Mechanisms of Calorie Restriction: A Review of Genes Required for the Life-Extending and Tumor-Inhibiting Effects of Calorie Restriction

 , , and
, , and 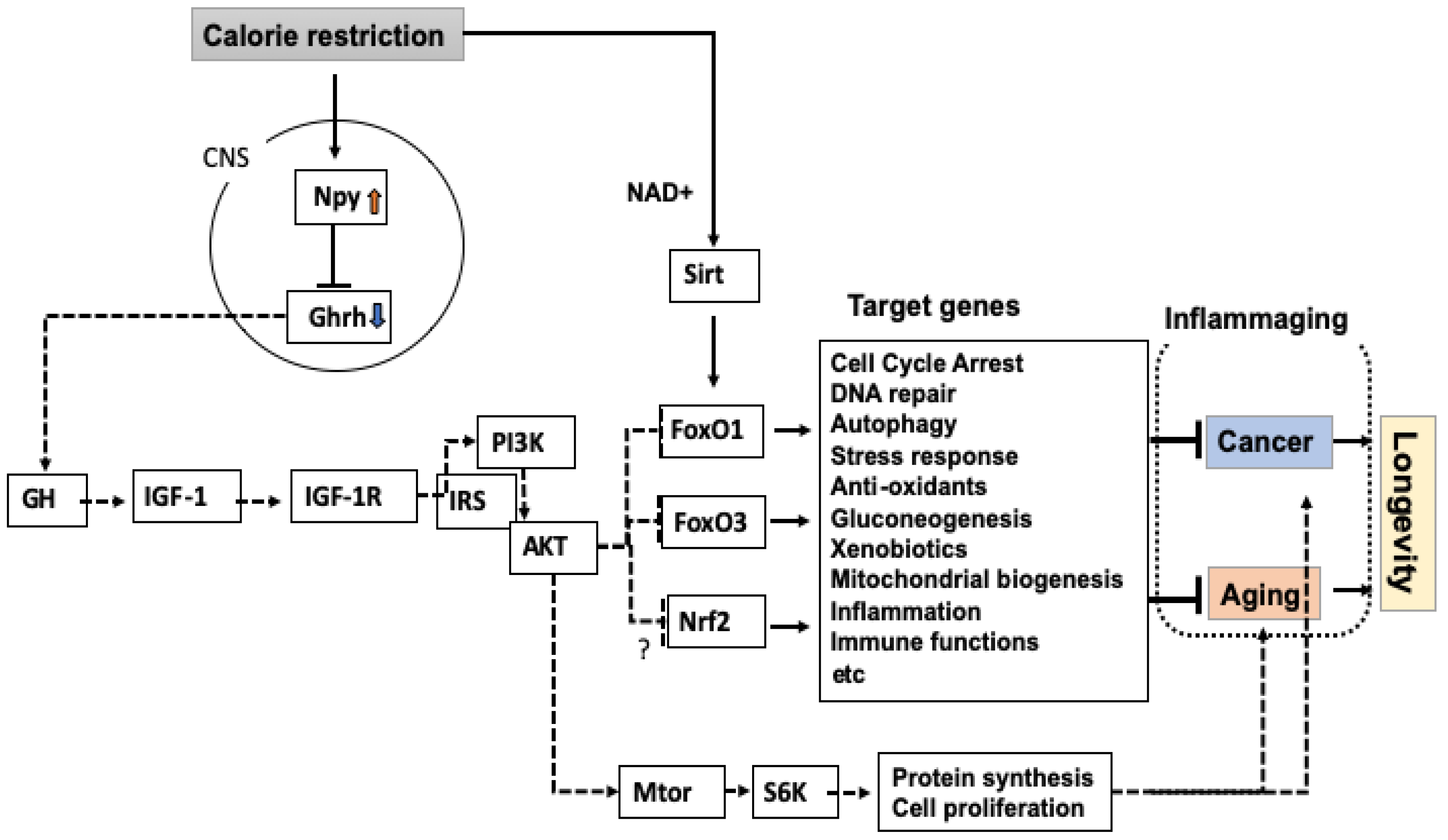{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
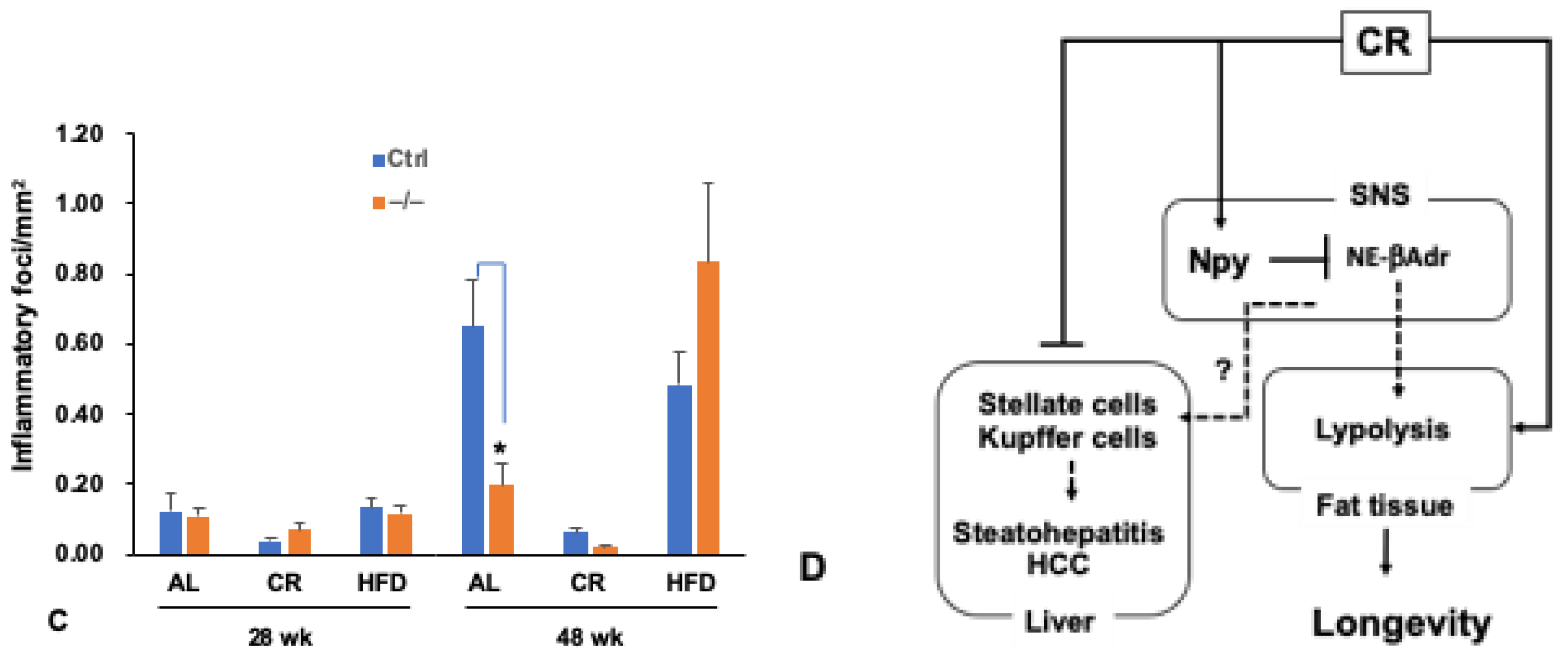
Abstract
:1. Introduction
2. A Central Role for GH and IGF-1 in the Regulation of Lifespan
3. Possible Relation of the GH-IGF-1 Axis with Inflammaging
4. Differential Regulation of Cancer and Lifespan by CR via FoxO Transcription Factors
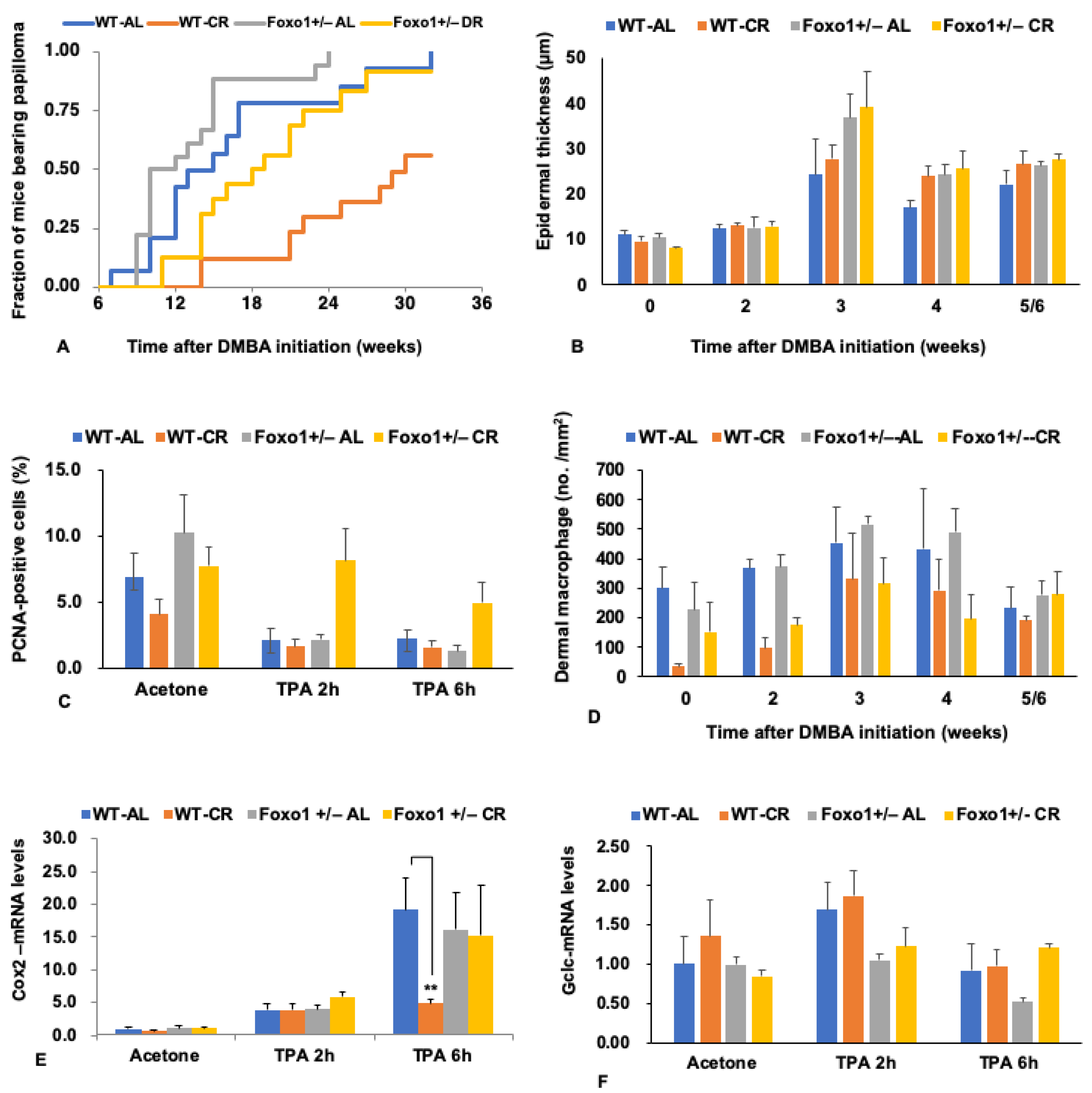
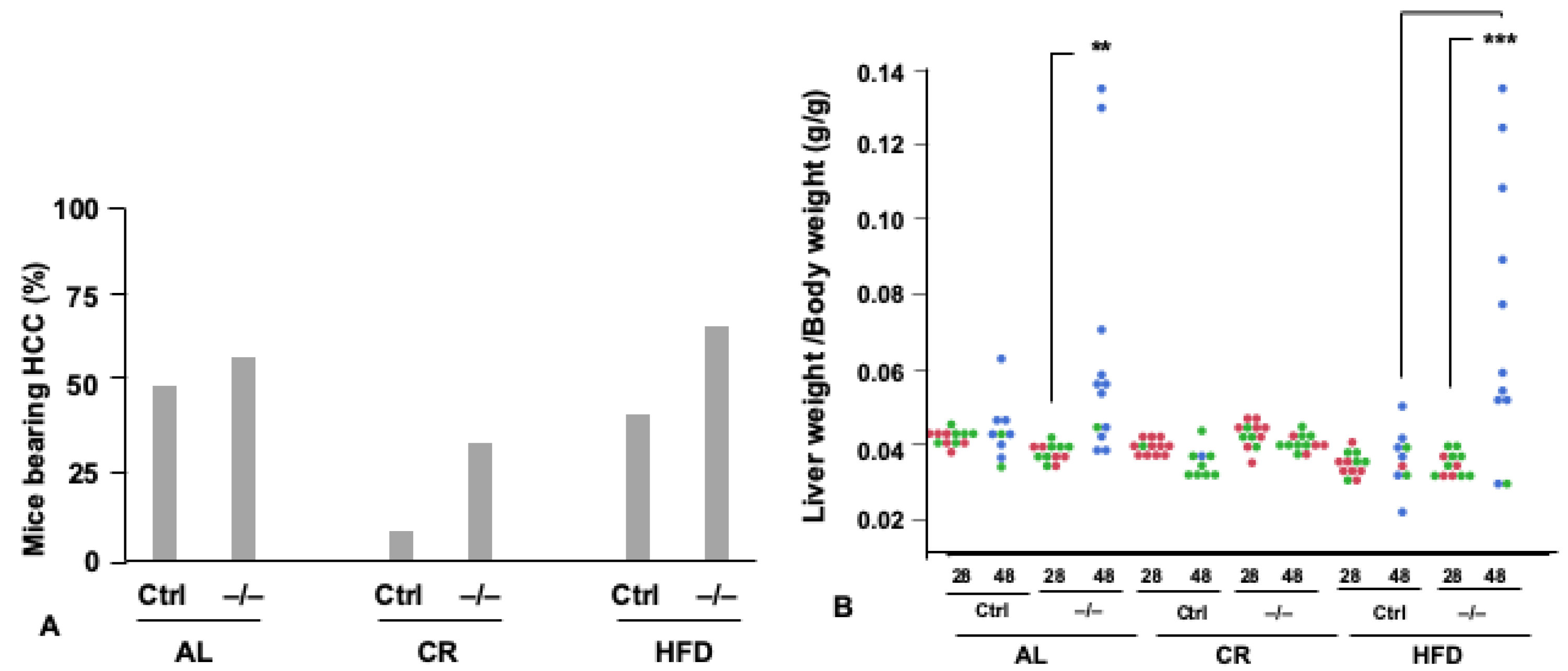
5. FoxO1 Mediates the Tumor-Inhibiting Effect of CR
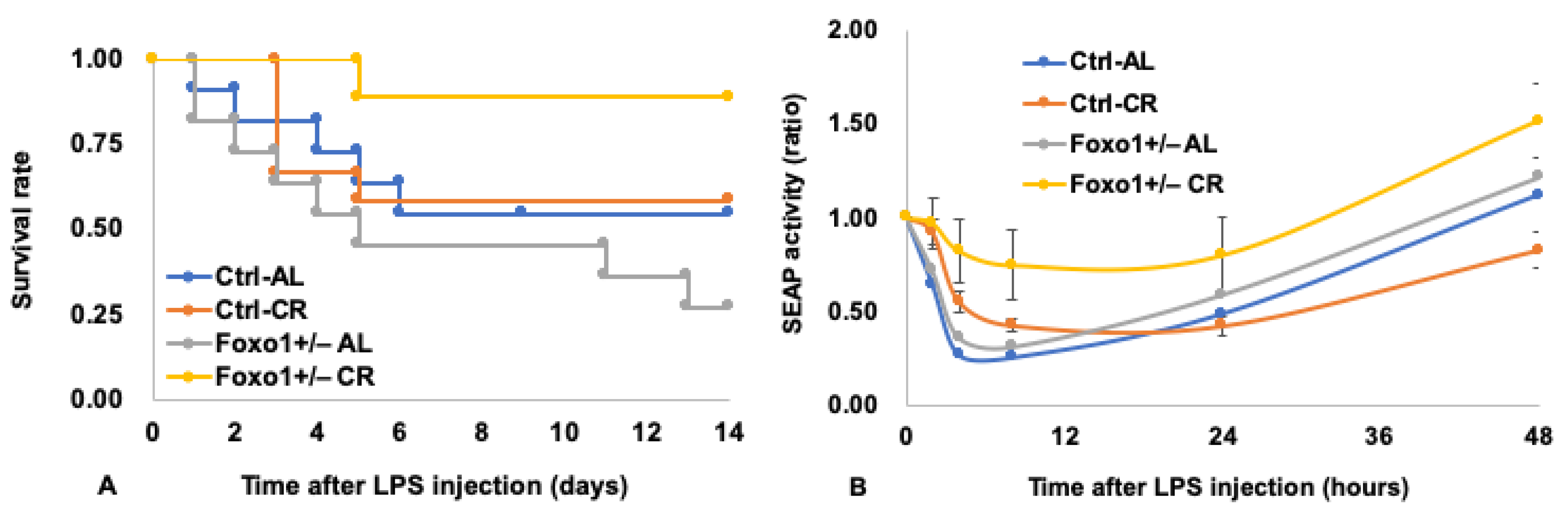
6. Pleiotropic Roles for Foxo1 in Disorders
7. Sirtuin as a Molecule Upstream of FoxO
8. Roles for Npy in the Effect of CR
9. Role for Npy in the Tumor-Inhibiting Effect of CR
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Microarray Analysis
Appendix A.2. The Multistage Skin Tumorigenesis Model
Appendix A.2.1. Experimental Protocol
Appendix A.2.2. Histology and Immunohistochemistry
Appendix A.2.3. RNA Isolation and Quantitative Polymerase Chain Reaction
Appendix A.2.4. Statistical Analysis
References
- Weindruch, R.; Walford, R.L. The Retardation of Aging and Disease by Dietary Restriction; Charles C Thomas Publisher: Springfield, IL, USA, 1988; pp. 31–115. [Google Scholar]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. J. Nutr. 1935, 10, 63–79. [Google Scholar] [CrossRef]
- Swindle, W.R. Dietary restriction in rats and mice: A meta-analysis and review of the evidence fro genotype-dependent effects on lifespan. Ageing Res. Rev. 2012, 11, 254–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baloni, P.; Sangar, V.; Yurkovich, J.; Robinson, M.; Taylor, S.; Karbowski, C.; Hamadeh, H.; He, Y.; Price, N. Genome-scale metabolic model of the rat liver predicts effects of diet restriction. Sci. Rep. 2019, 9, 9807. [Google Scholar]
- Mitchell, S.; Madrigal-Matute, J.; Scheibye-Knudsen, M.; Fang, E.; Aon, M.; González-Reyes, J.; Cortassa, S.; Kaushik, S.; Gonzalez-Freire, M.; Patel, B.; et al. Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell Metab. 2016, 23, 1093–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, A.; Sinclair, D.; Mitchell, J.; Mitchell, S. Sex differences in the response to dietary restriction in rodents. Curr. Opin. Physiol. 2018, 6, 28–34. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 2009, 8, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Shimokawa, I.; Chiba, T.; Yamaza, H.; Komatsu, T. Longevity genes: Insights from calorie restriction and genetic longevity models. Mol. Cells 2008, 26, 427–435. [Google Scholar]
- Longo, V.D.; Finch, C.E. Evolutionary medicine: From dwarf model systems to healthy centenarians? Science 2003, 299, 1342–1346. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://genomics.senescence.info/diet/ (accessed on 6 November 2019).
- Johnson, T.E. Increased life-span of age-1 mutants in Caenorhabditis elegans and lower Gompertz rate of aging. Science 1990, 249, 908–912. [Google Scholar] [CrossRef]
- Morris, J.Z.; Tissenbaum, H.A.; Ruvkun, G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 1996, 382, 536–539. [Google Scholar] [CrossRef]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Slack, C.; Foley, A.; Partridge, L. Activation of AMPK by the Putative Dietary Restriction Mimetic Metformin Is Insufficient to Extend Lifespan in Drosophila. PLoS ONE 2012, 7, e47699. [Google Scholar] [CrossRef] [PubMed]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L. Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef] [PubMed]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimokawa, I. Growth hormone and IGF-1 axis in aging and longevity. In Hormones in Ageing and Longevity, 1st ed.; Rattan, S., Sharma, R., Eds.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 91–106. [Google Scholar]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Selman, C.; Tullet, J.M.A.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal Protein S6 Kinase 1 Signaling Regulates Mammalian Life Span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.J.; Liu, J.; Chen, E.B.; Wang, J.J.; Cao, L.; Narayan, N.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013, 4, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.; Cervera, P.; Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Xu, J.; Gontier, G.; Chaker, Z.; Lacube, P.; Dupont, J.; Holzenberger, M. Longevity effect of IGF-1R(+/-) mutation depends on genetic background-specific receptor activation. Aging Cell 2014, 13, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Suh, Y.; Atzmon, G.; Cho, M.; Hwang, D.; Liu, B.; Leahy, D.; Barzilai, N.; Cohen, P. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl. Acad. Sci. USA 2008, 105, 3438–3442. [Google Scholar] [CrossRef] [Green Version]
- Milman, S.; Atzmon, G.; Huffman, D.; Wan, J.; Crandall, J.; Cohen, P.; Barzilai, N. Low insulin-like growth factor-1 level predicts survival in humans with exceptional longevity. Aging Cell 2014, 13, 769–771. [Google Scholar] [CrossRef] [PubMed]
- van Heemst, D.; Beekman, M.; Mooijaart, S.; Heijmans, B.; Brandt, B.; Zwaan, B.; Slagboom, P.; Westendorp, R. Reduced insulin/IGF-1 signalling and human longevity. Aging Cell 2005, 4, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Zaikin, A.; Gordleeva, S.; Ivanchenko, M.; Bonifazi, F.; Storci, G.; Bonafè, M. Inflammaging 2018: An update and a model. Semin. Immunol. 2018, 40, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- Huang, J.; Xie, Y.; Sun, X.; Zeh, H.J., 3rd; Kang, R.; Lotze, M.T.; Tang, D. DAMPs, ageing, and cancer: The ‘DAMP Hypothesis’. Ageing Res. Rev. 2015, 24, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Spadaro, O.; Goldberg, E.L.; Camell, C.D.; Youm, Y.-H.; Kopchick, J.J.; Nguyen, K.Y.; Bartke, A.; Sun, L.Y.; Dixit, V.D. Growth Hormone Receptor Deficiency Protects against Age-Related NLRP3 Inflammasome Activation and Immune Senescence. Cell Rep. 2016, 14, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [Green Version]
- Furuyama, T.; Kitayama, K.; Shimoda, Y.; Ogawa, M.; Sone, K.; Yoshida-Araki, K.; Hisatsune, H.; Nishikawa, S.-I.; Nakayama, K.; Nakayama, K.; et al. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J. Biol. Chem. 2004, 279, 34741–34749. [Google Scholar] [CrossRef] [Green Version]
- Yamaza, H.; Komatsu, T.; Wakita, S.; Kijogi, C.; Park, S.; Hayashi, H.; Chiba, T.; Mori, R.; Furuyama, T.; Mori, N.; et al. FoxO1 is involved in the antineoplastic effect of calorie restriction. Aging Cell 2010, 9, 372–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimokawa, I.; Komatsu, T.; Hayashi, N.; Kim, S.E.; Kawata, T.; Park, S.; Hayashi, H.; Yamaza, H.; Chiba, T.; Mori, R. The life-extending effect of dietary restriction requires Foxo3 in mice. Aging Cell 2015, 14, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.-H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.; Dorman, J.B.; Rodan, A.; Kenyon, C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 1997, 278, 1319–1322. [Google Scholar] [CrossRef] [Green Version]
- Ogg, S.; Paradis, S.; Gottlieb, S.; Patterson, G.I.; Lee, L.; Tissenbaum, H.A.; Ruvkun, G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 1997, 389, 994–999. [Google Scholar] [CrossRef]
- Kwon, E.-S.; Narasimhan, S.D.; Yen, K.; Tissenbaum, H.A. A new DAF-16 isoform regulates longevity. Nature 2010, 466, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Hsin, H.; Libina, N.; Kenyon, C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 2001, 28, 139–145. [Google Scholar] [CrossRef]
- Willcox, B.J.; Donlon, T.A.; He, Q.; Chen, R.; Grove, J.S.; Yano, K.; Masaki, K.H.; Willcox, D.C.; Rodriguez, B.; Curb, J.D. FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 2008, 105, 13987–13992. [Google Scholar] [CrossRef] [Green Version]
- Teumer, A.; Qi, Q.; Nethander, M.; Aschard, H.; Bandinelli, S.; Beekman, M.; Berndt, S.I.; Bidlingmaier, M.; Broer, L. CHARGE Longevity Working Group. Genomewide meta-analysis identifies loci associated with IGF-I and IGFBP-3 levels with impact on age-related traits. Aging Cell 2016, 15, 811–824. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Lee, B.G.; Lee, J.W.; Kim, M.E.; Lee, J.S.; Chung, J.H.; Yu, B.P.; Dong, H.H.; Chung, H.Y. FoxO6-mediated IL-1β induces hepatic insulin resistance and age-related inflammation via the TF/PAR2 pathway in aging and diabetic mice. Redox Biol. 2019, 24, 101184. [Google Scholar] [CrossRef]
- Litvak, V.; Ratushny, A.V.; Lampano, A.E.; Schmitz, F.; Huang, A.C.; Raman, A.; Rust, A.G.; Bergthaler, A.; Aitchison, J.D.; Aderem, A. A FOXO3–IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature 2012, 490, 421–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, K.J.; Lewis, K.N.; Price, N.L.; Chang, J.W.; Perez, E.; Cascajo, M.V.; Tamashiro, K.L.; Poosala, S.; Csiszar, A.; Ungvari, Z.; et al. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc. Natl. Acad. Sci. USA 2008, 105, 2325–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap“n”collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, M.; Münzel, P.A.; Braeuning, A. Non-melanoma skin cancer in mouse and man. Arch. Toxicol 2013, 87, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Passos, G.F.; Medeiros, R.; Marcon, R.; Nascimento, A.F.G.; Calixto, J.B.; Pianowski, L.F. The role of PKC/ERK1/2 signaling in the anti-inflammatory effect of tetracyclic triterpene euphol on TPA-induced skin inflammation in mice. Eur. J. Pharm. 2013, 698, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Przybysz, A.J.; Choe, K.P.; Roberts, L.J.; Strange, K. Increased age reduces DAF-16 and SKN-1 signaling and the hormetic response of Caenorhabditis elegans to the xenobiotic juglone. Mech. Ageing Dev. 2009, 130, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Mori, R.; Tanaka, K.; de Kerckhove, M.; Okamoto, M.; Kashiyama, K.; Tanaka, K.; Kim, S.; Kawata, T.; Komatsu, T.; Park, S.; et al. Reduced FOXO1 Accelerates Skin Wound Healing and Attenuates Scarring. Am. J. Pathol. 2014, 184, 2465–2467. [Google Scholar] [CrossRef] [Green Version]
- Nakae, J.; Oki, M.; Cao, Y. The FoxO transcription factors and metabolic regulation. FEBS Lett. 2008, 582, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Kamohara, R.; Yamaza, H.; Tsuchiya, T.; Komatsu, T.; Park, S.; Hayashi, H.; Chiba, T.; Mori, R.; Otabe, S.; Yamada, K.; et al. Overexpression of the adiponectin gene mimics the metabolic and stress resistance effects of calorie restriction, but not the anti-tumor effect. Exp. Gerontol. 2015, 64, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, M.; Hiramatsu, N. Real-time monitoring of ER stress in living cells and animals using ESTRAP assay. Method. Enzymol. 2011, 490, 93–106. [Google Scholar]
- Chalkiadaki, A.; Guarente, L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 2012, 8, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. SIRT1 and other sirtuins in metabolism. Genes. Dev. 2014, 25, 138–145. [Google Scholar]
- Banerjee, K.K.; Ayyub, C.; Zeeshan, A.S.; Mandot, V.; Prasad, G. dSir2 in the adult fat body, but not in muscles, regulates life span in a diet-dependent manner. Cell Rep. 2012, 2, 1485–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.-I.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercken, E.M.; Hu, J.; Krzysik-Walker, S.; Wei, M.; Li, Y.; McBurney, M.W.; de Cabo, R.; Longo, V.D. SIRT1 but not its increased expression is essential for lifespan extension in caloric-restricted mice. Aging Cell 2014, 13, 193–196. [Google Scholar] [CrossRef] [Green Version]
- Holliday, R. Food, reproduction and longevity: Is the extended lifespan of calorie-restricted animals an evolutionary adaptation? Bioessays 1989, 10, 125–127. [Google Scholar] [CrossRef]
- Bishop, N.A.; Guarente, L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 2007, 447, 545–549. [Google Scholar] [CrossRef]
- Bargmann, C.I.; Horvitz, H.R. Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Neuron 1991, 7, 729–742. [Google Scholar] [CrossRef]
- Shimokawa, I.; Higami, Y. Leptin signaling and aging: Insight from caloric restriction. Mech. Ageing Dev. 2001, 122, 1511–1519. [Google Scholar] [CrossRef]
- Sainsbury, A.; Zhang, L. Role of the hypothalamus in the neuroendocrine regulation of body weight and composition during energy deficit. Obes. Rev. 2011, 13, 234–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalkiewicz, M.; Knestaut, K.M.; Bytchkova, E.Y.; Michalkiewicz, T. Hypotension and reduced catecholamines in neuropeptide Y transgenic rats. Hypertension 2003, 41, 1056–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smiałowska, M.; Domin, H.; Zieba, B.; Koźniewska, E.; Michalik, R.; Piotrowski, P.; Kajta, M. Neuroprotective effects of neuropeptide Y-Y2 and Y5 receptor agonists in vitro and in vivo. Neuropeptides 2009, 43, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, I.; Fukuyama, T.; Yanagihara-Outa, K.; Tomita, M.; Komatsu, T.; Higami, Y.; Tuchiya, T.; Chiba, T.; Yamaza, Y. Effects of caloric restriction on gene expression in the arcuate nucleus. Neurobiol. Aging 2003, 24, 117–123. [Google Scholar] [CrossRef]
- Chiba, T.; Tamashiro, Y.; Park, D.; Kusudo, T.; Fujie, R.; Komatsu, T.; Kim, S.E.; Park, S.; Hayashi, H.; Mori, R.; et al. A key role for neuropeptide Y in lifespan extension and cancer suppression via dietary restriction. Sci. Rep. 2014, 4, 4517. [Google Scholar] [CrossRef] [Green Version]
- Luque, R.M.; Park, S.; Kineman, R.D. Severity of the catabolic condition differentially modulates hypothalamic expression of growth hormone-releasing hormone in the fasted mouse: Potential role of neuropeptide Y and corticotropin-releasing hormone. Endocrinology 2007, 148, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Fujishita, C.; Komatsu, T.; Kim, S.E.; Chiba, T.; Mori, R.; Shimokawa, I. NPY antagonism reduces adiposity and attenuates age-related imbalance of adipose tissue metabolism. FASEB J. 2014, 28, 5337–5348. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Komatsu, T.; Kim, S.E.; Tanaka, K.; Hayashi, H.; Mori, R.; Shimokawa, I. Neuropeptide Y resists excess loss of fat by lipolysis in calorie-restricted mice: A trait potential for the life-extending effect of calorie restriction. Aging Cell 2017, 16, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.-Y.; Rikke, B.A.; Johnson, T.E.; Gelfond, J.A.L.; Diaz, V.; Nelson, J.F. Fat maintenance is a predictor of the murine lifespan response to dietary restriction. Aging Cell 2011, 10, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Minor, R.K.; López, M.; Younts, C.M.; Jones, B.; Pearson, K.J.; Anson, M.R.; Dieguez, C.; de Cabo, R. The arcuate nucleus and neuropeptide Y contribute to the antitumorigenic effect of calorie restriction. Aging Cell 2011, 10, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Farzi, A.; Reichmann, F.; Holzer, P. The homeostatic role of neuropeptide Y in immune function and its impact on mood and behaviour. Acta Physiol. (Oxf.) 2015, 213, 603–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chida, Y.; Sudo, N.; Kubo, C. Does stress exacerbate liver diseases? J. Gastroenterol. Hepatol. 2006, 21, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Sigala, B.; McKee, C.; Soeda, J.; Pazienza, V.; Morgan, M.; Lin, C.I.; Selden, C.; Vander Borght, S.; Mazzoccoli, G.; Roskams, T.; et al. Sympathetic nervous system catecholamines and neuropeptide Y neurotransmitters are upregulated in human NAFLD and modulate the fibrogenic function of hepatic stellate cells. PLoS ONE 2013, 8, e72928. [Google Scholar] [CrossRef] [PubMed]
- Huan, H.-B.; Wen, X.-D.; Chen, X.-J.; Wu, L.; Wu, L.L.; Zhang, L.; Yang, D.P.; Zhang, X.; Bie, P.; Qian, C.; et al. Sympathetic nervous system promotes hepatocarcinogenesis by modulating inflammation through activation of alpha1-adrenergic receptors of Kupffer cells. Brain Behav. Immun. 2016, 59, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, A.; Hayashi, H.; Kusano, N.; Inoue, S.; Komatsu, T.; Mori, R.; Park, S.; Takatsuki, M.; Eguchi, S.; Shimokawa, I. Neuropeptide Y inhibits hepatocarcinogenesis in overnutrition in mice. Acta Medica Nagasakiensia 2019, 41, 711–722. [Google Scholar]
- Baldock, P.A.; Lin, S.; Zhang, L.; Karl, T.; Shi, Y.; Driessler, F.; Zengin, A.; Hörmer, B.; Lee, N.J.; Wong, I.P.; et al. Neuropeptide y attenuates stress-induced bone loss through suppression of noradrenaline circuits. J. Bone Miner. Res. 2014, 29, 2238–2249. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komatsu, T.; Park, S.; Hayashi, H.; Mori, R.; Yamaza, H.; Shimokawa, I. Mechanisms of Calorie Restriction: A Review of Genes Required for the Life-Extending and Tumor-Inhibiting Effects of Calorie Restriction. Nutrients 2019, 11, 3068. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11123068
Komatsu T, Park S, Hayashi H, Mori R, Yamaza H, Shimokawa I. Mechanisms of Calorie Restriction: A Review of Genes Required for the Life-Extending and Tumor-Inhibiting Effects of Calorie Restriction. Nutrients. 2019; 11(12):3068. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11123068
Chicago/Turabian StyleKomatsu, Toshimitsu, Seongjoon Park, Hiroko Hayashi, Ryoichi Mori, Haruyoshi Yamaza, and Isao Shimokawa. 2019. "Mechanisms of Calorie Restriction: A Review of Genes Required for the Life-Extending and Tumor-Inhibiting Effects of Calorie Restriction" Nutrients 11, no. 12: 3068. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11123068