Maternal Selenium Deficiency in Mice Alters Offspring Glucose Metabolism and Thyroid Status in a Sexually Dimorphic Manner

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Procedures
2.2. Post-Mortem Tissue Collection
2.3. Glucose Tolerance Testing
2.4. Hormone Analysis
2.5. Quantitative PCR
2.6. Western Immunoblotting
2.7. Statistical Analysis
3. Results
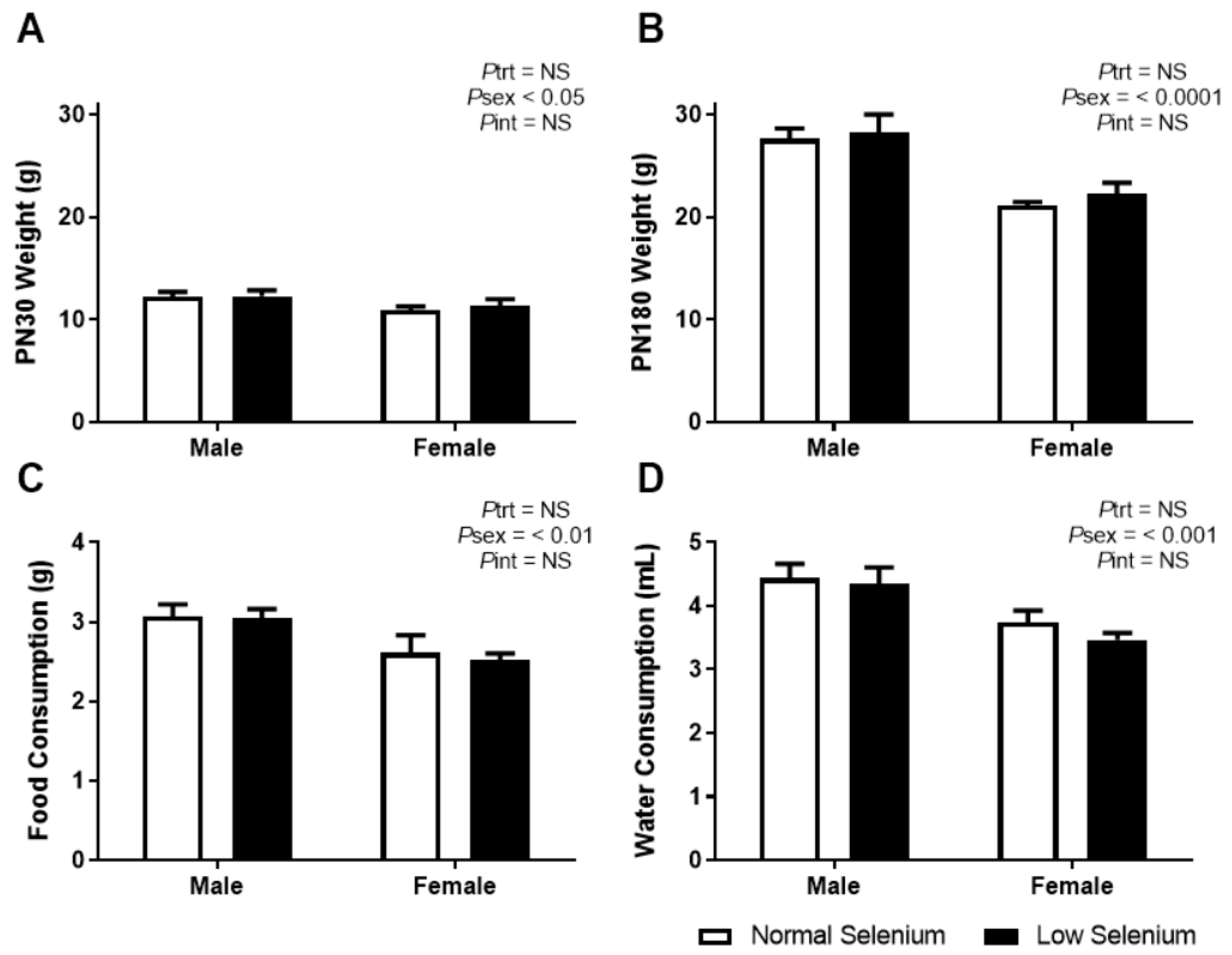
3.1. Offspring Weights, Food and Water Consumption
3.2. PN30 Offspring Allometry
3.3. PN180 Offspring Allometry
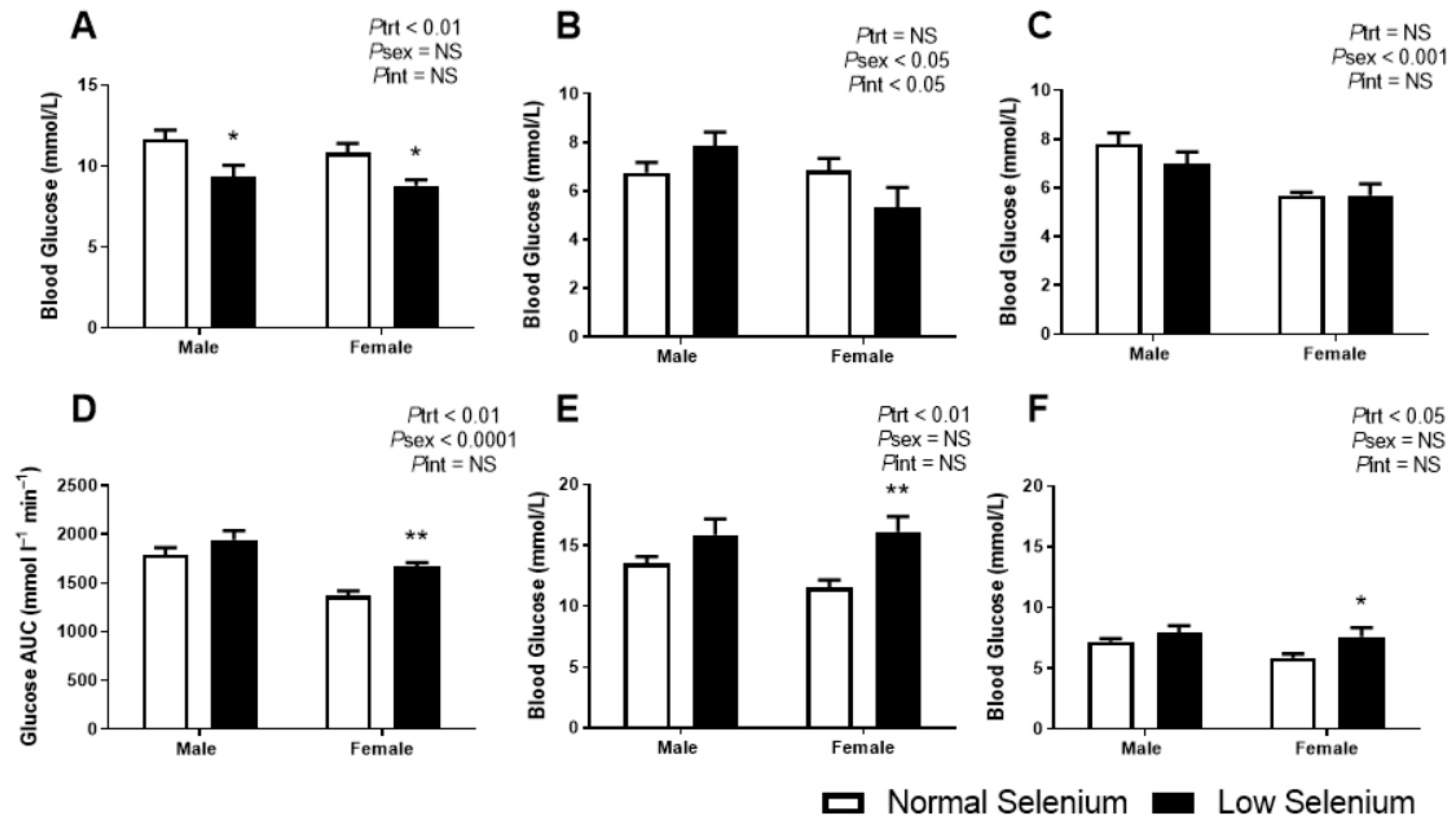
3.4. Glucose Metabolism
3.5. Endocrine Analysis
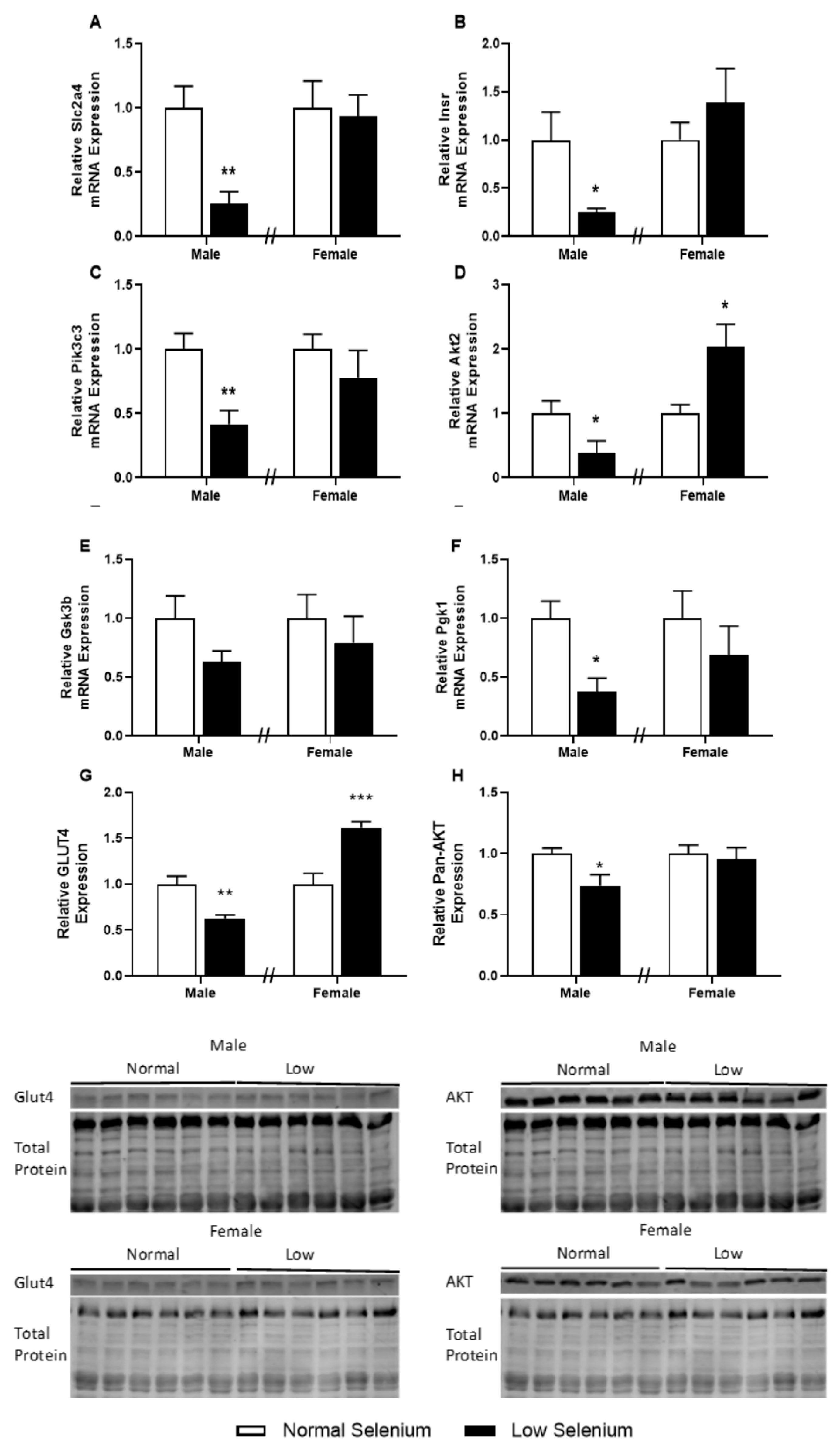
3.6. Skeletal Muscle Metabolic Protein and Gene Expression
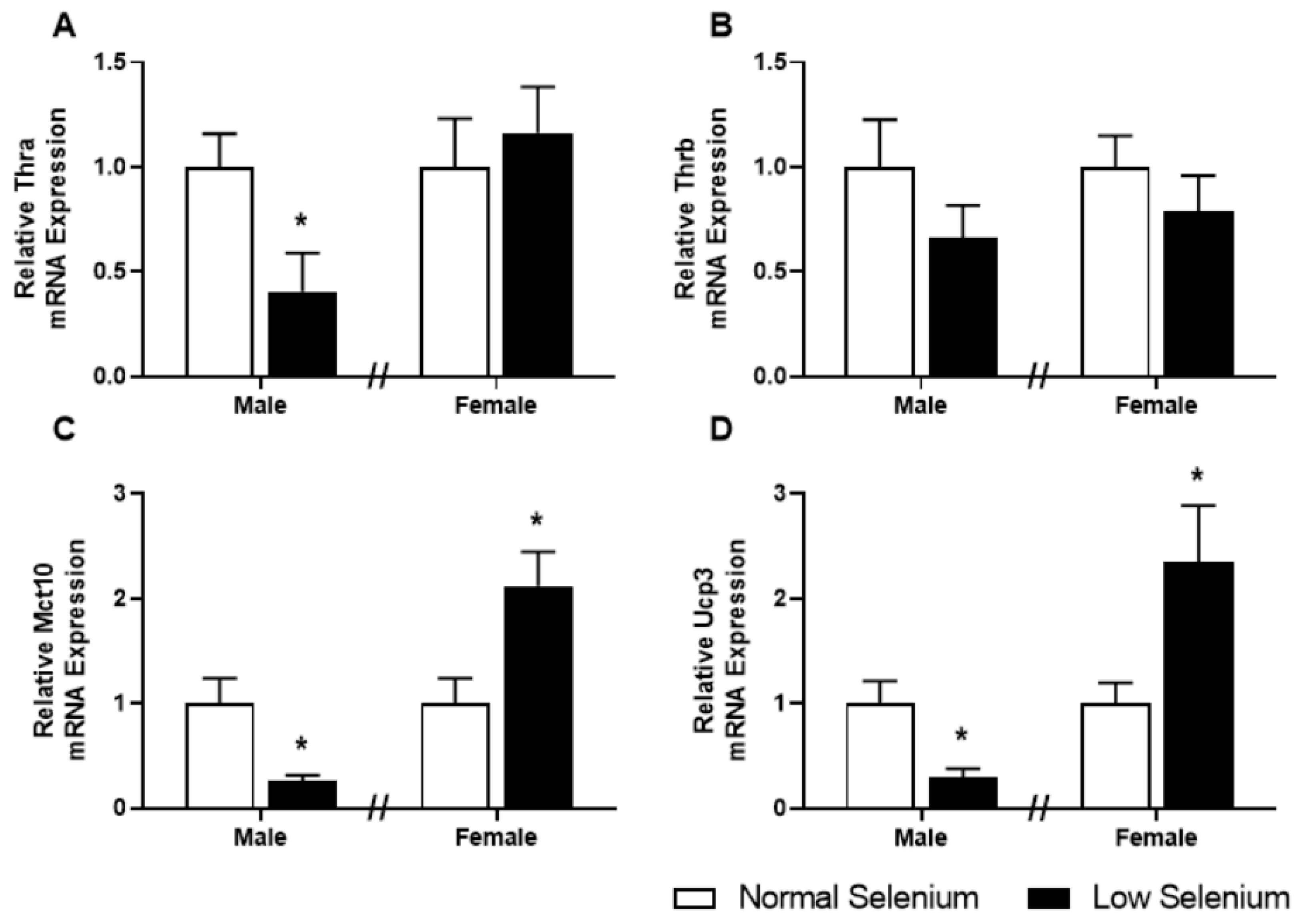
3.7. Skeletal Muscle Thyroid Hormone Gene Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hsu, C.-N.; Tain, Y.-L. The Good, the Bad, and the Ugly of Pregnancy Nutrients and Developmental Programming of Adult Disease. Nutrients 2019, 11, 894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Hofstee, P.; McKeating, D.; Perkins, A.V.; Cuffe, J.S. Placental adaptations to micronutrient dysregulation in the programming of chronic disease. Clin. Exp. Pharmacol. Physiol. 2018, 45, 871–884. [Google Scholar] [CrossRef]
- Pieczyńska, J.; Grajeta, H. The role of selenium in human conception and pregnancy. J. Trace Elem. Med. Biol. 2015, 29, 31–38. [Google Scholar] [CrossRef]
- Rayman, M.P.; Wijnen, H.; Vader, H.; Kooistra, L.; Pop, V. Maternal selenium status during early gestation and risk for preterm birth. CMAJ 2011, 183, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandowska, M.; Sajdak, S.; Lubinski, J. Serum Selenium Level in Early Healthy Pregnancy as a Risk Marker of Pregnancy Induced Hypertension. Nutrients 2019, 11, 1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayman, M.P.; Bath, S.C.; Westaway, J.; Williams, P.; Mao, J.; Vanderlelie, J.J.; Perkins, A.V.; Redman, C.W. Selenium status in UK pregnant women and its relationship with hypertensive conditions of pregnancy. Br. J. Nutr. 2015, 113, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, F.J.; Ma, L.L.; Chen, S.P.; Li, G.; Zhou, J.Q. Serum selenium level and gestational diabetes mellitus: a systematic review and meta-analysis. Nutr. J. 2016, 15, 94. [Google Scholar] [CrossRef] [Green Version]
- Lewandowska, M.; Sajdak, S.; Lubinski, J. The Role of Early Pregnancy Maternal Selenium Levels on the Risk for Small-for-Gestational Age Newborns. Nutrients 2019, 11, 2298. [Google Scholar] [CrossRef] [Green Version]
- Palmsten, K.; Buka, S.L.; Michels, K.B. Maternal pregnancy-related hypertension and risk for hypertension in offspring later in life. Obstet. Gynecol. 2010, 116, 858. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Yang, H. Gestational diabetes mellitus, programing and epigenetics. J. Matern.-Fetal Neonatal Med. 2014, 27, 1266–1269. [Google Scholar] [CrossRef] [PubMed]
- Bogden, J.D.; Kemp, F.W.; Chen, X.; Stagnaro-Green, A.; Stein, T.P.; Scholl, T.O. Low-normal serum selenium early in human pregnancy predicts lower birth weight. Nutr. Res. 2006, 26, 497–502. [Google Scholar] [CrossRef]
- Ojeda, M.L.; Nogales, F.; del Valle, P.M.; Díaz-Castro, J.; Murillo, M.L.; Carreras, O. Metabolic syndrome and selenium in fetal programming: gender differences. Food Funct. 2016, 7, 3031–3038. [Google Scholar] [CrossRef] [PubMed]
- Rederstorff, M.; Krol, A.; Lescure, A. Understanding the importance of selenium and selenoproteins in muscle function. Cell. Mol. Life Sci. 2006, 63, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Augusto, V.; Padovani, C.R.; Campos, G.R. Skeletal muscle fiber types in C57BL6J mice. Braz. J. Morphol. Sci. 2004, 21, 89–94. [Google Scholar]
- Köhrle, J. Selenium and the control of thyroid hormone metabolism. Thyroid 2005, 15, 841–853. [Google Scholar] [CrossRef]
- Marsili, A.; Zavacki, A.M.; Harney, J.W.; Larsen, P.R. Physiological role and regulation of iodothyronine deiodinases: A 2011 update. J. Endocrinol. Investig. 2011, 34, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Hofstee, P.; Bartho, L.A.; McKeating, D.R.; Radenkovic, F.; McEnroe, G.; Fisher, J.J.; Holland, O.J.; Vanderlelie, J.J.; Perkins, A.V.; Cuffe, J.S. Maternal selenium deficiency during pregnancy in mice increases thyroid hormone concentrations, alters placental function and reduces fetal growth. J. Physiol. 2019, 597, 5597–5671. [Google Scholar] [CrossRef]
- Delitala, A.P.; Capobianco, G.; Cherchi, P.L.; Dessole, S.; Delitala, G. Thyroid function and thyroid disorders during pregnancy: A review and care pathway. Arch. Gynecol. Obstet. 2019, 299, 327–338. [Google Scholar] [CrossRef]
- Bartho, L.A.; Holland, O.J.; Moritz, K.M.; Perkins, A.V.; Cuffe, J.S. Maternal corticosterone in the mouse alters oxidative stress markers, antioxidant function and mitochondrial content in placentas of female fetuses. J. Physiol. 2019, 597, 3053–3067. [Google Scholar] [CrossRef]
- Richard, K.; Holland, O.; Landers, K.; Vanderlelie, J.J.; Hofstee, P.; Cuffe, J.S.; Perkins, A.V. Effects of maternal micronutrient supplementation on placental function. Placenta 2017, 54, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappas, A.C.; Zoidis, E.; Chadio, S.E. Maternal Selenium and Developmental Programming. Antioxidants 2019, 8, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, W.W., Jr. Placental-fetal glucose exchange and fetal glucose metabolism. Trans. Am. Clin. Climatol. Assoc. 2006, 117, 321. [Google Scholar] [PubMed]
- Michelsen, T.M.; Holme, A.M.; Holm, M.B.; Roland, M.C.; Haugen, G.; Powell, T.L.; Jansson, T.; Henriksen, T. Uteroplacental glucose uptake and fetal glucose consumption: A quantitative study in human pregnancies. J. Clin. Endocrinol. Metab. 2018, 104, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Thorn, S.R.; Rozance, P.J.; Brown, L.D.; Hay, W.W. The intrauterine growth restriction phenotype: Fetal adaptations and potential implications for later life insulin resistance and diabetes. Semin. Reprod. Med. 2011, 29, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. BioMed Res. Int. 2010, 2010, 476279. [Google Scholar] [CrossRef] [Green Version]
- Ravelli, A.C.; van der Meulen, J.H.; Michels, R.; Osmond, C.; Barker, D.J.; Hales, C.; Bleker, O.P. Glucose tolerance in adults after prenatal exposure to famine. Lancet 1998, 351, 173–177. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Ozanne, S.; Wang, C.; Coleman, N.; Smith, G. Altered muscle insulin sensitivity in the male offspring of protein-malnourished rats. Am. J. Physiol. Endocrinol. Metab. 1996, 271, E1128–E1134. [Google Scholar] [CrossRef]
- Castets, P.; Lescure, A.; Guicheney, P.; Allamand, V. Selenoprotein N in skeletal muscle: from diseases to function. J. Mol. Med. 2012, 90, 1095–1107. [Google Scholar] [CrossRef]
- Clarke, N.F.; Kidson, W.; Quijano-Roy, S.; Estournet, B.; Ferreiro, A.; Guicheney, P.; Manson, J.I.; Kornberg, A.J.; Shield, L.K.; North, K.N. SEPN1: Associated with congenital fiber-type disproportion and insulin resistance. Ann. Neurol. 2006, 59, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Devaskar, S.U.; Thamotharan, M. Metabolic programming in the pathogenesis of insulin resistance. Rev. Endocr. Metab. Disord. 2007, 8, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Goodyear, L.J. Exercise and type 2 diabetes: molecular mechanisms regulating glucose uptake in skeletal muscle. Adv. Physiol. Educ. 2014, 38, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiken, C.E.; Ozanne, S.E. Sex differences in developmental programming models. Reproduction 2013, 145, R1–R13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, D.A.; Ertl, A.C.; Richardson, M.A.; Tate, D.B.; Davis, S.N. Estrogen blunts neuroendocrine and metabolic responses to hypoglycemia. Diabetes 2003, 52, 1749–1755. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Lin, H.; Jiao, H.; Song, Z. Corticosterone suppresses insulin-and NO-stimulated muscle glucose uptake in broiler chickens (Gallus gallus domesticus). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2009, 149, 448–454. [Google Scholar] [CrossRef]
- Ventura, M.; Melo, M.; Carrilho, F. Selenium and thyroid disease: From pathophysiology to treatment. Int. J. Endocrinol. 2017, 2017, 1297658. [Google Scholar] [CrossRef] [Green Version]
- Korevaar, T.I.; Muetzel, R.; Medici, M.; Chaker, L.; Jaddoe, V.W.; de Rijke, Y.B.; Steegers, E.A.; Visser, T.J.; White, T.; Tiemeier, H. Association of maternal thyroid function during early pregnancy with offspring IQ and brain morphology in childhood: A population-based prospective cohort study. Lancet Diabetes Endocrinol. 2016, 4, 35–43. [Google Scholar] [CrossRef]
- Alonso, M.; Goodwin, C.; Liao, X.; Page, D.; Refetoff, S.; Weiss, R.E. Effects of maternal levels of thyroid hormone (TH) on the hypothalamus-pituitary-thyroid set point: studies in TH receptor β knockout mice. Endocrinology 2007, 148, 5305–5312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Real, J.-M.; López-Bermejo, A.; Castro, A.; Casamitjana, R.; Ricart, W. Thyroid function is intrinsically linked to insulin sensitivity and endothelium-dependent vasodilation in healthy euthyroid subjects. J. Clin. Endocrinol. Metab. 2006, 91, 3337–3343. [Google Scholar] [CrossRef]
- Vyakaranam, S.; Vanaparthy, S.; Nori, S.; Palarapu, S.; Bhongir, A.V. Study of insulin resistance in subclinical hypothyroidism. Int. J. Health Sci. Res. 2014, 4, 147. [Google Scholar] [PubMed]
- Dimitriadis, G.; Raptis, S. Thyroid hormone excess and glucose intolerance. Exp. Clin. Endocrinol. Diabetes 2001, 109, S225–S239. [Google Scholar] [CrossRef] [PubMed]
- Bjoro, T.; Holmen, J.; Kruger, O.; Midthjell, K.; Hunstad, K.; Schreiner, T.; Sandnes, L.; Brochmann, H. Prevalence of thyroid disease, thyroid dysfunction and thyroid peroxidase antibodies in a large, unselected population. The Health Study of Nord-Trondelag (HUNT). Eur. J. Endocrinol. 2000, 143, 639–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, M.; Glenn, T.; Pilhatsch, M.; Pfennig, A.; Whybrow, P.C. Gender differences in thyroid system function: relevance to bipolar disorder and its treatment. Bipolar Disord. 2014, 16, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, L.; Romero, F.; Charli, J.-L.; Joseph-Bravo, P. Sex Dimorphic Responses of the Hypothalamus-Pituitary-Thyroid Axis to Maternal Separation and Palatable Diet. Frontiers in endocrinol. 2019, 10, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapp, J.F., III. Effects of diet and exercise on insulin resistance during pregnancy. Metab. Syndr. Relat. Disord. 2006, 4, 84–90. [Google Scholar] [CrossRef]
- Borghouts, L.; Keizer, H. Exercise and insulin sensitivity: a review. Int. J. Sports Med. 2000, 21, 1–12. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Gene Name | Gene Acronym | Accession Number | Primer Sequence |
|---|---|---|---|---|
| Housekeepers | Hypoxanthine Phosphoribosyl transferase 1 | Hprt1 | NM_013556 | F′ AGGGATTTGAATCACGTTTG R′ TTTACTGGCAACATCAACAG |
| Beta-2-Microglobulin | B2m | NM_009735 | F′ GTATGCTATCCAGAAAACCC R′ CTGAAGGACATATCTGACATC | |
| Insulin Receptor Pathway | Solute Carrier Family 2 Member 4 (GLUT4) | Slc2a4 | NM_009204 | F′ CAATGGTTGGGAAGGAAAAG R′ AATGAGTATCTCATAGGAGGC |
| Insulin Receptor | Insr | NM_010568 | F′ AAGACCTTGGTTACCTTCTC R′ GGATTAGTGGCATCTGTTTG | |
| Phosphatidylinositol 3-Kinase Catalytic Subunit Type 3 | Pik3c3 | NM_181414 | F′ CTATGGAGAATTTAGTGGAGAG R′ CTTCATATGTGAGTTGCTTGG | |
| Thymoma Viral Proto-Oncogene 2 | Akt2 | NM_007434 | F′ GAAAGGAGACTGTAAAAAGTGG R′ ATACAGTATCGTCTTGGGTC | |
| Glycogen Synthase Kinase 3 Beta | Gsk3b | NM_019827 | F′ CACTCTTCAACTTTACCACTC R′ ATTAGTATCTGAGGCTGCTG | |
| Phosphoglycerate Kinase 1 | Pgk1 | NM_008828 | F′ CTATCATAGGTGGTGGAGAC R′ ACACTAGGTTGACTTAGGAG | |
| Uncoupling Protein 3 | Ucp3 | NM_009464 | F′ CAAGAAATGCCATTGTCAAC R′ GAAGTTGTCAGTAAACAGGTG | |
| Thyroid Transporters and Receptors | Solute Carrier Family 16 Member 10 (Mct10) | Slc16a10 | NM_001114332 | F′ TCCTATTGCAGGGTTACTTC R′ GATCTTTCTTTGCTTCTTGC |
| Thyroid Hormone Receptor α | Thra | NM_178060 | F′ CATGGACTTGGTTCTAGATG R′ CTGTAGCAACATGTATCAGG | |
| Thyroid Hormone Receptor β | Thrb | NM_001113417 | F′ GAGACTCTAACTTTGAATGGG R′ CGATCTGAAGACATTAGCAG |
| Male | Female | Ptrt | Psex | Pint | |||
|---|---|---|---|---|---|---|---|
| (mg) | Control | Low | Control | Low | |||
| Liver | 717.37 ± 46.21 | 785.03 ± 24.44 | 665.82 ± 38.67 | 714.83 ± 19.39 | 0.1507 | 0.1344 | 0.8147 |
| Adrenal | 2.11 ± 0.24 | 1.96 ± 0.24 | 2.03 ± 0.11 | 1.95 ± 0.24 | 0.5711 | 0.8387 | 0.8722 |
| Brain | 374.55 ± 10.92 | 376.12 ± 10.65 | 380.90 ± 6.79 | 377.98 ± 8.97 | 0.9454 | 0.6793 | 0.8208 |
| Gastrocnemius | 50.87 ± 1.75 | 57.05 ± 3.11 | 48.50 ± 2.01 | 46.36 ± 3.36 | 0.4315 | 0.0160 | 0.1123 |
| Tibialis Anterior | 16.55 ± 0.70 | 20.80 ± 1.87 * | 14.49 ± 0.61 | 15.76 ± 0.95 | 0.0231 | 0.0047 | 0.2017 |
| Soleus | 3.64 ± 0.39 | 3.63 ± 0.24 | 3.10 ± 0.13 | 3.01 ± 0.25 | 0.8641 | 0.0534 | 0.8855 |
| EDL | 4.19 ± 0.33 | 4.93 ± 0.26 * | 4.33 ± 0.20 | 3.59 ± 0.15 | 0.9992 | 0.0257 | 0.0076 |
| Testes | 35.17 ± 1.96 | 33.71 ± 1.63 | - | - | |||
| Ovaries | - | - | 4.98 ± 0.88 | 4.01 ± 0.77 | |||
| Male | Female | Ptrt | Psex | Pint | |||
|---|---|---|---|---|---|---|---|
| (mg) | Control | Low | Control | Low | |||
| Liver | 1431.69 ± 55.10 | 1347.10 ± 54.17 | 1080.80 ± 53.76 | 998.97 ± 37.44 | 0.9797 | <0.0001 | 0.1311 |
| Adrenal | 2.09 ± 0.29 | 1.82 ± 0.30 | 3.59 ± 0.25 | 3.55 ± 0.57 | 0.6604 | <0.0001 | 0.7542 |
| Brain | 425.30 ± 8.64 | 428.70 ± 4.09 | 426.46 ± 5.48 | 443.66 ± 1.52 | 0.1205 | 0.2199 | 0.2918 |
| Gastrocnemius | 146.76 ± 3.80 | 148.15 ± 3.18 | 115.42 ± 1.85 | 114.70 ± 1.75 | 0.9092 | <0.0001 | 0.7183 |
| Tibialis Anterior | 41.80 ± 1.51 | 42.58 ± 2.11 | 35.86 ± 1.22 | 35.45 ± 0.77 | 0.3946 | <0.0001 | 0.2460 |
| Soleus | 8.52 ± 0.35 | 8.04 ± 0.33 | 6.98 ± 0.33 | 6.19 ± 0.33 | 0.1338 | <0.0001 | 0.7474 |
| EDL | 9.84 ± 0.63 | 9.57 ± 0.19 | 8.38 ± 0.42 | 7.92 ± 0.22 | 0.0721 | <0.0001 | 0.6543 |
| Testes | 95.11 ± 2.80 | 91.06 ± 1.73 | - | - | |||
| Ovaries | - | - | 3.47 ± 0.35 | 4.83 ± 1.28 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofstee, P.; McKeating, D.R.; Bartho, L.A.; Anderson, S.T.; Perkins, A.V.; Cuffe, J.S.M. Maternal Selenium Deficiency in Mice Alters Offspring Glucose Metabolism and Thyroid Status in a Sexually Dimorphic Manner. Nutrients 2020, 12, 267. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12010267
Hofstee P, McKeating DR, Bartho LA, Anderson ST, Perkins AV, Cuffe JSM. Maternal Selenium Deficiency in Mice Alters Offspring Glucose Metabolism and Thyroid Status in a Sexually Dimorphic Manner. Nutrients. 2020; 12(1):267. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12010267
Chicago/Turabian StyleHofstee, Pierre, Daniel R. McKeating, Lucy A. Bartho, Stephen T. Anderson, Anthony V. Perkins, and James S. M. Cuffe. 2020. "Maternal Selenium Deficiency in Mice Alters Offspring Glucose Metabolism and Thyroid Status in a Sexually Dimorphic Manner" Nutrients 12, no. 1: 267. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12010267