Atorvastatin and Vitamin E Accelerates NASH Resolution by Dietary Intervention in a Preclinical Guinea Pig Model

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
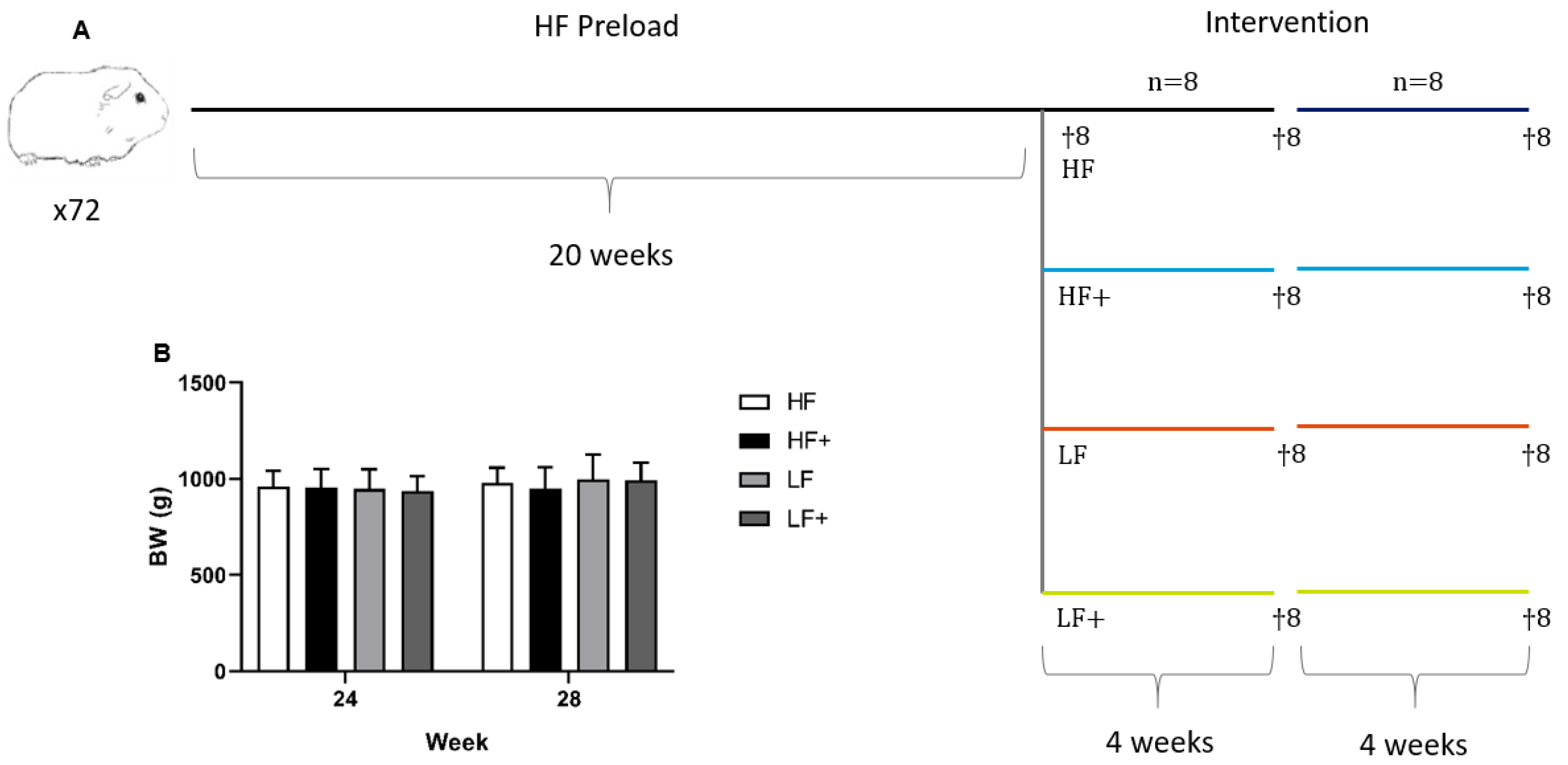
2.1. Animals and Experimental Design
2.2. Plasma Samples
2.3. Liver Samples
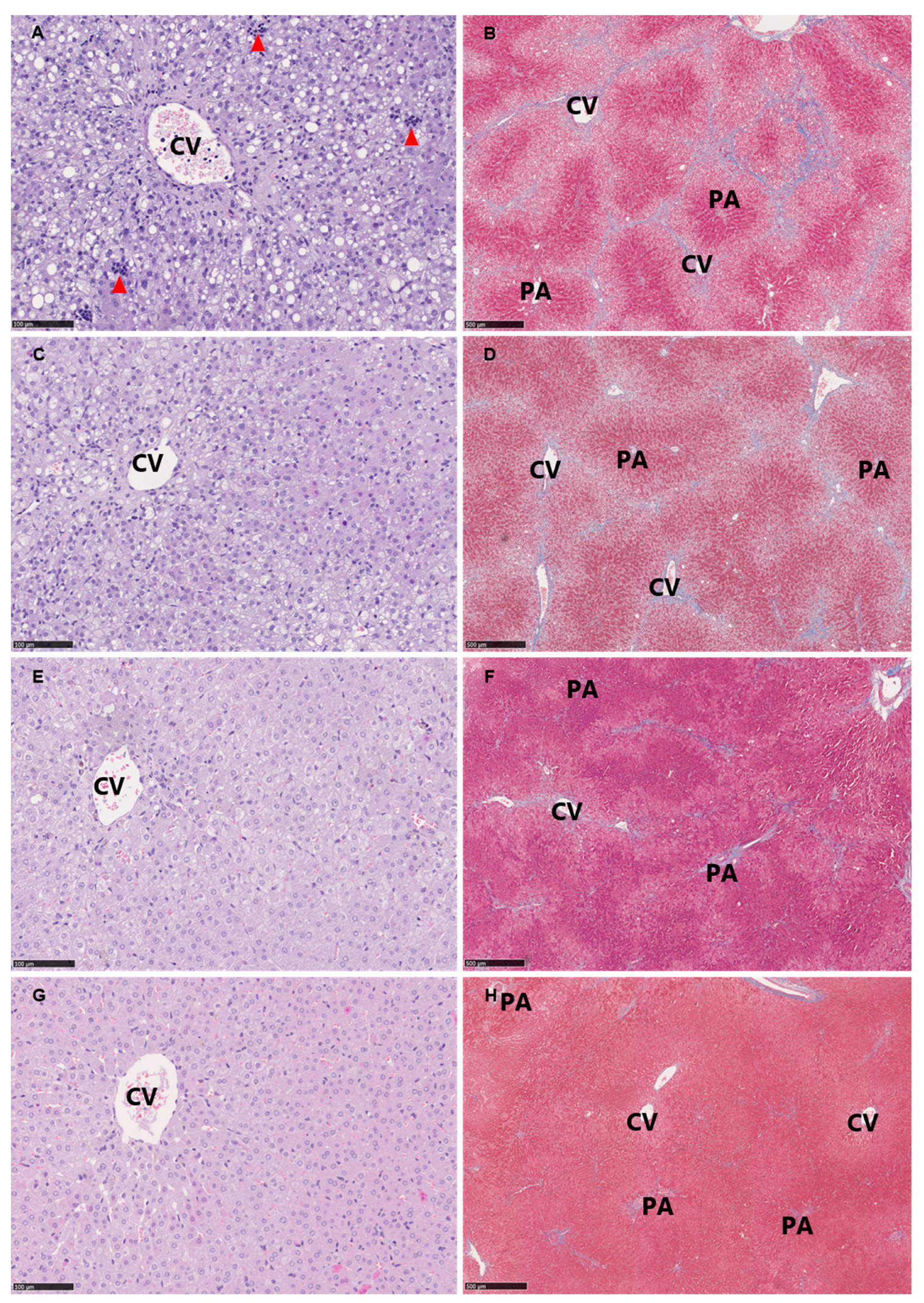
2.4. Histology
2.5. qPCR
2.6. Statistical Analyses
3. Results
3.1. Plasma and Hepatic Vitamin E Status
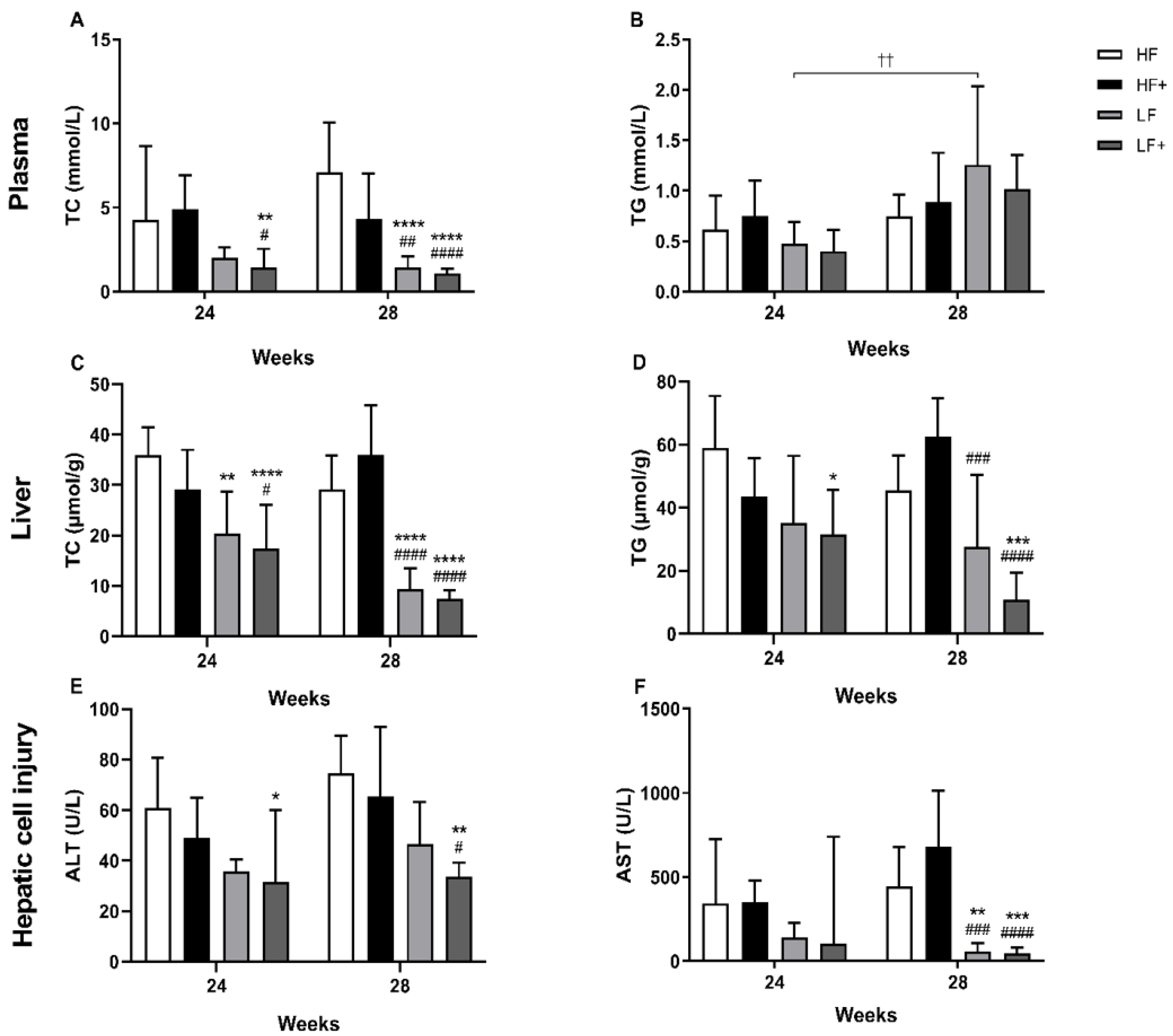
3.2. Dyslipidemia
3.3. Biochemical Markers
3.4. Liver Status
3.5. Histopathological Evaluation
3.6. Expression of Target Genes/qPCR
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.; Arrese, T.M.F.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.; Yilmaz, Y.; George, J.; Fan, J.; Vos, M.B. Global Perspectives on Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis. Hepatology 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (Nafld). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Birck, M.M.; Ipsen, D.H.; Thiessen, T.; Feldmann, L.B.; Lindblad, M.M.; Jensen, H.E.; Lykkesfeldt, J. Diet-Induced Dyslipidemia Leads to Nonalcoholic Fatty Liver Disease and Oxidative Stress in Guinea Pigs. Transl. Res. 2016, 168, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of Nash. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis Stage Is the Strongest Predictor for Disease-Specific Mortality in Nafld after up to 33 Years of Follow-Up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Rafiq, N.; Makhlouf, H.; Younoszai, Z.; Agrawal, R.; Goodman, Z. Pathologic Criteria for Nonalcoholic Steatohepatitis: Interprotocol Agreement and Ability to Predict Liver-Related Mortality. Hepatology 2011, 53, 1874–1882. [Google Scholar] [CrossRef]
- Kim, D.; Kim, W.R.; Kim, H.J.; Therneau, T.M. Association between Noninvasive Fibrosis Markers and Mortality among Adults with Nonalcoholic Fatty Liver Disease in the United States. Hepatology 2013, 57, 1357–1365. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. Diseases American Association for the Study of Liver, Gastroenterology American College of, and Association American Gastroenterological. The Diagnosis and Management of Non-Alcoholic Fatty Liver Disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Am. J. Gastroenterol. 2012, 107, 811–826. [Google Scholar] [CrossRef]
- Catapano, A.L.; Graham, I.; De Backer, G.; Wiklund, O.; Chapman, M.J.; Drexel, H.; Hoes, A.W.; Jennings, C.S.; Landmesser, U.; Pedersen, T.R.; et al. 2016 Esc/Eas Guidelines for the Management of Dyslipidaemias. Rev. Esp. Cardiol. 2017, 70, 115. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). Easl-Easd-Easo Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Banini, B.A.; Sanyal, A.J. Nonalcoholic Fatty Liver Disease: Epidemiology, Pathogenesis, Natural History, Diagnosis, and Current Treatment Options. Clin. Med. Insights Ther. 2016, 8, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Hyogo, H.; Tazuma, S.; Arihiro, K.; Iwamoto, K.; Nabeshima, Y.; Inoue, M.; Ishitobi, T.; Nonaka, M.; Chayama, K. Efficacy of Atorvastatin for the Treatment of Nonalcoholic Steatohepatitis with Dyslipidemia. Metabolism 2008, 57, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Athyros, V.G.; Boutari, C.; Stavropoulos, K.; Anagnostis, P.; Imprialos, K.P.; Doumas, M.; Karagiannis, A. Statins: An under-Appreciated Asset for the Prevention and the Treatment of Nafld or Nash and the Related Cardiovascular Risk. Curr. Vasc. Pharmacol. 2018, 16, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Nagashimada, M.; Ota, T. Role of Vitamin E in Nonalcoholic Fatty Liver Disease. IUBMB Life 2019, 71, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Gosho, M.; Yamamoto, T.; Kobayashi, Y.; Ishii, N.; Ohashi, T.; Nakade, Y.; Ito, K.; Fukuzawa, Y.; Yoneda, M. Vitamin E Has a Beneficial Effect on Nonalcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. Nutrition 2015, 31, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Tao, A.; Zhang, S.; Deng, Y.; Chen, G. Association between Vitamin E and Non-Alcoholic Steatohepatitis: A Meta-Analysis. Int. J. Clin. Exp. Med. 2015, 8, 3924–3934. [Google Scholar]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of Nafld. Diseases 2018, 6, 86. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Tveden-Nyborg, P.; Rolin, B.; Rakipovski, G.; Beck, M.; Mortensen, L.W.; Faerk, L.; Heegaard, P.M.; Moller, P.; Lykkesfeldt, J. High-Fat but Not Sucrose Intake Is Essential for Induction of Dyslipidemia and Non-Alcoholic Steatohepatitis in Guinea Pigs. Nutr. Metab. 2016, 13, 51. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Skat-Rordam, J.; Tsamouri, M.M.; Latta, M.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Drivers of Non-Alcoholic Steatohepatitis Are Sustained in Mild-to-Late Fibrosis Progression in a Guinea Pig Model. Mol. Genet Genom. 2019, 294, 649–661. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Ascorbate and Dehydroascorbic Acid as Reliable Biomarkers of Oxidative Stress: Analytical Reproducibility and Long-Term Stability of Plasma Samples Subjected to Acidic Deproteinization. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2513–2516. [Google Scholar] [CrossRef]
- Reimann, M.J.; Haggstrom, J.; Mortensen, A.; Lykkesfeldt, J.; Moller, J.E.; Falk, T.; Olsen, L.H. Biopterin Status in Dogs with Myxomatous Mitral Valve Disease Is Associated with Disease Severity and Cardiovascular Risk Factors. J. Vet. Intern. Med. 2014, 28, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, A.; Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Guinea Pig Ascorbate Status Predicts Tetrahydrobiopterin Plasma Concentration and Oxidation Ratio in Vivo. Nutr. Res. 2013, 33, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Ekeloef, S.; Larsen, M.H.; Schou-Pedersen, A.M.; Lykkesfeldt, J.; Rosenberg, J.; Gogenur, I. Endothelial Dysfunction in the Early Postoperative Period after Major Colon Cancer Surgery. Br. J. Anaesth. 2017, 118, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Hissin, P.J.; Hilf, R. A Fluorometric Method for Determination of Oxidized and Reduced Glutathione in Tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Determination of Malondialdehyde as Dithiobarbituric Acid Adduct in Biological Samples by Hplc with Fluorescence Detection: Comparison with Ultraviolet-Visible Spectrophotometry. Clin. Chem. 2001, 47, 1725–1727. [Google Scholar]
- Burton, G.W.; Webb, A.; Ingold, K.U. A Mild, Rapid, and Efficient Method of Lipid Extraction for Use in Determining Vitamin E/Lipid Ratios. Lipids 1985, 20, 29–39. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. And Network Nonalcoholic Steatohepatitis Clinical Research. Design and Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Hasselholt, S.; Miyashita, N.; Moos, T.; Poulsen, H.E.; Lykkesfeldt, J. Chronic Vitamin C Deficiency Does Not Accelerate Oxidative Stress in Ageing Brains of Guinea Pigs. Basic Clin. Pharmacol. Toxicol. 2012, 110, 524–529. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of Nafld Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and Future Pharmacological Therapies for Nafld/Nash. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Hadzi-Petrushev, N.; Dimovska, K.; Jankulovski, N.; Mitrov, D.; Mladenov, M. Supplementation with Alpha-Tocopherol and Ascorbic Acid to Nonalcoholic Fatty Liver Disease’s Statin Therapy in Men. Adv. Pharmacol. Sci. 2018, 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costache, I.I.; Garleanu, I.; Aursulesei, V.; Namat, R.A.; Ion, A.; Miftode, R.S.; Tesloianu, D.; Iliescu, D.; Petris, A.O.; Costache, A.D.; et al. Atorvastatin in the Treatment of Dyslipidemic Patients with Very High Cardiovascular Risk and Nonalcoholic Fatty Liver Disease. Rev. Chim. 2019, 70, 2159–2163. [Google Scholar]
- Sigler, M.A.; Congdon, L.; Edwards, K.L. An Evidence-Based Review of Statin Use in Patients with Nonalcoholic Fatty Liver Disease. Clin. Med. Insights Gastroenterol. 2018, 11, 1179552218787502. [Google Scholar] [CrossRef] [Green Version]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. And Network Nonalcoholic Steatohepatitis Clinical Research. Effect of Vitamin E or Metformin for Treatment of Nonalcoholic Fatty Liver Disease in Children and Adolescents: The Tonic Randomized Controlled Trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [Green Version]
- Athyros, V.G.; Tziomalos, K.; Gossios, T.D.; Griva, T.; Anagnostis, P.; Kargiotis, K.; Pagourelias, E.D.; Theocharidou, E.; Karagiannis, A.; Mikhailidis, D.P. Greace Study Collaborative Group. Safety and Efficacy of Long-Term Statin Treatment for Cardiovascular Events in Patients with Coronary Heart Disease and Abnormal Liver Tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (Greace) Study: A Post-Hoc Analysis. Lancet 2010, 376, 1916–1922. [Google Scholar] [CrossRef]
- Oshakbayev, K.; Bimbetov, B.; Manekenova, K.; Bedelbayeva, G.; Mustafin, K.; Dukenbayeva, B. Severe Nonalcoholic Steatohepatitis and Type 2 Diabetes: Liver Histology after Weight Loss Therapy in a Randomized Clinical Trial. Curr. Med. Res. Opin. 2019, 35, 157–165. [Google Scholar] [CrossRef]
- Chalasani, N.P.; Sanyal, A.J.; Kowdley, K.V.; Robuck, P.R.; Hoofnagle, J.; Kleiner, D.E.; Unalp, A.; Tonascia, J. And Nash Crn Research Group. Pioglitazone Versus Vitamin E Versus Placebo for the Treatment of Non-Diabetic Patients with Non-Alcoholic Steatohepatitis: Pivens Trial Design. Contemp. Clin. Trials 2009, 30, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Connolly, J.J.; Ooka, K.; Lim, J.K. Future Pharmacotherapy for Non-Alcoholic Steatohepatitis (Nash): Review of Phase 2 and 3 Trials. J. Clin. Transl. Hepatol. 2018, 6, 264–275. [Google Scholar] [CrossRef]
- Okada, Y.; Yamaguchi, K.; Nakajima, T.; Nishikawa, T.; Jo, M.; Mitsumoto, Y.; Kimura, H.; Nishimura, T.; Tochiki, N.; Yasui, K.; et al. Rosuvastatin Ameliorates High-Fat and High-Cholesterol Diet-Induced Nonalcoholic Steatohepatitis in Rats. Liver Int. 2013, 33, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Yoneda, M.; Nakamura, K.; Makino, I.; Terano, A. Plasma Transforming Growth Factor-Beta1 Level and Efficacy of Alpha-Tocopherol in Patients with Non-Alcoholic Steatohepatitis: A Pilot Study. Aliment. Pharmacol. Ther. 2001, 15, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. Tgf-Beta in Progression of Liver Disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemes, K.; Aberg, F. Interpreting Lipoproteins in Nonalcoholic Fatty Liver Disease. Curr. Opin. Lipidol. 2017, 28, 355–360. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, K.L.; Ruan, X.Z.; Liu, B.C. Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury. Int. J. Biol. Sci. 2016, 12, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Kayden, H.J.; Traber, M.G. Lipoprotein Transport, and Regulation of Plasma Concentrations of Vitamin E in Humans. J. Lipid Res. 1993, 34, 343–358. [Google Scholar]
- Jeanes, Y.M.; Hall, W.L.; Ellard, S.; Lee, E.; Lodge, J.K. The Absorption of Vitamin E Is Influenced by the Amount of Fat in a Meal and the Food Matrix. Br. J. Nutr. 2004, 92, 575–579. [Google Scholar] [CrossRef] [Green Version]
- Traber, M.G.; Leonard, S.W.; Ebenuwa, I.; Violet, P.C.; Wang, Y.; Niyyati, M.; Padayatty, S.; Tu, H.; Courville, A.; Bernstein, S.; et al. Vitamin E Absorption and Kinetics in Healthy Women, as Modulated by Food and by Fat, Studied Using 2 Deuterium-Labeled Alpha-Tocopherols in a 3-Phase Crossover Design. Am. J. Clin. Nutr. 2019, 110, 1148–1167. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nutrient | HF | HF+ | LF | LF+ |
|---|---|---|---|---|
| Protein (%) | 16.7 | 16.7 | 16.8 | 16.8 |
| Carbohydrates (%) | 37.9 | 37.9 | 47.1 | 47.1 |
| Fat (%) | 20 | 20 | 4 | 4 |
| Cholesterol (%) | 0.35 | 0.35 | - | - |
| Sucrose (%) | 15 | 15 | - | - |
| Vitamin E (all-rac-alpha-tocopheryl acetate (mg/kg feed)) | 125 | 375 | 125 | 375 |
| Atorvastatin (mg/kg feed) | - | 20 | - | 20 |
| Gene | Accession no. | Forward (5’-3’) | Reverse (3’-5’) | Product (bp) |
|---|---|---|---|---|
| HMGR | XM_003461336.3 | TACAGACATGGGCATTGG GT | GGCAGGGAAAGTGTTGAGTG | 184 |
| LDLR | XM_013149927.1 | GACGTGTCCCAGAGG AAGAT | CGAGTCGGTCCAGTAGATGTT | 144 |
| SREBP2 | XM_003470391.3 | GGGGCTCAAAGGTCTTCTCT | AGGACCCCATCAAAGTGAGG | 189 |
| ABCG5 | XM_003472925.3 | ATCCTGAGGCTGCTCGATTT | CCAGATCCAATCAGCAACCC | 163 |
| ABCG8 | XM_003472926.2 | GTCTCAACTCCCACCCTCTC | CTGAAGGGTCTTCTCCGAGG | 170 |
| CYP7a1 | GQ507494.1 | CTGGAGAAGGCAGGTCAACA | CTCCTTAGCTGTCCGGATGT | 150 |
| IL-8 | NM_001173399.2 | GGCAGCCTTCCTGCTCTCT | CAGCTCCGAGACCAACTTTGT | 67 |
| MCP-1 | NM_001172926.1 | TGCCAAACTGGACCAGAGAA | CGAATGTTCAAAGGCTTTGAAGT | 75 |
| TNF-α | NM_001173025.1 | GCCGTCTCCTACCCGGAAAA | TAGATCTGCCCGGAATCGGC | 203 |
| TGF-β | NM_001173023.1 | AACCCGAGCCGGACTACTATG | TGCTTTTATAGATATTGTGGCTGT TGT | 78 |
| Col1a1 | XM_003466865.2 | CTGGACAGCGTGGTGTAGTC | TCCAGAAGGACCTTGTTTGC | 104 |
| α-sma | ENSCPOT00000011693.2 | GACATCAAGGAGAAGCTGTG | GCTGTTGTAGGTGGTTTCAT | 273 |
| Pre-intervention | Week 24 | Week 28 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HF | HF+ | LF | LF+ | HF | HF+ | LF | LF+ | ||
| FFA (mmol/L) | 0.41 ± 0.04 | 0.64 ± 0.17 | 0.40 ± 0.10 | 0.63 ± 0.20 | 0.61 ±0.16 | 0.50 ± 0.21 | 0.42 ± 0.14 | 0.58 ± 0.20 | 0.42 ± 0.17 |
| ALP (U/L) | 46.13 ± 9.73 | 37.38 ± 7.76 | 40.0 ± 7.60 | 48.0 ± 9.32 | 47.25 ± 12.61 | 41.90 ± 8.36 | 38.5 ± 7.35 | 51.88 ± 10.66 | 50.13 ± 13.81 |
| LW (%) j | 4.15 (3.44–4.86) | 5.15 (4.58−5.72) | 4.82 (4.10−5.54) | 3.34 (2.91−3.77) f, h | 3.18 (2.73−3.63) f, i | 4.60 (3.64−5.56) | 5.96 (4.76−7.16) | 3.15 (2.81−3.50) e, i | 2.85 (2.53−3.16) b, f, i |
| BH2/BH4 | 0.05 ± 0.02 | 0.13 ± 0.037 c | 0.11 ± 0.03 a | 0.13 ± 0.03 c | 0.12 ± 0.04 b | 0.08 ± 0.04 | 0.10 ± 0.04 | 0.08 ± 0.03 | 0.07 ± 0.03 |
| TAA (µM) | 40.56 ± 17.09 | 37.39 ± 14.40 | 32.28 ± 12.93 | 70.73 ± 27.94 a, e, i | 57.64 ± 19.34 | 28.69 ± 6.92 | 33.85 ± 15.51 | 55.05 ± 11.11 d | 56.51 ± 10.56 d |
| αToc (µM) j | 4.52 (2.84−6.20) | 3.10 (1.91−4.29) | 6.70 (3.37−10.03) d | 1.35 (0.89−1.82) b, i | 1.89 (1.32−2.46) i | 2.70 (1.73−3.68) | 6.63 (2.88−10.38) d | 1.59 (0.94−2.23) a, h | 2.69 (1.62−3.77) g |
| Pre-intervention | HF | HF+ | LF | LF+ | |
|---|---|---|---|---|---|
| AA (nmol/g) | 1283.5 ± 379.51 | 1343 ± 533.44 | 1230.88 ± 474.33 | 1381.5 ± 518.21 | 1663.13 ± 598.48 |
| BH2/BH4 | 0.21 ± 0.05 | 0.15 ± 0.10 | 0.23 ± 0.11 | 0.20 ± 0.12 | 0.23 ± 0.12 |
| L-arg (nmol/g) | 32.96 ± 8.73 | 30.47 ± 11.83 | 27.54 ± 7.10 | 29.14 ± 13.40 | 21.60 ± 6.54 |
| ADMA (nmol/g) | 0.73 ± 0.92 | 0.40 ± 0.53 | 0.92 ± 1.20 | 0.50 ± 0.53 | 0.17 ± 0.50 |
| GSH (nmol/g) | 3160.63 ± 503.63 | 3131.5 ± 316.22 | 2967.75 ± 336.71 | 3305.90 ± 498.19 | 3118.00 ± 424.33 |
| GSSG (nmol/g) | 317.72 ± 49.40 | 331.40 ± 51.99 | 293.81 ± 83.70 | 315.01 ± 61.90 | 307.70 ± 49.51 |
| SOD (U/mg) | 2489.5 ± 432.10 | 2621.40 ± 343.50 | 2495.00 ± 782.92 | 2449.40 ± 405.90 | 3107.80 ± 952.75 |
| αToc (nmol/g) j | 1.71 (1.22–2.19) | 3.47 (0.92–6.03) | 5.14 (2.02–8.26) a | 3.39 (1.88–4.91) | 3.30 (1.53–5.06) |
| Pre-intervention | HF | HF+ | LF | LF+ | |
|---|---|---|---|---|---|
| AA (nmol/g) | 1283.5 ± 379.51 | 1404.5 ± 497.99 | 1357 ± 457.91 | 1287.75 ± 573.73 | 1770 ± 380.30 |
| BH2/BH4 | 0.21 ± 0.05 | 0.20 ± 0.12 | 0.30 ± 0.16 | 0.23 ± 0.10 | 0.23 ± 0.20 |
| L-arg (nmol/g) | 32.96 ± 8.73 | 22.10 ± 9.56 | 24.95 ± 6.47 | 23.60 ± 6.50 | 36.74 ± 10.80 |
| ADMA (nmol/g) | 0.73 ± 0.92 | 0.99 ± 0.80 | 0.70 ± 0.60 | 0.50 ± 0.60 | 0.91 ± 1.10 |
| GSH (nmol/g) | 3160.63 ± 503.63 | 3440.40 ± 487.40 | 3138.63 ± 301.32 | 3072.25 ± 305.80 | 3303.25 ± 170.97 |
| GSSG (nmol/g) | 317.72 ± 49.40 | 300.52 ± 69.90 | 295.40 ± 68.50 | 322.70 ± 32.30 | 324.80 ± 61.96 |
| SOD (U/mg) | 2489.5 ± 432.10 | 2783.90 ± 786.60 | 2512.00 ± 642.80 | 3206.40 ± 1059.80 | 2899.13 ± 502.82 |
| αToc (nmol/g) j | 1.71 (1.22–2.19) | 2.87 (0.90–4.84) | 9.43 (3.68–15.18) a, b, c | 3.49 (1.95–5.02) | 8.61 (4.98–12.24) a, b, c |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klaebel, J.H.; Skjødt, M.; Skat-Rørdam, J.; Rakipovski, G.; Ipsen, D.H.; Schou-Pedersen, A.M.V.; Lykkesfeldt, J.; Tveden-Nyborg, P. Atorvastatin and Vitamin E Accelerates NASH Resolution by Dietary Intervention in a Preclinical Guinea Pig Model. Nutrients 2019, 11, 2834. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11112834
Klaebel JH, Skjødt M, Skat-Rørdam J, Rakipovski G, Ipsen DH, Schou-Pedersen AMV, Lykkesfeldt J, Tveden-Nyborg P. Atorvastatin and Vitamin E Accelerates NASH Resolution by Dietary Intervention in a Preclinical Guinea Pig Model. Nutrients. 2019; 11(11):2834. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11112834
Chicago/Turabian StyleKlaebel, Julie Hviid, Mia Skjødt, Josephine Skat-Rørdam, Günaj Rakipovski, David H. Ipsen, Anne Marie V. Schou-Pedersen, Jens Lykkesfeldt, and Pernille Tveden-Nyborg. 2019. "Atorvastatin and Vitamin E Accelerates NASH Resolution by Dietary Intervention in a Preclinical Guinea Pig Model" Nutrients 11, no. 11: 2834. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11112834