Psychiatry 2019, submitted. [Google Scholar]
- Van Dongen, J. Epigenome-Wide Association Study Meta-Analysis of Aggressive Behavior. In Proceedings of the ECS Workshop on Aggression, Nijmegen, The Netherlands, 31 October 2016. [Google Scholar]
- Boyd, A.; Golding, J.; Macleod, J.; Lawlor, D.A.; Fraser, A.; Henderson, J.; Molloy, L.; Ness, A.; Ring, S.; Davey Smith, G. Cohort Profile: The ‘Children of the 90s’—The Index Offspring of the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 2013, 42, 111–127. [Google Scholar] [CrossRef] [Green Version]
- Fraser, A.; Macdonald-Wallis, C.; Tilling, K.; Boyd, A.; Golding, J.; Davey Smith, G.; Henderson, J.; Macleod, J.; Molloy, L.; Ness, A.; et al. Cohort Profile: The Avon Longitudinal Study of Parents and Children: ALSPAC Mothers Cohort. Int. J. Epidemiol. 2013, 42, 97–110. [Google Scholar] [CrossRef] [Green Version]
- Relton, C.L.; Gaunt, T.; McArdle, W.; Ho, K.; Duggirala, A.; Shihab, H.; Woodward, G.; Lyttleton, O.; Evans, D.M.; Reik, W.; et al. Data Resource Profile: Accessible Resource for Integrated Epigenomic Studies (ARIES). Int. J. Epidemiol. 2015, 44, 1181–1190. [Google Scholar] [CrossRef] [Green Version]
- CBS. Standard Classification of Occupations; Central Bureau of Statistics: Heerlen/Voorburg, The Netherlands, 2001.
- Erikson, R.; Goldthorpe, J.H.; Portocarero, L. Intergenerational class mobility and the convergence thesis: England, France and Sweden. Br. J. Sociol. 2010, 61, 185–219. [Google Scholar] [CrossRef]
- Dolan, C.V.; Geels, L.; Vink, J.M.; van Beijsterveldt, C.E.M.; Neale, M.C.; Bartels, M.; Boomsma, D.I. Testing Causal Effects of Maternal Smoking during Pregnancy on Offspring’s Externalizing and Internalizing Behavior. Behav. Genet. 2016, 46, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Van Beijsterveldt, C.E.M.T.; Hoekstra, C.; Schats, R.; Montgomery, G.W.; Willemsen, G.; Boomsma, D.I. Mode of Conception of Twin Pregnancies: Willingness to Reply to Survey Items and Comparison of Survey Data to Hospital Records. Twin Res. Hum. Genet. 2008, 11, 349–351. [Google Scholar] [CrossRef]
- Odintsova, V.V.; Dolan, C.V.; Van Beijsterveldt, C.E.M.; De Zeeuw, E.L.; Van Dongen, J.; Boomsma, D.I. Pre- and Perinatal Characteristics Associated with Apgar Scores in a Review and in a New Study of Dutch Twins. Twin Res. Hum. Genet. 2019, 22, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Meulenbelt, I.; Droog, S.; Trommelen, G.J.; Boomsma, D.I.; Slagboom, P.E. High-yield noninvasive human genomic DNA isolation method for genetic studies in geographically dispersed families and populations. Am. J. Hum. Genet. 1995, 57, 1252–1254. [Google Scholar] [PubMed]
- Moran, S.; Arribas, C.; Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 2016, 8, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinke, L.; van Iterson, M.; Cats, D.; Slieker, R.; Heijmans, B. DNAmArray: Streamlined Workflow for the Quality Control, Normalization, and Analysis of Illumina Methylation Array Data. Zenodo 2019. [Google Scholar] [CrossRef]
- Van Iterson, M.; Tobi, E.W.; Slieker, R.C.; Hollander, W.D.; Luijk, R.; Slagboom, P.; Heijmans, B.T. MethylAid: Visual and interactive quality control of large Illumina 450k datasets. Bioinformatics 2014, 30, 3435–3437. [Google Scholar] [CrossRef]
- Van Iterson, M.; Cats, D.; Hop, P.; Heijmans, B.T.; BIOS Consortium. omicsPrint: Detection of data linkage errors in multiple omics studies. Bioinformatics 2018, 34, 2142–2143. [Google Scholar] [CrossRef] [Green Version]
- Min, J.L.; Hemani, G.; Smith, G.D.; Relton, C.; Suderman, M. Meffil: Efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics 2018, 34, 3983–3989. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Boil. 2016, 17, 208. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.C.; Webster, A.P.; Dong, D.; Feber, A.; Graham, D.G.; Sullivan, R.; Jevons, S.; Lovat, L.B.; Beck, S.; Widschwendter, M.; et al. A novel cell-type deconvolution algorithm reveals substantial contamination by immune cells in saliva, buccal and cervix. Epigenomics 2018, 10, 925–940. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [Green Version]
- Van Iterson, M.; Van Zwet, E.W.; BIOS Consortium; Heijmans, B.T. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Boil. 2017, 18, 19. [Google Scholar] [CrossRef] [Green Version]
- Tukey, J.W. Exploratory Data Analysis. Biom. J. 1981, 23, 413–414. [Google Scholar]
- Leek, J.T.; Storey, J.D. Capturing Heterogeneity in Gene Expression Studies by Surrogate Variable Analysis. PLoS Genet. 2007, 3, e161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zou, D.; Li, Z.; Gao, R.; Sang, J.; Zhang, Y.; Li, R.; Xia, L.; Zhang, T.; Niu, G.; et al. EWAS Atlas: A Curated Knowledgebase of Epigenome-Wide Association Studies. Nucleic Acids Res. 2019, 47, D983–D988. [Google Scholar] [CrossRef]
- Yousefi, P.; Huen, K.; Davé, V.; Barcellos, L.; Eskenazi, B.; Holland, N. Sex differences in DNA methylation assessed by 450 K BeadChip in newborns. BMC Genom. 2015, 16, 911. [Google Scholar] [CrossRef] [Green Version]
- Husquin, L.T.; Rotival, M.; Fagny, M.; Quach, H.; Zidane, N.; McEwen, L.M.; MacIsaac, J.L.; Kobor, M.S.; Aschard, H.; Patin, E.; et al. Exploring the genetic basis of human population differences in DNA methylation and their causal impact on immune gene regulation. Genome Boil. 2018, 19, 222. [Google Scholar] [CrossRef] [Green Version]
- Marioni, R.E.; Harris, S.E.; Zhang, Q.; McRae, A.F.; Hagenaars, S.P.; Hill, W.D.; Davies, G.; Ritchie, C.W.; Gale, C.R.; Starr, J.M.; et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 2018, 8, 99. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Gharaibeh, R.Z.; Pietryk, E.W.; Brouwer, C.; Tarantino, L.M.; Valdar, W.; Ideraabdullah, F.Y. Impact of vitamin D depletion during development on mouse sperm DNA methylation. Epigenetics 2018, 13, 959–974. [Google Scholar] [CrossRef] [Green Version]
- Sujit, K.M.; Sarkar, S.; Singh, V.; Pandey, R.; Agrawal, N.K.; Trivedi, S.; Singh, K.; Gupta, G.; Rajender, S. Genome-wide differential methylation analyses identifies methylation signatures of male infertility. Hum. Reprod. 2018, 33, 2256–2267. [Google Scholar] [CrossRef]
- Kim, S.K. Identification of 613 new loci associated with heel bone mineral density and a polygenic risk score for bone mineral density, osteoporosis and fracture. PLoS ONE 2018, 13, e0200785. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.A.; Kemp, J.P.; Youlten, S.E.; Laurent, L.; Logan, J.G.; Chai, R.C.; Vulpescu, N.A.; Forgetta, V.; Kleinman, A.; Mohanty, S.T.; et al. An Atlas of Genetic Influences on Osteoporosis in Humans and Mice. Nat. Genet. 2019, 51, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Kemp, J.P.; Morris, J.A.; Medina-Gomez, C.; Forgetta, V.; Warrington, N.M.; Youlten, S.E.; Zheng, J.; Gregson, C.L.; Grundberg, E.; Trajanoska, K.; et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat. Genet. 2017, 49, 1468–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, K.; Styrkarsdottir, U.; Evangelou, E.; Hsu, Y.-H.; Duncan, E.L.; Ntzani, E.E.; Oei, L.; Albagha, O.M.E.; Amin, N.; Kemp, J.P.; et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 2012, 44, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Gómez, C.; Kemp, J.P.; Trajanoska, K.; Luan, J.; Chesi, A.; Ahluwalia, T.S.; Mook-Kanamori, D.O.; Ham, A.; Hartwig, F.P.; Evans, D.S.; et al. Life-Course Genome-wide Association Study Meta-analysis of Total Body BMD and Assessment of Age-Specific Effects. Am. J. Hum. Genet. 2018, 102, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, V.; Ilkov, M.; Lamb, J.R.; Finkel, N.; Gudmundsson, E.F.; Pitts, R.; Hoover, H.; Gudmundsdottir, V.; Horman, S.R.; Aspelund, T.; et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 2018, 361, 769–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.B.; Maranville, J.C.; Peters, J.E.; Stacey, D.; Staley, J.R.; Blackshaw, J.; Burgess, S.; Jiang, T.; Paige, E.; Surendran, P.; et al. Genomic atlas of the human plasma proteome. Nature 2018, 558, 73–79. [Google Scholar] [CrossRef] [PubMed]
- De Vries, P.S.; Brown, M.R.; Bentley, A.R.; Sung, Y.J.; Winkler, T.W.; Ntalla, I.; Schwander, K.; Kraja, A.T.; Guo, X.; Franceschini, N.; et al. Multiancestry Genome-Wide Association Study of Lipid Levels Incorporating Gene-Alcohol Interactions. Am. J. Epidemiol. 2019, 188, 1033–1054. [Google Scholar] [CrossRef] [Green Version]
- Imaizumi, A.; Adachi, Y.; Kawaguchi, T.; Higasa, K.; Tabara, Y.; Sonomura, K.; Sato, T.-A.; Takahashi, M.; Mizukoshi, T.; Yoshida, H.-O.; et al. Genetic basis for plasma amino acid concentrations based on absolute quantification: A genome-wide association study in the Japanese population. Eur. J. Hum. Genet. 2019, 27, 621–630. [Google Scholar] [CrossRef]
- Shin, S.-Y.; The Multiple Tissue Human Expression Resource (MuTHER) Consortium; Fauman, E.B.; Petersen, A.-K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Johnston, K.J.A.; Adams, M.J.; Nicholl, B.I.; Ward, J.; Strawbridge, R.J.; Ferguson, A.; McIntosh, A.M.; Bailey, M.E.S.; Smith, D.J. Genome-wide association study of multisite chronic pain in UK Biobank. PLoS Genet. 2019, 15, e1008164. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sekula, P.; Wuttke, M.; Wahrheit, J.; Hausknecht, B.; Schultheiss, U.T.; Gronwald, W.; Schlosser, P.; Tucci, S.; Ekici, A.B.; et al. Genome-Wide Association Studies of Metabolites in Patients with CKD Identify Multiple Loci and Illuminate Tubular Transport Mechanisms. J. Am. Soc. Nephrol. 2018, 29, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Case Control Consortium. Genome-Wide Association Study of 14,000 Cases of Seven Common Diseases and 3000 Shared Controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Jiang, Y.; Wedow, R.; Li, Y.; Brazel, D.M.; Chen, F.; Datta, G.; Davila-Velderrain, J.; McGuire, D.; Tian, C.; et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 2019, 51, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Linnér, R.K.; Biroli, P.; Kong, E.; Meddens, S.F.W.; Wedow, R.; Fontana, M.A.; Lebreton, M.; Tino, S.P.; Abdellaoui, A.; Hammerschlag, A.R.; et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat. Genet. 2019, 51, 245–257. [Google Scholar] [CrossRef]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a Novel Mammalian Autophagy Protein Interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Ahola-Olli, A.V.; Würtz, P.; Havulinna, A.S.; Aalto, K.; Pitkänen, N.; Lehtimäki, T.; Kähönen, M.; Lyytikäinen, L.-P.; Raitoharju, E.; Seppälä, I.; et al. Genome-Wide Association Study Identifies 27 Loci Influencing Concentrations of Circulating Cytokines and Growth Factors. Am. J. Hum. Genet. 2017, 100, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Astle, W.J.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.L.; Mead, D.; Bouman, H.; Riveros-McKay, F.; Kostadima, M.A.; et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016, 167, 1415–1429. [Google Scholar] [CrossRef] [Green Version]
- Rojas, D.; Rager, J.E.; Smeester, L.; Bailey, K.A.; Drobná, Z.; Rubio-Andrade, M.; Stýblo, M.; García-Vargas, G.; Fry, R.C. Prenatal Arsenic Exposure and the Epigenome: Identifying Sites of 5-Methylcytosine Alterations That Predict Functional Changes in Gene Expression in Newborn Cord Blood and Subsequent Birth Outcomes. Toxicol. Sci. 2015, 143, 97–106. [Google Scholar] [CrossRef]
- McNally, K.E.; Faulkner, R.; Steinberg, F.; Gallon, M.; Ghai, R.; Pim, D.; Langton, P.; Pearson, N.; Danson, C.M.; Nägele, H.; et al. Retriever is a multiprotein complex for retromer-independent endosomal cargo recycling. Nature 2017, 19, 1214–1225. [Google Scholar] [CrossRef]
- Johansson, Å.; Enroth, S.; Gyllensten, U. Continuous Aging of the Human DNA Methylome Throughout the Human Lifespan. PLoS ONE 2013, 8, e67378. [Google Scholar] [CrossRef] [Green Version]
- Gervin, K.; Andreassen, B.K.; Hjorthaug, H.S.; Carlsen, K.C.L.; Carlsen, K.-H.; Undlien, D.E.; Lyle, R.; Munthe-Kaas, M.C. Intra-individual changes in DNA methylation not mediated by cell-type composition are correlated with aging during childhood. Clin. Epigenet. 2016, 8, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, E.C.; Soare, T.W.; Simpkin, A.J.; Suderman, M.J.; Zhu, Y.; Klengel, T.; Smith, A.D.A.C.; Ressler, K.; Relton, C.L. Sensitive Periods for the Effect of Childhood Adversity on DNA Methylation: Results from a Prospective, Longitudinal Study. bioRxiv 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpkin, A.J.; Suderman, M.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Tilling, K.; Smith, G.D.; Relton, C.L. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum. Mol. Genet. 2015, 24, 3752–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agha, G.; Hajj, H.; Rifas-Shiman, S.L.; Just, A.C.; Hivert, M.-F.; Burris, H.H.; Lin, X.; Litonjua, A.A.; Oken, E.; DeMeo, D.L.; et al. Birth weight-for-gestational age is associated with DNA methylation at birth and in childhood. Clin. Epigenet. 2016, 8, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfano, R.; Guida, F.; Galobardes, B.; Chadeau-Hyam, M.; Delpierre, C.; Ghantous, A.; Henderson, J.; Herceg, Z.; Jain, P.; Nawrot, T.S.; et al. Socioeconomic position during pregnancy and DNA methylation signatures at three stages across early life: Epigenome-wide association studies in the ALSPAC birth cohort. Int. J. Epidemiol. 2019, 48, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Richmond, R.C.; Simpkin, A.J.; Woodward, G.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Smith, A.D.A.C.; Timpson, N.J.; Tilling, K.; et al. Prenatal Exposure to Maternal Smoking and Offspring DNA Methylation across the Lifecourse: Findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum. Mol. Genet. 2015, 24, 2201–2217. [Google Scholar] [CrossRef] [Green Version]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef]
- Muniz, L.C.; Menezes, A.M.B.; Buffarini, R.; Wehrmeister, F.C.; Assunção, M.C.F. Effect of Breastfeeding on Bone Mass from Childhood to Adulthood: A Systematic Review of the Literature. Int. Breastfeed. J. 2015, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Dessì, A.; Murgia, A.; Agostino, R.; Pattumelli, M.G.; Schirru, A.; Scano, P.; Fanos, V.; Caboni, P. Exploring the Role of Different Neonatal Nutrition Regimens during the First Week of Life by Urinary GC-MS Metabolomics. Int. J. Mol. Sci. 2016, 17, 265. [Google Scholar] [CrossRef] [Green Version]
- San-Cristobal, R.; Navas-Carretero, S.; Milagro, F.I.; Riezu-Boj, J.I.; Guruceaga, E.; Celis-Morales, C.; Livingstone, K.M.; Brennan, L.; Lovegrove, J.A.; Daniel, H.; et al. Gene methylation parallelisms between peripheral blood cells and oral mucosa samples in relation to overweight. J. Physiol. Biochem. 2016, 73, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Van Dongen, J.; Nivard, M.G.; Willemsen, G.; Hottenga, J.-J.; Helmer, Q.; Dolan, C.V.; Ehli, E.A.; Davies, G.E.; Van Iterson, M.; Breeze, C.E.; et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun. 2016, 7, 11115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colodro-Conde, L.; Sánchez-Romera, J.F.; Ordoñana, J.R. Heritability of Initiation and Duration of Breastfeeding Behavior. Twin Res. Hum. Genet. 2013, 16, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Merjonen, P.; Dolan, C.V.; Bartels, M.; Boomsma, D.I. Does Breastfeeding Behavior Run in Families? Evidence from Twins, Their Sisters and Their Mothers in the Netherlands. Twin Res. Hum. Genet. 2015, 18, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, N.; Benton, G.M.; Do, C.B.; Kiefer, A.K.; Mountain, J.L.; Hinds, D.A.; Francke, U.; Tung, J.Y. Genetic variants associated with breast size also influence breast cancer risk. BMC Med. Genet. 2012, 13, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, B.; Bass, S.; Fuh, G.; Wells, J. Zinc mediation of the binding of human growth hormone to the human prolactin receptor. Science 1990, 250, 1709–1712. [Google Scholar] [CrossRef]
- Sukonina, V.; Ma, H.; Zhang, W.; Bartesaghi, S.; Subhash, S.; Heglind, M.; Foyn, H.; Betz, M.J.; Nilsson, D.; Lidell, M.E.; et al. FOXK1 and FOXK2 regulate aerobic glycolysis. Nature 2019, 566, 279–283. [Google Scholar] [CrossRef]
- Hannenhalli, S.; Kaestner, K.H. The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 2009, 10, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Tachmazidou, I.; Süveges, D.; Min, J.L.; Ritchie, G.R.; Steinberg, J.; Walter, K.; Iotchkova, V.; Schwartzentruber, J.; Huang, J.; Memari, Y.; et al. Whole-Genome Sequencing Coupled to Imputation Discovers Genetic Signals for Anthropometric Traits. Am. J. Hum. Genet. 2017, 100, 865–884. [Google Scholar] [CrossRef] [Green Version]
- Crujeiras, A.B.; Pissios, P.; Moreno-Navarrete, J.M.; Díaz-Lagares, A.; Sandoval, J.; Gómez, A.; Ricart, W.; Esteller, M.; Casanueva, F.F.; Fernandez-Real, J.M. An Epigenetic Signature in Adipose Tissue Is Linked to Nicotinamide N-Methyltransferase Gene Expression. Mol. Nutr. Food Res. 2018, 62, 1700933. [Google Scholar] [CrossRef]
- Bardanzellu, F.; Fanos, V.; Strigini, F.A.L.; Artini, P.G.; Peroni, D.G. Human Breast Milk: Exploring the Linking Ring Among Emerging Components. Front. Pediatr. 2018, 6, 215. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, G.M.; LaKind, J.S.; Davis, M.H.; Hines, E.P.; Marchitti, S.A.; Alcala, C.; Lorber, M. Environmental Chemicals in Breast Milk and Formula: Exposure and Risk Assessment Implications. Environ. Health Perspect. 2018, 126, 096001. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gallego, C.; Morales, J.M.; Monleón, D.; Du Toit, E.; Kumar, H.; Linderborg, K.M.; Zhang, Y.; Yang, B.; Isolauri, E.; Salminen, S.; et al. Human Breast Milk NMR Metabolomic Profile across Specific Geographical Locations and Its Association with the Milk Microbiota. Nutrients 2018, 10, 1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermansson, H.; Kumar, H.; Collado, M.C.; Salminen, S.; Isolauri, E.; Rautava, S. Breast Milk Microbiota Is Shaped by Mode of Delivery and Intrapartum Antibiotic Exposure. Front. Nutr. 2019, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Breastfeeding Never (n = 265) | Breastfeeding Ever (n = 741) | Total | ||||
|---|---|---|---|---|---|---|
| n | % | n | % | n | % | |
| Sex | ||||||
| male | 138 | 52.1% | 349 | 47.1% | 487 | 48.4% |
| female | 127 | 47.9% | 392 | 52.9% | 519 | 51.6% |
| Zygosity | ||||||
| Monozygotic (MZ) | 226 | 85.3% | 613 | 82.7% | 839 | 83.4% |
| Dizygotic (DZ) | 39 | 14.7% | 128 | 17.3% | 167 | 16.6% |
| Chorionicity | ||||||
| MCMA | 4 | 3.5% | 21 | 7.3% | 25 | 6.2% |
| MCDA | 61 | 53.0% | 144 | 50.3% | 205 | 51.1% |
| DCDA | 50 | 43.5% | 121 | 42.3% | 171 | 42.6% |
| Gestational Age (Weeks) | ||||||
| Mean (SD) | 36.2 | (22.2) | 35.8 | (25.9) | 35.9 | (25.1) |
| ≤ 32 | 12 | 4.8% | 61 | 8.4% | 73 | 7.5% |
| 33–36 | 128 | 51.4% | 359 | 49.4% | 487 | 49.9% |
| ≥ 37 | 109 | 43.8% | 306 | 42.1% | 415 | 42.6% |
| Mother’s Age at Birth (Years) | ||||||
| Mean (SD) | 31.9 | (4.5) | 31.2 | (4.2) | 31.4 | (4.3) |
| 19–29 | 76 | 29.0% | 288 | 39.0% | 364 | 36.4% |
| 30–39 | 175 | 66.8% | 435 | 58.9% | 610 | 61.0% |
| >40 | 11 | 4.2% | 15 | 2.0% | 26 | 2.6% |
| Mother’s BMI Before Pregnancy | ||||||
| Mean (SD) | 24.3 | (4.0) | 24.2 | (4.11) | 24.2 | (4.1) |
| <25 | 149 | 61.3% | 470 | 66.1% | 619 | 64.9% |
| 25–29 | 70 | 28.8% | 169 | 23.8% | 239 | 25.1% |
| 30–39 | 24 | 9.9% | 65 | 9.1% | 89 | 9.3% |
| >40 | 0 | 0.0% | 7 | 1.0% | 7 | 0.7% |
| Father’s Age at Birth (Years) | ||||||
| Mean (SD) | 33.2 | (4.4) | 33.9 | (5.4) | 33.7 | (5.2) |
| 20–29 | 53 | 22.0% | 146 | 20.3% | 199 | 20.8% |
| 30–39 | 163 | 67.6% | 482 | 67.1% | 645 | 67.3% |
| >40 | 25 | 10.4% | 90 | 12.5% | 115 | 12.0% |
| Mode of Conception | ||||||
| naturally | 227 | 92.7% | 623 | 86.5% | 850 | 88.1% |
| stimulated | 4 | 1.6% | 26 | 3.6% | 30 | 3.1% |
| IVF/ICSI | 14 | 5.7% | 71 | 9.9% | 85 | 8.8% |
| Maternal Smoking | ||||||
| no smoking | 205 | 86.1% | 631 | 92.9% | 836 | 91.2% |
| smoking | 33 | 13.9% | 48 | 7.1% | 81 | 8.8% |
| Parental SES | ||||||
| low skill level | 0 | 0.0% | 8 | 1.2% | 8 | 0.9% |
| lower secondary educational level | 30 | 11.9% | 41 | 6.1% | 71 | 7.6% |
| upper secondary education level | 99 | 39.3% | 203 | 30.0% | 302 | 32.5% |
| higher vocational level | 89 | 35.3% | 234 | 34.6% | 323 | 34.8% |
| scientific level | 34 | 13.5% | 191 | 28.2% | 225 | 24.2% |
| Mode of Delivery | ||||||
| vaginal | 141 | 56.6% | 416 | 57.1% | 557 | 57.0% |
| caesarean planned | 43 | 17.3% | 97 | 13.3% | 140 | 14.3% |
| urgent intervention (forceps, vacuum extraction) | 20 | 8.0% | 75 | 10.3% | 95 | 9.7% |
| urgent caesarean section | 45 | 18.1% | 140 | 19.2% | 185 | 18.9% |
| Birth Weight | ||||||
| Mean (SD) | 2435.7 | (444.8) | 2394.6 | (558) | 2405 | (531.7) |
| <1500 | 8 | 3.2% | 52 | 7.1% | 60 | 6.2% |
| 1500–2500 | 123 | 49.8% | 338 | 46.4% | 461 | 47.3% |
| >2500 | 116 | 47.0% | 338 | 46.4% | 454 | 46.6% |
| Apgar Score at 1st Minute | ||||||
| 0–6 | 17 | 12.3% | 48 | 12.8% | 65 | 12.7% |
| 7–9 | 103 | 74.6% | 290 | 77.5% | 393 | 76.8% |
| 10 | 18 | 13.0% | 36 | 9.6% | 54 | 10.5% |
| Apgar Score at 5th Minute | ||||||
| 0–6 | 1 | 0.8% | 14 | 3.9% | 15 | 3.1% |
| 7–9 | 41 | 31.1% | 130 | 36.3% | 171 | 34.9% |
| 10 | 90 | 68.2% | 214 | 59.8% | 304 | 62.0% |
| Breastfeeding Duration | ||||||
| no | 265 | 100.0% | 0 | 265 | 26.3% | |
| less than 2 weeks | 75 | 10.1% | 75 | 7.5% | ||
| 2–6 weeks | 189 | 25.5% | 189 | 18.8% | ||
| 6 weeks to 3 months | 181 | 24.4% | 181 | 18.0% | ||
| 3–6 months | 148 | 20.0% | 148 | 14.7% | ||
| more than 6 months | 148 | 20.0% | 148 | 14.7% | ||
| cgID | Chromosome | Position | Gene | Gene Region | Discovery Study | Discovery Study Without Outliers | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | SE | p-Value | Estimate | SE | p-Value | |||||
| Basic Model (1). Sub-Sample < 10 years (n = 517) | ||||||||||
| cg25178826 | chr5 | 35165447 | PRLR | 5’UTR | −0.026 | 0.004 | 8.04 × 10–12 | −6.04 × 10–5 | 0.001 | 0.98 |
| cg12087956 | chr15 | 43022167 | CDAN1 | Body | −0.031 | 0.005 | 1.18 × 10–8 | 4.92 × 10–5 | 0.001 | 0.50 |
| cg24192772 | chr17 | 80536920 | FOXK2 | Body | −0.024 | 0.004 | 2.52 × 10–8 | 9.17 × 10–4 | 0.001 | 0.15 |
| cg10142656 | chr9 | 37753047 | TRMT10B | TSS1500 | −0.019 | 0.004 | 6.28 × 10–8 | 9.98 × 10–5 | 0.0007 | 0.14 |
| Adjusted Model (2). Discovery Sample (n = 1006) | ||||||||||
| cg22491379 | chr2 | 120553625 | PTPN4 | 5’UTR | −0.007 | 0.001 | 1.30 × 10–9 | −0.005 | 0.001 | 5.78 × 10–3 |
| Adjusted Model (2). Sub-Sample < 10 Years (n = 517) | ||||||||||
| cg03463465 | chr6 | 164143581 | 0.360 | 0.034 | 4.51 × 10–26 | −0.003 | 0.001 | 0.01 | ||
| cg07670516 | chr17 | 5019840 | ZNF232 | 5’UTR | 0.249 | 0.032 | 1.40 × 10–14 | 0.006 | 0.014 | 0.65 |
| cg20820810 | chr11 | 71850130 | FOLR3 | Body | −0.300 | 0.054 | 2.82 × 10–8 | −0.001 | 0.001 | 0.21 |
| cg16279140 | chr14 | 103981749 | −0.411 | 0.052 | 3.50 × 10–15 | no outliers | ||||
| cg05823759 | chr7 | 149646627 | 0.205 | 0.032 | 2.35 × 10–10 | no outliers | ||||
| cg27284194 | chr4 | 1044797 | 0.638 | 0.107 | 2.90 × 10–9 | no outliers | ||||
| cg03995300 | chr17 | 5019989 | ZNF232 | 5’UTR | 0.229 | 0.040 | 1.02 × 10–8 | no outliers | ||
| cgID | Direction of Effect in Discovery Study <10 Years | NTR Replication Study (n = 98) | ALSPAC Replication Study (n = 938) | ||||
|---|---|---|---|---|---|---|---|
| Estimate | SE | P-Value a | Estimate | SE | P-Value | ||
| cg16279140 | − | −0.0326 | 0.0412 | 0.43 | NA | NA | NA |
| cg05823759 | + | 0.0329 | 0.0332 | 0.32 | NA | NA | NA |
| cg27284194 | + | 0.0668 | 0.0542 | 0.21 | 0.0047 | 0.016 | 0.77 |
| cg03995300 | + | −0.0502 | 0.0334 | 0.13 | 0.0140 | 0.011 | 0.19 |
| cgID | Chromosome | Position | Gene | Gene Region | ESTIMATE | SE | P-Value |
|---|---|---|---|---|---|---|---|
| Total Sample (n = 1006) | |||||||
| cg16387046 | chr12 | 55248207 | MUCL1 | TSS200 | 0.027 | 0.005 | 4.93 × 10–7 |
| Sub-Sample <10 (n = 517) | |||||||
| cg11287055 | chr21 | 38630234 | VPS26C (DSCR3) | Body | 0.056 | 0.012 | 4.93 × 10–6 |
| cg16704958 | chr21 | 38630728 | VPS26C (DSCR3) | Body | 0.009 | 0.002 | 8.03 × 10–6 |
| cg26479305 | chr12 | 52470979 | ATG10 (C12orf44) | 3’UTR | 0.338 | 0.077 | 1.11 × 10–5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odintsova, V.V.; Hagenbeek, F.A.; Suderman, M.; Caramaschi, D.; van Beijsterveldt, C.E.M.; Kallsen, N.A.; Ehli, E.A.; Davies, G.E.; Sukhikh, G.T.; Fanos, V.; et al. DNA Methylation Signatures of Breastfeeding in Buccal Cells Collected in Mid-Childhood. Nutrients 2019, 11, 2804. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11112804
Odintsova VV, Hagenbeek FA, Suderman M, Caramaschi D, van Beijsterveldt CEM, Kallsen NA, Ehli EA, Davies GE, Sukhikh GT, Fanos V, et al. DNA Methylation Signatures of Breastfeeding in Buccal Cells Collected in Mid-Childhood. Nutrients. 2019; 11(11):2804. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11112804
Chicago/Turabian StyleOdintsova, Veronika V., Fiona A. Hagenbeek, Matthew Suderman, Doretta Caramaschi, Catharina E. M. van Beijsterveldt, Noah A. Kallsen, Erik A. Ehli, Gareth E. Davies, Gennady T. Sukhikh, Vassilios Fanos, and et al. 2019. "DNA Methylation Signatures of Breastfeeding in Buccal Cells Collected in Mid-Childhood" Nutrients 11, no. 11: 2804. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11112804