Impact of the Apolipoprotein E (epsilon) Genotype on Cardiometabolic Risk Markers and Responsiveness to Acute and Chronic Dietary Fat Manipulation


Abstract
:1. Introduction
2. Methods
2.1. Study Participants and Design
2.2. Baseline Associations Between APOE Genotype with Established and Novel Cardiometabolic Risk Markers
2.3. Impact of the APOE Genotype on the Responsiveness of Cardiometabolic Risk Markers
2.3.1. Chronic Dietary Fat Composition
2.3.2. Acute Meal Fat Composition
2.4. Vascular Reactivity Measurements and Blood Pressure
2.5. Biochemical Analysis, Estimates of Insulin Sensitivity/Resistance and CVD Risk Score
2.6. DNA Extraction and Genotyping
2.7. Statistical Analysis
3. Results
3.1. Baseline Associations between the APOE Genotype with Established and Novel Cardiometabolic Risk Markers
3.2. Effect of Dietary Fat Manipulation and the APOE Genotype on Cardiometabolic Risk Markers
3.2.1. Chronic Dietary Fat Composition (DIVAS)
3.2.2. Acute Meal Fat Composition (DIVAS-2)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| %TE | Total energy |
| ANCOVA | Analysis of covariance |
| ANOVA | Analysis of variance |
| apo | Apolipoprotein |
| AU | Arbitrary units |
| AUC | Area under the curve |
| BMI | Body mass index |
| CRP | C-reactive protein |
| CVD | Cardiovascular disease |
| DBP | Diastolic blood pressure |
| DIVAS | Dietary intervention and vascular function |
| DVP | Digital volume pulse |
| DVP-RI | DVP reflection index |
| DVP-SI | DVP stiffness index |
| FMD | Flow mediated dilatation |
| HDL-C | High-density lipoprotein cholesterol |
| HOMA-IR | Quantitative insulin resistance index |
| IAUC | Incremental AUC |
| LDI | Laser Doppler imaging |
| LDL-C | Low-density lipoprotein cholesterol |
| MUFA | Monounsaturated fatty acids |
| NEFA | Non-esterified fatty acids |
| NOx | Sum of nitrite and nitrate concentrations |
| PUFA | Polyunsaturated fatty acids |
| rQUICKI | Revised quantitative insulin sensitivity check index |
| SBP | Systolic blood pressure |
| SFA | Saturated fatty acids |
| sICAM-1 | Soluble intercellular cell adhesion molecule-1 |
| SNP | Sodium nitroprusside |
| sVCAM-1 | Soluble vascular cell adhesion molecule-1 |
| TAG | Triacylglycerol |
| TC | Total cholesterol |
References
- Wilson, P.W.F.; Schaefer, E.J.; Larson, M.G.; Ordovas, J.M. Apolipoprotein E alleles and risk of coronary disease. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Waterworth, D.M.; Ricketts, S.L.; Song, K.; Chen, L.; Zhao, J.H.; Ripatti, S.; Aulchenko, Y.S.; Zhang, W.; Yuan, X.; Lim, N. Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2264–2276. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Stampfer, M.J.; Liu, S. Meta-analysis: Apolipoprotein E genotypes and risk for coronary heart disease. Ann. Intern. Med. 2004, 141, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Fallaize, R.; Celis-Morales, C.; Macready, A.L.; Marsaux, C.F.M.; Forster, H.; O’Donovan, C.; Woolhead, C.; San-Cristobal, R.; Kolossa, S.; Hallmann, J. The effect of the apolipoprotein E genotype on response to personalized dietary advice intervention: Findings from the Food4Me randomized controlled trial. Am. J. Clin. Nutr. 2016, 104, 827–836. [Google Scholar] [PubMed]
- Bennet, A.M.; Di Angelantonio, E.; Ye, Z.; Wensley, F.; Dahlin, A.; Ahlbom, A.; Keavney, B.; Collins, R.; Wiman, B.; de Faire, U. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 2007, 298, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Miranda, C.; Cancelas, P.; Sanz, M.; Porres, A.; Gerique, J.G. Influence of apolipoprotein-E phenotypes on postprandial lipoprotein metabolism after three different fat loads. Nutrition 2001, 17, 529–533. [Google Scholar] [CrossRef]
- Jackson, K.G.; Lockyer, S.; Carvalho-Wells, A.L.; Williams, C.M.; Minihane, A.M.; Lovegrove, J.A. Dietary fat manipulation has a greater impact on postprandial lipid metabolism than the apolipoprotein E (epsilon) genotype–insights from the SATgenε study. Mol. Nutr. Food Res. 2012, 56, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Leigh-Firbank, E.C.; Minihane, A.M.; Leake, D.S.; Wright, J.W.; Murphy, M.C.; Griffin, B.A.; Williams, C.M. Eicosapentaenoic acid and docosahexaenoic acid from fish oils: Differential associations with lipid responses. Br. J. Nutr. 2002, 87, 435–445. [Google Scholar] [CrossRef]
- Moreno, J.A.; Pérez-Jiménez, F.; Marín, C.; Gómez, P.; Pérez-Martínez, P.; Moreno, R.; Bellido, C.; Fuentes, F.; López-Miranda, J. Apolipoprotein E gene promoter −219G→ T polymorphism increases LDL-cholesterol concentrations and susceptibility to oxidation in response to a diet rich in saturated fat. Am. J. Clin. Nutr. 2004, 80, 1404–1409. [Google Scholar] [CrossRef]
- Olano-Martin, E.; Anil, E.; Caslake, M.J.; Packard, C.J.; Bedford, D.; Stewart, G.; Peiris, D.; Williams, C.M.; Minihane, A.M. Contribution of apolipoprotein E genotype and docosahexaenoic acid to the LDL-cholesterol response to fish oil. Atherosclerosis 2010, 209, 104–110. [Google Scholar] [CrossRef]
- Wei, M.Y.; Jacobson, T.A. Effects of eicosapentaenoic acid versus docosahexaenoic acid on serum lipids: A systematic review and meta-analysis. Curr. Atheroscler. Rep. 2011, 13, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Bowman, R.; Welch, A.A.; Luben, R.N.; Wareham, N.; Khaw, K.T.; Bingham, S.A. Apolipoprotein E polymorphisms, dietary fat and fibre, and serum lipids: The EPIC Norfolk study. Eur. Heart J. 2007, 28, 2930–2936. [Google Scholar] [CrossRef] [PubMed]
- Schächinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000, 101, 1899–1906. [Google Scholar] [CrossRef]
- Jackson, K.G.; Poppitt, S.D.; Minihane, A.M. Postprandial lipemia and cardiovascular disease risk: Interrelationships between dietary, physiological and genetic determinants. Atherosclerosis 2012, 220, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Miranda, J.; Williams, C.; Lairon, D. Dietary, physiological, genetic and pathological influences on postprandial lipid metabolism. Br. J. Nutr. 2007, 98, 458–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Martinez, P.; Delgado-Lista, J.; Perez-Jimenez, F.; Lopez-Miranda, J. Update on genetics of postprandial lipemia. Atheroscler. Suppl. 2010, 11, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Irvin, M.R.; Kabagambe, E.K.; Tiwari, H.K.; Parnell, L.D.; Straka, R.J.; Tsai, M.; Ordovas, J.M.; Arnett, D.K. Apolipoprotein E polymorphisms and postprandial triglyceridemia before and after fenofibrate treatment in the Genetics of Lipid Lowering and Diet Network (GOLDN) Study. Circ. Cardiovasc. Genet. 2010, 3, 462–467. [Google Scholar] [CrossRef]
- Carvalho-Wells, A.L.; Jackson, K.G.; Gill, R.; Olano-Martin, E.; Lovegrove, J.A.; Williams, C.M.; Minihane, A.M. Interactions between age and apoE genotype on fasting and postprandial triglycerides levels. Atherosclerosis 2010, 212, 481–487. [Google Scholar] [CrossRef]
- Cardona, F.; Morcillo, S.; Gonzalo-Marin, M.; Tinahones, F.J. The apolipoprotein E genotype predicts postprandial hypertriglyceridemia in patients with the metabolic syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 2972–2975. [Google Scholar] [CrossRef]
- Griffin, B.; Walker, C.; Jebb, S.; Moore, C.; Frost, G.; Goff, L.; Sanders, T.; Lewis, F.; Griffin, M.; Gitau, R.; et al. APOE4 genotype exerts greater benefit in lowering plasma cholesterol and apolipoprotein B than wild type (E3/E3), after replacement of dietary saturated fats with low glycaemic index carbohydrates. Nutrients 2018, 10, 1524. [Google Scholar] [CrossRef]
- Vafeiadou, K.; Weech, M.; Altowaijri, H.; Todd, S.; Yaqoob, P.; Jackson, K.G.; Lovegrove, J.A. Replacement of saturated with unsaturated fats had no impact on vascular function but beneficial effects on lipid biomarkers, E-selectin, and blood pressure: Results from the randomized, controlled Dietary Intervention and VAScular function (DIVAS) study. Am. J. Clin. Nutr. 2015, 102, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Rathnayake, K.M.; Weech, M.; Jackson, K.G.; Lovegrove, J.A. Meal Fatty Acids Have Differential Effects on Postprandial Blood Pressure and Biomarkers of Endothelial Function but Not Vascular Reactivity in Postmenopausal Women in the Randomized Controlled Dietary Intervention and VAScular function (DIVAS)-2 Study. J. Nutr. 2018, 148, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Weech, M.; Vafeiadou, K.; Hasaj, M.; Todd, S.; Yaqoob, P.; Jackson, K.G.; Lovegrove, J.A. Development of a food-exchange model to replace saturated fat with MUFAs and n–6 PUFAs in adults at moderate cardiovascular risk. J. Nutr. 2014, 144, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Newens, K.J.; Thompson, A.K.; Jackson, K.G.; Wright, J.; Williams, C.M. DHA-rich fish oil reverses the detrimental effects of saturated fatty acids on postprandial vascular reactivity. Am. J. Clin. Nutr. 2011, 94, 742–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar]
- Ignarro, L.J.; Fukuto, J.M.; Griscavage, J.M.; Rogers, N.E.; Byrns, R.E. Oxidation of nitric oxide in aqueous solution to nitrite but not nitrate: Comparison with enzymatically formed nitric oxide from L-arginine. Proc. Natl. Acad. Sci. USA 1993, 90, 8103–8107. [Google Scholar] [CrossRef] [PubMed]
- Rassaf, T.; Bryan, N.S.; Kelm, M.; Feelisch, M. Concomitant presence of N-nitroso and S-nitroso proteins in human plasma. Free Radic. Biol. Med. 2002, 33, 1590–1596. [Google Scholar] [CrossRef]
- Brady, L.M.; Gower, B.A.; Lovegrove, S.S.; Williams, C.M.; Lovegrove, J.A. Revised QUICKI provides a strong surrogate estimate of insulin sensitivity when compared with the minimal model. Int. J. Obes. 2004, 28, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, D.E.; El-Sohemy, A. A randomized trial of genetic information for personalized nutrition. Genes Nutr. 2012, 7, 559. [Google Scholar] [CrossRef]
- Joost, H.G.; Gibney, M.J.; Cashman, K.D.; Görman, U.; Hesketh, J.E.; Mueller, M.; Van Ommen, B.; Williams, C.M.; Mathers, J.C. Personalised nutrition: Status and perspectives. Br. J. Nutr. 2007, 98, 26–31. [Google Scholar] [CrossRef]
- Carvalho-Wells, A.L.; Jackson, K.G.; Lockyer, S.; Lovegrove, J.A.; Minihane, A.M. APOE genotype influences triglyceride and C-reactive protein responses to altered dietary fat intake in UK adults. Am. J. Clin. Nutr. 2012, 96, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Caslake, M.J.; Miles, E.A.; Kofler, B.M.; Lietz, G.; Curtis, P.; Armah, C.K.; Kimber, A.C.; Grew, J.P.; Farrell, L.; Stannard, J. Effect of sex and genotype on cardiovascular biomarker response to fish oils: The FINGEN Study. Am. J. Clin. Nutr. 2008, 88, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Fallaize, R.; Carvalho-Wells, A.L.; Tierney, A.C.; Marin, C.; Kieć-Wilk, B.; Dembińska-Kieć, A.; Drevon, C.A.; DeFoort, C.; Lopez-Miranda, J.; Risérus, U.; et al. APOE genotype influences insulin resistance, apolipoprotein CII and CIII according to plasma fatty acid profile in the Metabolic Syndrome. Sci. Rep. 2017, 7, 6274. [Google Scholar] [CrossRef] [PubMed]
- Weisgraber, K.H. Apolipoprotein E distribution among human plasma lipoproteins: Role of the cysteine-arginine interchange at residue 112. J. Lipid Res. 1990, 31, 1503–1511. [Google Scholar] [PubMed]
- Jackson, K.G.; Maitin, V.; Leake, D.S.; Yaqoob, P.; Williams, C.M. Saturated fat-induced changes in Sf 60–400 particle composition reduces uptake of LDL by HepG2 cells. J. Lipid Res. 2006, 47, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, M.S.; Eisenberg, S.; Breslow, J.L. Dietary fat clearance in normal subjects is regulated by genetic variation in apolipoprotein E. J. Clin. Investig. 1987, 80, 1571. [Google Scholar] [CrossRef] [PubMed]
- Gregg, R.E.; Zech, L.A.; Schaefer, E.J.; Stark, D.; Wilson, D.; Brewer, H.B., Jr. Abnormal in vivo metabolism of apolipoprotein E4 in humans. J. Clin. Investig. 1986, 78, 815. [Google Scholar] [CrossRef]
- Rontu, R.; Ojala, P.; Hervonen, A.; Goebeler, S.; Karhunen, P.J.; Nikkilä, M.; Kunnas, T.; Jylhä, M.; Eklund, C.; Hurme, M. Apolipoprotein E genotype is related to plasma levels of C-reactive protein and lipids and to longevity in nonagenarians. Clin. Endocrinol. 2006, 64, 265–270. [Google Scholar] [CrossRef]
- Mänttäri, M.; Manninen, V.; Palosuo, T.; Ehnholm, C. Apolipoprotein E polymorphism and C-reactive protein in dyslipidemic middle-aged men. Atherosclerosis 2001, 156, 237–238. [Google Scholar] [CrossRef]
- Judson, R.; Brain, C.; Dain, B.; Windemuth, A.; Ruaño, G.; Reed, C. New and confirmatory evidence of an association between APOE genotype and baseline C-reactive protein in dyslipidemic individuals. Atherosclerosis 2004, 177, 345–351. [Google Scholar] [CrossRef]
- Grönroos, P.; Raitakari, O.T.; Kähönen, M.; Hutri-Kähönen, N.; Marniemi, J.; Viikari, J.; Lehtimäki, T. Association of high sensitive C-reactive protein with apolipoprotein E polymorphism in children and young adults: The Cardiovascular Risk in Young Finns Study. Clin. Chem. Lab. Med. 2008, 46, 179–186. [Google Scholar] [CrossRef]
- Golledge, J.; Biros, E.; Cooper, M.; Warrington, N.; Palmer, L.J.; Norman, P.E. Apolipoprotein E genotype is associated with serum C-reactive protein but not abdominal aortic aneurysm. Atherosclerosis 2010, 209, 487–491. [Google Scholar] [CrossRef] [Green Version]
- Eiriksdottir, G.; Aspelund, T.; Bjarnadottir, K.; Olafsdottir, E.; Launer, L.J.; Harris, T.B.; Gudnason, V. Apolipoprotein E genotype and statins affect CRP levels through independent and different mechanisms: AGES-Reykjavik Study. Atherosclerosis 2006, 186, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Austin, M.A.; Zhang, C.; Humphries, S.E.; Chandler, W.L.; Talmud, P.J.; Edwards, K.L.; Leonetti, D.L.; McNeely, M.J.; Fujimoto, W.Y. Heritability of C-Reactive Protein and Association with Apolipoprotein E Genotypes in Japanese Americans. Ann. Hum. Genet. 2004, 68, 179–188. [Google Scholar] [CrossRef]
- Angelopoulos, T.J.; Miles, M.P.; Lowndes, J.; Sivo, S.A.; Seip, R.L.; Pescatello, L.S.; Zoeller, R.F.; Visich, P.S.; Gordon, P.M.; Moyna, N.M. Apolipoprotein E genotype and sex influence C-reactive protein levels regardless of exercise training status. Metabolism 2008, 57, 1204–1210. [Google Scholar] [CrossRef] [Green Version]
- Pearson, T.A.; Mensah, G.A.; Alexander, R.W.; Anderson, J.L.; Cannon, R.O.; Criqui, M.; Fadl, Y.Y.; Fortmann, S.P.; Hong, Y.; Myers, G.L. Markers of inflammation and cardiovascular disease. Circulation 2003, 107, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Danesh, J.; Wheeler, J.G.; Hirschfield, G.M.; Eda, S.; Eiriksdottir, G.; Rumley, A.; Lowe, G.D.O.; Pepys, M.B.; Gudnason, V. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N. Engl. J. Med. 2004, 350, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R. C-reactive protein reassessed. Mass. Med. Soc. 2004, 350, 1450–1452. [Google Scholar] [CrossRef]
- Eltoft, A.; Arntzen, K.A.; Hansen, J.B.; Wilsgaard, T.; Mathiesen, E.B.; Johnsen, S.H. C-reactive protein in atherosclerosis–A risk marker but not a causal factor? A 13-year population-based longitudinal study: The Tromsø study. Atherosclerosis 2017, 263, 293–300. [Google Scholar] [CrossRef]
- Bonacina, F.; Coe, D.; Wang, G.; Longhi, M.P.; Baragetti, A.; Moregola, A.; Garlaschelli, K.; Uboldi, P.; Pellegatta, F.; Grigore, L.; et al. Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nat. Commun. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Hodson, L.; Skeaff, C.M.; Fielding, B.A. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog. Lipid Res. 2008, 47, 348–380. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Pérez-Jiménez, F.; Moreno-Luna, R.; Pérez-Martínez, P.; Fuentes-Jiménez, F.; Marín, C.; Portugal, H.; Lairon, D.; López-Miranda, J. The effect of apoE genotype and sex on ApoE plasma concentration is determined by dietary fat in healthy subjects. Br. J. Nutr. 2008, 101, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B. Blood pressure as a cardiovascular risk factor: Prevention and treatment. JAMA 1996, 275, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Mattace-Raso, F.U.S.; van der Cammen, T.J.M.; Hofman, A.; van Popele, N.M.; Bos, M.L.; Schalekamp, M.A.D.H.; Asmar, R.; Reneman, R.S.; Hoeks, A.P.G.; Breteler, M.M.B. Arterial stiffness and risk of coronary heart disease and stroke the Rotterdam study. Circulation 2006, 113, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, J.; Crouse, J.R.; Hsu, F.C.; Burke, G.L.; Herrington, D.M. Brachial flow-mediated dilation predicts incident cardiovascular events in older adults the cardiovascular health study. Circulation 2007, 115, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Urbina, E.M.; Khoury, P.R.; McCoy, C.E.; Dolan, L.M.; Daniels, S.R.; Kimball, T.R. Triglyceride to HDL-C ratio and increased arterial stiffness in children, adolescents, and young adults. Pediatrics 2013, 131, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Sanders, T.A.B.; Lewis, F.J.; Goff, L.M.; Chowienczyk, P.J. SFAs do not impair endothelial function and arterial stiffness. Am. J. Clin. Nutr. 2013, 98, 677–683. [Google Scholar] [Green Version]
- Fielding, B.A.; Callow, J.; Owen, R.M.; Samra, J.S.; Matthews, D.R.; Frayn, K.N. Postprandial lipemia: The origin of an early peak studied by specific dietary fatty acid intake during sequential meals. Am. J. Clin. Nutr. 1996, 63, 36–41. [Google Scholar] [CrossRef]
- Evans, K.; Kuusela, P.J.; Cruz, M.L.; Wilhelmova, I.; Fielding, B.A.; Frayn, K.N. Rapid chylomicron appearance following sequential meals: Effects of second meal composition. Br. J. Nutr. 1998, 79, 425–429. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| All (n = 216) | E2 Carriers (n = 30) | E3/E3 (n = 128) | E4 Carriers (n = 58) | P (Genotype) 1 | |
|---|---|---|---|---|---|
| Genotype Frequency (%) | - | 14 | 59 | 27 | |
| Characteristics | |||||
| Sex, M/F | 84/132 | 13/17 | 43/85 | 28/30 | |
| Age, y | 46 ± 1 | 48 ± 2 | 45 ± 1 | 46 ± 2 | 0.537 |
| Weight, kg | 76.5 ± 1.0 | 78.2 ± 2.8 | 75.0 ± 1.2 | 78.9 ± 1.8 | 0.082 |
| BMI, kg/m2 | 26.6 ± 0.3 | 27.7 ± 0.8 | 26.4 ± 0.3 | 26.5 ± 0.5 | 0.341 |
| Waist circumference, cm | 91.3 ± 0.8 | 96.2 ± 2.7 | 90.1 ± 1.0 | 91.3 ± 1.5 | 0.091 |
| Waist:hip ratio | 0.87 ± 0.01 | 0.90 ± 0.02 | 0.86 ± 0.01 | 0.88 ± 0.01 | 0.152 |
| Clinic blood pressure | |||||
| Systolic, mm Hg | 119 ± 1 | 122 ± 3 | 119 ± 1 | 118 ± 2 | 0.432 |
| Diastolic, mm Hg | 74 ± 1 | 77 ± 1 | 74 ± 1 | 73 ± 1 | 0.193 |
| Pulse pressure, mm Hg | 45 ± 1 | 45 ± 2 | 45 ± 1 | 45 ± 1 | 0.873 |
| Biochemical profile and CVD risk | |||||
| TC, mmol/L | 5.49 ± 0.07 | 4.77 ± 0.20 a | 5.49 ± 0.09 b | 5.88 ± 0.13 c | 0.0001 |
| HDL-C, mmol/L | 1.49 ± 0.02 | 1.43 ± 0.06 | 1.52 ± 0.03 | 1.47 ± 0.05 | 0.606 |
| LDL-C, mmol/L | 3.42 ± 0.06 | 2.73 ± 0.16 a | 3.42 ± 0.07 b | 3.77 ± 0.11 c | 0.0001 |
| TC: HDL-C ratio | 3.84 ± 0.07 | 3.49 ± 0.19 a | 3.77 ± 0.09 b | 4.20 ± 0.16 c | 0.002 |
| LDL-C: HDL-C ratio | 2.41 ± 0.06 | 2.02 ± 0.15 a | 2.37 ± 0.07 b | 2.72 ± 0.12 c | 0.0001 |
| TAG, mmol/L | 1.27 ± 0.04 | 1.34 ± 0.13 | 1.21 ± 0.05 | 1.34 ± 0.10 | 0.551 |
| NEFA, µmol/L | 502 ± 12 | 525 ± 28 | 509 ± 17 | 472 ± 22 | 0.413 |
| Glucose, mmol/L | 5.09 ± 0.03 | 5.12 ± 0.07 | 5.05 ± 0.04 | 5.18 ± 0.07 | 0.527 |
| Insulin, pmol/L | 31.2 ± 1.3 | 36.4 ± 4.6 | 30.4 ± 1.6 | 30.2 ± 2.3 | 0.619 |
| HOMA-IR | 1.19 ± 0.05 | 1.41 ± 0.19 | 1.14 ± 0.06 | 1.18 ± 0.10 | 0.605 |
| rQUICKI | 0.45 ± 0.01 | 0.43 ± 0.01 | 0.45 ± 0.01 | 0.46 ± 0.01 | 0.238 |
| QRISK®2, 2 % | 2.8 ± 0.2 | 3.3 ± 0.7 | 2.4 ± 0.2 | 3.3 ± 0.4 | 0.142 |
| Vascular function | |||||
| %FMD response | 6.2 ± 0.2 | 5.8 ± 0.5 | 6.4 ± 0.3 | 6.0 ± 0.4 | 0.698 |
| LDI-Ach, AUC, PU | 1548 ± 59 | 1529 ± 164 | 1523 ± 77 | 1601 ± 114 | 0.588 |
| LDI-SNP, AUC, PU | 1464 ± 50 | 1327 ± 106 | 1448 ± 64 | 1557 ± 105 | 0.370 |
| DVP-RI, % | 63.2 ± 0.9 | 64.8 ± 2.5 | 62.1 ± 1.1 | 64.8 ± 1.7 | 0.649 |
| DVP-SI, m/s | 6.9 ± 0.1 | 7.7 ± 0.5 a | 6.6 ± 0.1 b | 7.2 ± 0.2 a,b | 0.027 |
| Biomarkers of inflammation and endothelial activation | |||||
| C-reactive protein, mg/L | 2.23 ± 0.23 | 3.20 ± 0.70 a | 2.27 ± 0.29 a | 1.66 ± 0.41 b | 0.002 |
| sVCAM-1, ng/mL | 661 ± 11 | 653 ± 23 | 652 ± 16 | 685 ± 20 | 0.400 |
| sICAM-1, ng/mL | 218 ± 3 | 228 ± 9 | 220 ± 4 | 207 ± 5 | 0.120 |
| E-selectin, ng/mL | 34.2 ± 1.0 | 31.4 ± 2.5 | 34.5 ± 1.3 | 35.0 ± 1.9 | 0.189 |
| P-selectin, ng/mL | 40.7 ± 1.0 | 37.3 ± 2.5 | 41.0 ± 1.3 | 41.8 ± 1.9 | 0.078 |
| E3/E3 (n = 107) | E4 Carriers (E3/E4 and E4/E4, n = 52) | p Value 1 | ||||||
|---|---|---|---|---|---|---|---|---|
| SFA | MUFA | n-6 PUFA | SFA | MUFA | n-6 PUFA | Genotype | Diet × Genotype | |
| N | 35 | 36 | 36 | 17 | 17 | 18 | ||
| Age, y | 44 ± 1 | 42 ± 2 | 43 ± 2 | 44 ± 3 | 46 ± 3 | 47 ± 3 | ||
| BMI, kg/m2 | 25.8 ± 0.8 | 26.5 ± 0.8 | 26.9 ± 0.6 | 27.5 ± 0.9 | 26.1 ± 1.0 | 25.9 ± 0.8 | ||
| Biochemical profile and estimates of insulin sensitivity/resistance | ||||||||
| TC, mmol/L | 0.42 ± 0.10 | −0.03 ± 0.13 | −0.01 ± 0.13 | 0.21 ± 0.19 | −0.29 ± 0.17 | −0.19 ± 0.20 | 0.165 | 0.760 |
| HDL-C, mmol/L | 0.06 ± 0.03 | 0.04 ± 0.03 | 0.12 ± 0.04 | 0.03 ± 0.05 | −0.05 ± 0.05 | −0.02 ± 0.07 | 0.015 | 0.473 |
| LDL-C, mmol/L | 0.35 ± 0.09 | −0.09 ± 0.11 | −0.11 ± 0.10 | 0.17 ± 0.15 | −0.19 ± 0.15 | −0.18 ± 0.14 | 0.401 | 0.984 |
| TC: HDL-C ratio | 0.20 ± 0.08 | −0.11 ± 0.07 | −0.25 ± 0.07 | 0.01 ± 0.13 | −0.13 ± 0.17 | −0.05 ± 0.08 | 0.703 | 0.263 |
| LDL-C: HDL-C ratio | 0.19 ± 0.08 | −0.12 ± 0.07 | −0.23 ± 0.07 | 0.02 ± 0.11 | −0.10 ± 0.15 | −0.07 ± 0.08 | 0.652 | 0.324 |
| TAG, mmol/L | −0.00 ± 0.05 | 0.10 ± 0.06 a | −0.07 ± 0.05 | 0.06 ± 0.14 | −0.23 ± 0.10 b | 0.08 ± 0.18 | 0.160 | 0.010 |
| NEFA, µmol/L | −17.6 ± 35.1 | −13.6 ± 22.1 | −11.1 ± 21.5 | −64.8 ± 40.2 | 46.5 ± 66.3 | 87.7 ± 26.2 | 0.413 | 0.082 |
| Glucose, mmol/L | 0.04 ± 0.05 | 0.07 ± 0.04 | 0.10 ± 0.06 | 0.08 ± 0.09 | 0.01 ± 0.06 | 0.01 ± 0.11 | 0.957 | 0.614 |
| Insulin, pmol/L | 1.10 ± 2.73 | 1.23 ± 1.86 | 2.67 ± 2.31 | 0.97 ± 2.77 | 0.49 ± 1.55 | 0.71 ± 1.67 | 0.851 | 0.857 |
| HOMA-IR | 0.07 ± 0.11 | 0.05 ± 0.08 | 0.12 ± 0.10 | 0.05 ± 0.12 | 0.03 ± 0.07 | 0.06 ± 0.07 | 0.821 | 0.930 |
| rQUICKI | 0.00 ± 0.01 | −0.00 ± 0.01 | −0.01 ± 0.01 | 0.00 ± 0.01 | −0.01 ± 0.01 | −0.02 ± 0.01 | 0.620 | 0.420 |
| Vascular function | ||||||||
| %FMD response | −0.55 ± 0.33 | 0.33 ± 0.44 | −0.09 ± 0.36 | −0.40 ± 0.49 | −0.20 ± 0.69 | −0.95 ± 0.73 | 0.918 | 0.368 |
| LDI 2 | ||||||||
| LDI-Ach AUC, PU | −460.3 ± 198.1 | −2.1 ± 154.6 | 91.1 ± 119.9 | 38.5 ± 164.6 | −40.7 ± 232.8 | −115.4 ± 142.2 | 0.438 | 0.134 |
| LDI-SNP AUC, PU | −283 ± 173 | −187 ± 158 | 131 ± 127 | −28 ± 212 | −296 ± 287 | 187 ± 174 | 0.233 | 0.601 |
| DVP-RI, % | −1.98 ± 2.34 | 4.33 ± 2.57 | 4.29 ± 2.27 | −0.27 ± 2.57 | 1.47 ± 1.91 | −4.92 ± 3.52 | 0.078 | 0.171 |
| DVP-SI, m/s | 0.23 ± 0.32 | 0.76 ± 0.31 | 0.22 ± 0.37 | 0.37 ± 0.37 | −0.15 ± 0.32 | −0.93 ± 0.43 | 0.125 | 0.562 |
| Ambulatory blood pressure 3 | ||||||||
| 24-h blood pressure | ||||||||
| SBP, mm Hg | 1.7 ± 1.3 | −1.1 ± 1.3 | 0.2 ± 1.8 | 0.5 ± 2.7 | −0.9 ± 2.4 | −1.3 ± 2.0 | 0.681 | 0.860 |
| DBP, mm Hg | 1.6 ± 0.9 | −0.4 ± 0.9 | −0.3 ± 1.1 | 1.1 ± 1.9 | 0.9 ± 1.7 | −1.4 ± 1.3 | 0.921 | 0.813 |
| Pulse pressure, mm Hg | 0.2 ± 1.4 | −0.7 ± 0.8 | 0.6 ± 1.0 | −0.6 ± 1.1 | −1.7 ± 1.4 | 0.1 ± 1.5 | 0.502 | 0.974 |
| Heart rate, bpm | 0.8 ± 1.0 | 0.7 ± 1.1 | 0.6 ± 1.1 | 2.8 ± 1.3 | 1.9 ± 1.8 | −1.6 ± 1.6 | 0.565 | 0.292 |
| Biomarkers of inflammation and endothelial activation | ||||||||
| C-reactive protein, mg/L | 0.60 ± 0.60 | 0.04 ± 0.28 | 0.024 ± 0.51 | −0.14 ± 1.14 | −0.25 ± 0.57 | −0.79 ± 0.63 | 0.036 | 0.786 |
| NOx, µmol/L | 0.51 ± 3.22 | −1.89 ± 1.52 | −1.78 ± 1.87 | 1.70 ± 3.48 | 4.43 ± 2.97 | −2.27 ± 1.62 | 0.208 | 0.073 |
| sVCAM-1, ng/mL | −40.0 ± 16.8 | 11.4 ± 22.2 | 2.3 ± 13.6 | −2.1 ± 16.2 | 3.9 ± 35.4 | 25.1 ± 25.2 | 0.063 | 0.451 |
| sICAM-1, ng/mL | −1.4 ± 4.2 | 1.3 ± 3.6 | 1.5 ± 6.3 | 5.2 ± 6.6 | 15.3 ± 8.2 | 12.8 ± 5.1 | 0.100 | 0.836 |
| E-selectin, ng/mL | 0.41 ± 1.39 | −2.90 ± 1.15 | −0.63 ± 1.03 | 0.70 ± 2.01 | −3.03 ± 1.86 | −0.24 ± 1.43 | 0.827 | 0.881 |
| P-selectin, ng/mL | −0.94 ± 1.70 | −1.94 ± 1.06 | −3.49 ± 1.23 | 3.63 ± 1.74 | −1.18 ± 2.32 | 0.21 ± 1.44 | 0.026 | 0.403 |
| E3/E3 (n = 22) | E3/E4 (n = 5) | p Value 1 | ||||||
|---|---|---|---|---|---|---|---|---|
| SFA | MUFA | n-6 PUFA | SFA | MUFA | n-6 PUFA | Genotype | Test Fat × Genotype | |
| Biochemical measures 2 | ||||||||
| TAG, mmol/L | ||||||||
| AUC | 942 ± 90 | 1022 ± 87 | 1035 ± 124 | 1063 ± 186 | 1022 ± 164 | 1125 ± 299 | 0.702 | 0.683 |
| IAUC | 320 ± 52 | 360 ± 53 | 361 ± 77 | 402 ± 94 | 403 ± 126 | 386 ± 115 | 0.702 | 0.828 |
| NEFA, μmol/L | ||||||||
| AUC | 143.5 ± 8.4 | 136.1 ± 11.3 | 126.9 ± 7.3 | 108.6 ± 16.1 | 102.4 ± 13.3 | 116.9 ± 21.1 | 0.147 | 0.382 |
| IAUC | −73.5 ± 12.3 | −87.9 ± 12.7 | −83.7 ± 12.7 | −75.2 ± 29.6 | −12.6 ± 16.9 | −62.0 ± 29.8 | 0.802 | 0.111 |
| Apo B, mg/mL | ||||||||
| AUC | 473 ± 18 | 469 ± 22 | 480 ± 25 | 549 ± 32 | 561 ± 50 | 512 ± 34 | 0.329 | 0.134 |
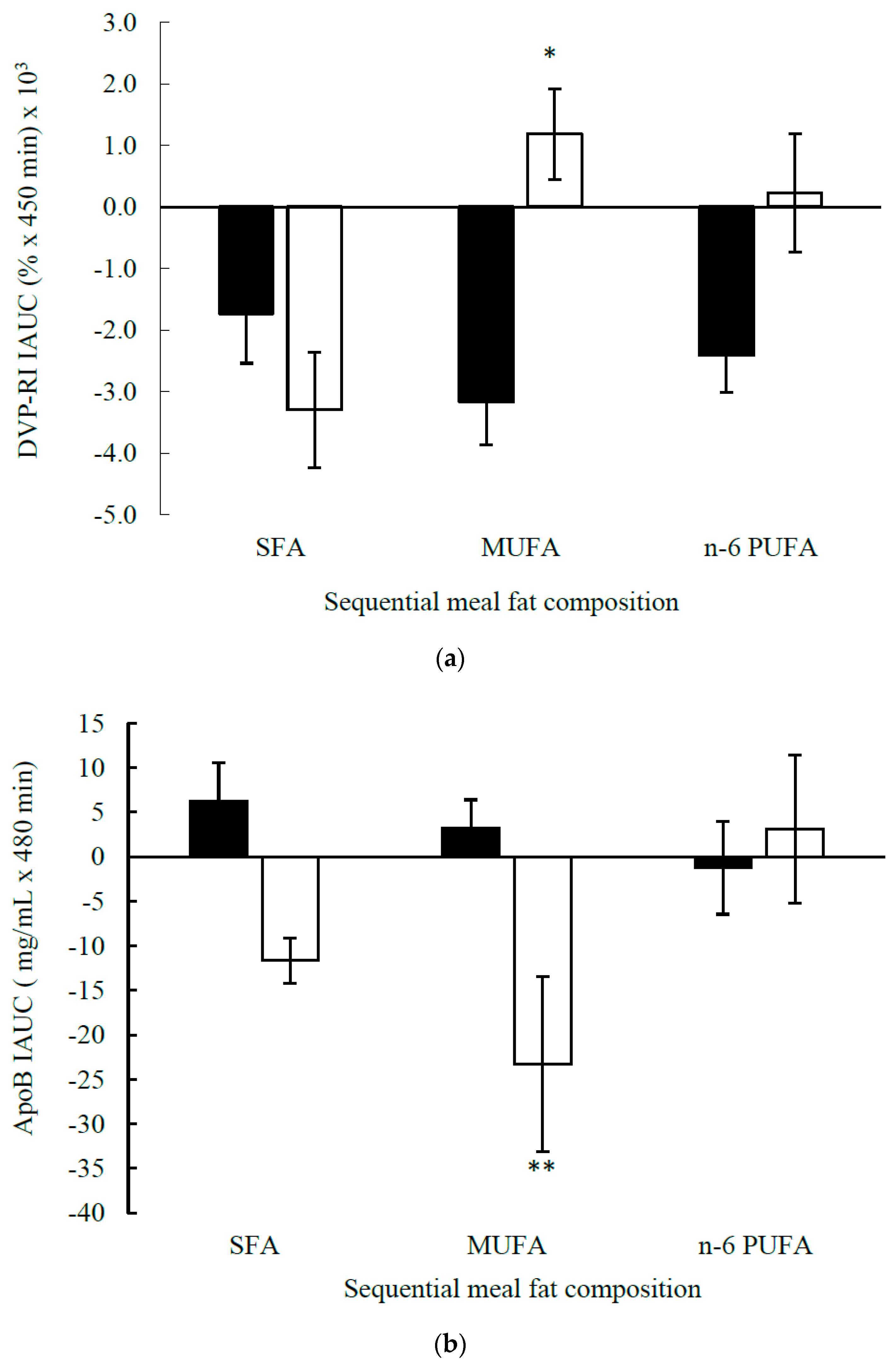
| IAUC | 62.4 ± 4.3 | 50.7 ± 3.2 | −12.4 ± 5.2 | −11.6 ± 2.5 | −23.3 ± 9.8 | 31.3 ± 8.3 | 0.197 | 0.022 3 |
| Glucose, mmol/L | ||||||||
| AUC | 942 ± 90 | 1022 ± 87 | 1035 ± 124 | 1063 ± 186 | 1022 ± 164 | 1125 ± 299 | 0.666 | 0.299 |
| IAUC | 320 ± 52 | 360 ± 53 | 361 ± 77 | 402 ± 94 | 403 ± 126 | 386 ± 115 | 0.342 | 0.523 |
| Insulin, μmol/L | ||||||||
| AUC | 913 ± 8 | 877 ± 11 | 821 ± 9 | 948 ± 19 | 951 ± 15 | 952 ± 18 | 0.640 | 0.814 |
| IAUC | 881 ± 8 | 831 ± 9 | 779 ± 6 | 795 ± 16 | 783 ± 12 | 753 ± 14 | 0.610 | 0.750 |
| Vascular function 2 | ||||||||
| % FMD response | ||||||||
| AUC | 1939 ± 148 | 2305 ± 191 | 2131 ± 191 | 2105 ± 235 | 2629 ± 486 | 2329 ± 218 | 0.510 | 0.898 |
| IAUC | −149 ± 183 | 156 ± 261 | 114 ± 189 | 345 ± 335 | 431 ± 279 | 79 ± 281 | 0.433 | 0.683 |
| LDI-Ach 4, AU × 103 | ||||||||
| AUC | 742 ± 47 | 766 ± 56 | 772 ± 50 | 871 ± 101 | 973 ± 320 | 941 ± 160 | 0.888 | 0.931 |
| IAUC | 7.8 ± 35.1 | −8.1 ± 62.2 | −11.0 ± 53.9 | −46.4 ± 101.6 | −70.0 ± 132.3 | −21.8 ± 127.9 | 0.088 | 0.975 |
| LDI-SNP 4, AU × 103 | ||||||||
| AUC | 755 ± 57 | 807 ± 66 | 652 ± 33 | 802 ± 99 | 957 ± 264 | 113 ± 227 | 0.690 | 0.083 |
| IAUC | 318 ± 60 | −101 ± 59 | −331 ± 61 | −118 ± 74 | −286 ± 117 | −505 ± 171 | 0.754 | 0.624 |
| DVP-RI, % × 103 | ||||||||
| AUC | 26.1 ± 1.0 | 24.7 ± 0.7 | 25.5 ± 0.9 | 25.9 ± 0.8 | 27.6 ± 1.5 | 27.1 ± 2.7 | 0.407 | 0.392 |
| IAUC | −1.7 ± 0.8 | −3.2 ± 0.7 | −2.4 ± 0.6 | −3.3 ± 0.9 | 1.2 ± 0.7 | 0.2 ± 1.0 | 0.055 | 0.030 3 |
| DVP-SI, m/s | ||||||||
| AUC | 3193 ± 121 | 3074 ± 110 | 3169 ± 114 | 3463 ± 178 | 3505 ± 277 | 3617 ± 415 | 0.177 | 0.735 |
| IAUC | 14.6 ± 97.6 | −181.2 ± 110.1 | 40.5 ± 120.4 | 171.3 ± 178.4 | −44.0 ± 241.3 | 226.5 ± 154.4 | 0.373 | 0.987 |
| SBP, mmHg × 103 | ||||||||
| AUC | 56.3 ± 1.5 | 57.1 ± 1.4 | 57.1 ± 1.6 | 58.9 ± 3.5 | 56.7 ± 2.5 | 58.3 ± 3.5 | 0.716 | 0.267 |
| IAUC | −2.5 ± 0.6 | −4.8 ± 0.6 | −3.3 ± 0.7 | −3.6 ± 0.7 | −3.2 ± 1.5 | −4.2 ± 1.0 | 0.914 | 0.178 |
| DBP, mmHg × 103 | ||||||||
| AUC | 32.9 ± 3.3 | 33.1 ± 3.3 | 33.3 ± 3.3 | 32.9 ± 1.2 | 31.9 ± 1.0 | 32.0 ± 1.4 | 0.686 | 0.341 |
| IAUC | −1.1 ± 0.4 | −2.2 ± 0.3 | −1.5 ± 0.3 | −1.0 ± 0.6 | −1.7 ± 0.3 | −9.7 ± 0.7 | 0.442 | 0.867 |
| Biomarkers of endothelial activation 2 | ||||||||
| NOx, μmol/L | ||||||||
| AUC | 5820 ± 482 | 5962 ± 708 | 5119 ± 389 | 6308 ± 258 | 6278 ± 258 | 6084 ± 1330 | 0.288 | 0.996 |
| IAUC | −1618 ± 231 | −1446 ± 370 | −1057 ± 219 | −2165 ± 521 | −3015 ± 695 | −1762 ± 393 | 0.078 | 0.318 |
| sVCAM-1, µg/mL | ||||||||
| AUC | 263.0 ± 7.1 | 270.7 ± 9.2 | 256.9 ± 7.3 | 263.4 ± 22.2 | 243.0 ± 14.6 | 243.5 ± 16.4 | 0.431 | 0.118 |
| IAUC | 4.2 ± 5.0 | 3.1 ± 4.6 | −4.7 ± 2.9 | 3.9 ± 4.9 | −8.5 ± 9.2 | −9.6 ± 8.1 | 0.360 | 0.707 |
| sICAM-1, µg/mL | ||||||||
| AUC | 86.3 ± 3.1 | 876.9 ± 3.8 | 702.5 ± 5.7 | 715.3 ± 3.5 | 717.9 ± 2.9 | 588.9 ± 6.1 | 0.141 | 0.775 |
| IAUC | −3.2 ± 1.3 | −0.2 ± 1.5 | −18.1 ± 4.1 | 0.4 ± 6.4 | 0.6 ± 2.5 | −15.9 ± 10.0 | 0.581 | 0.881 |
| E-selectin, µg/mL | ||||||||
| AUC | 10.7 ± 0.9 | 11.0 ± 0.9 | 10.6 ± 0.8 | 11.4 ± 1.6 | 11.9 ± 1.5 | 10.6 ± 1.6 | 0.694 | 0.407 |
| IAUC | −0.4 ± 0.3 | −0.01 ± 0.2 | −0.2 ± 0.1 | −0.3 ± 0.3 | 0.6 ± 0.4 | 0.5 ± 0.01 | 0.065 | 0.472 |
| P-selectin, µg/mL | ||||||||
| AUC | 13.0 ± 0.7 | 13.0 ± 0.9 | 13.1 ± 0.8 | 14.7 ± 1.8 | 16.5 ± 2.9 | 14.8 ± 2.1 | 0.300 | 0.131 |
| IAUC | −0.6 ± 0.3 | 0.01 ± 0.2 | −0.01 ± 0.3 | −0.07 ± 0.0 | 1.3 ± 1.3 | 0.3 ± 0.4 | 0.078 | 0.737 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rathnayake, K.M.; Weech, M.; Jackson, K.G.; Lovegrove, J.A. Impact of the Apolipoprotein E (epsilon) Genotype on Cardiometabolic Risk Markers and Responsiveness to Acute and Chronic Dietary Fat Manipulation. Nutrients 2019, 11, 2044. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11092044
Rathnayake KM, Weech M, Jackson KG, Lovegrove JA. Impact of the Apolipoprotein E (epsilon) Genotype on Cardiometabolic Risk Markers and Responsiveness to Acute and Chronic Dietary Fat Manipulation. Nutrients. 2019; 11(9):2044. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11092044
Chicago/Turabian StyleRathnayake, Kumari M., Michelle Weech, Kim G. Jackson, and Julie A. Lovegrove. 2019. "Impact of the Apolipoprotein E (epsilon) Genotype on Cardiometabolic Risk Markers and Responsiveness to Acute and Chronic Dietary Fat Manipulation" Nutrients 11, no. 9: 2044. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11092044