Enhancement of Ketone Supplements-Evoked Effect on Absence Epileptic Activity by Co-Administration of Uridine in Wistar Albino Glaxo Rijswijk Rats


 ,
,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Implantation of Screw Electrodes for Detection of EEG Signals
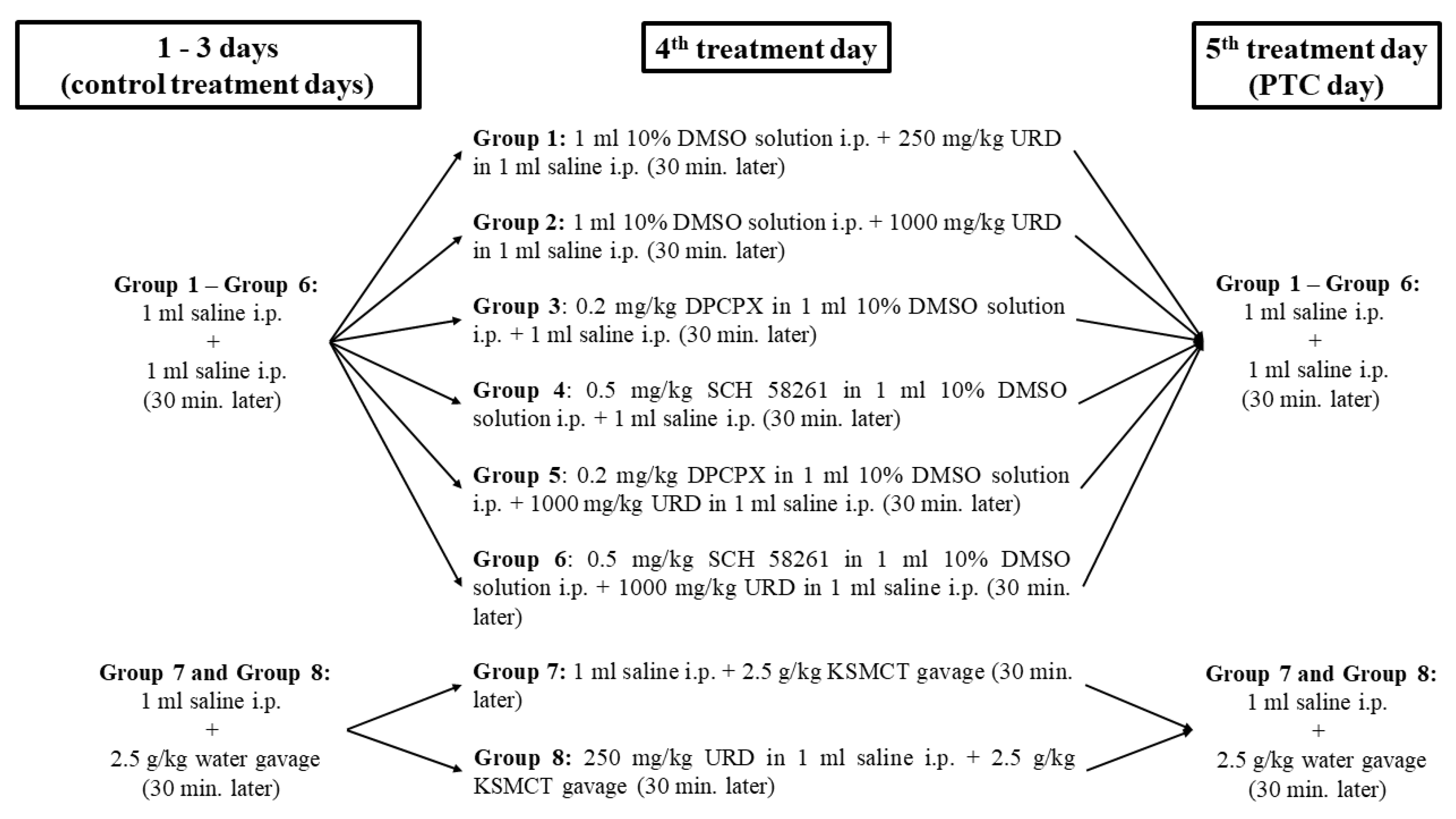
2.2. Treatment Groups
2.3. Detection of R-βHB and Glucose Levels
2.4. Statistical Analysis
3. Results
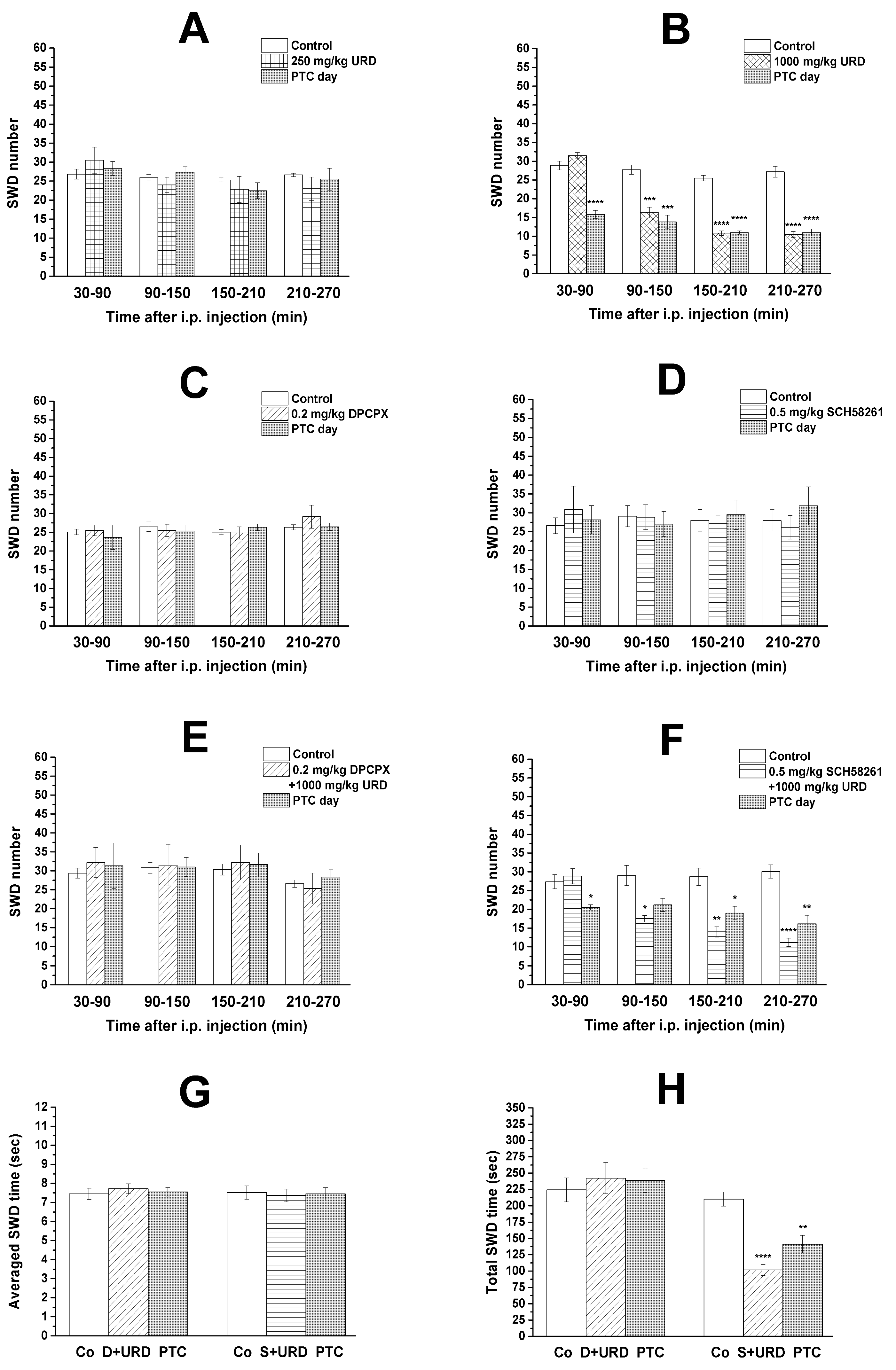
3.1. Effect of Uridine, DPCPX, and SCH 58261 Alone on SWD Number
3.2. Effect of Combined Administration of DPCPX and SCH 58261 on Uridine-Evoked Decrease in SWD Number and SWD Time
3.3. KSMCT-Evoked Changes in Blood R-βHB and Glucose Levels and SWD Number
3.4. Effect of Combined Administration of Uridine and KSMCT on SWD Number and SWD Time
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A1Rs | A1 type of Ado receptors |
| A2ARs | A2A type of Ado receptors |
| AcAc | acetoacetate |
| CNS | central nervous system |
| DMSO | dimethyl sulfoxide |
| DPCPX | 1,3-dipropyl-8-cyclopentylxanthine |
| EKSs | exogenous ketone supplements |
| IL-1β | interleukin-1β |
| i.p. | intraperitoneal |
| KD | ketogenic diet |
| KS | ketone salt |
| KSMCT | mix of ketone salt (KS) and medium chain triglyceride (MCT) oil in a 1:1 ratio |
| LPS | lipopolysaccharide |
| MCT | medium chain triglyceride |
| NF-κB | nuclear factor-κB |
| PTC day | post-treatment control day |
| R-βHB | R-beta-hydroxybutyrate |
| SCH 58261 | (7-(2-phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3-e]-1,2,4-triazolo [1,5-c]pyrimidine) |
| SWD | spike-wave discharge |
| WAG/Rij | Wistar Albino Glaxo/Rijswijk |
References
- Kovács, Z.; Juhász, G.; Palkovits, M.; Dobolyi, A.; Kékesi, K.A. Area, age and gender dependence of the nucleoside system in the brain: A review of current literature. Curr. Top. Med. Chem. 2011, 11, 1012–1033. [Google Scholar] [CrossRef] [PubMed]
- Haskó, G.; Pacher, P.; Vizi, E.S.; Illes, P. Adenosine receptor signaling in the brain immune system. Trends Pharmacol. Sci. 2005, 26, 511–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Shatskikh, T.; Marolewski, A.; Rusche, J.R.; Holmes, G.L. Effects of uridine on kindling. Epilepsy Behav. 2008, 13, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Kovács, Z.; Kékesi, K.A.; Juhász, G.; Dobolyi, A. The antiepileptic potential of nucleosides. Curr. Med. Chem. 2014, 21, 788–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, C.A. Anticonvulsant effects of uridine: Comparative analysis of metrazol and penicillin induced foci. Brain Res. 1973, 55, 291–308. [Google Scholar] [CrossRef]
- Zhao, Q.; Marolewski, A.; Rusche, J.R.; Holmes, G.L. Effects of uridine in models of epileptogenesis and seizures. Epilepsy Res. 2006, 70, 73–82. [Google Scholar] [CrossRef]
- Kovács, Z.; Slézia, A.; Bali, Z.K.; Kovács, P.; Dobolyi, A.; Szikra, T.; Hernádi, I.; Juhász, G. Uridine modulates neuronal activity and inhibits spike-wave discharges of absence epileptic Long Evans and Wistar Albino Glaxo/Rijswijk rats. Brain Res. Bull. 2013, 97, 16–23. [Google Scholar] [CrossRef]
- Kovács, Z.; Kékesi, K.A.; Dobolyi, Á.; Lakatos, R.; Juhász, G. Absence epileptic activity changing effects of non-adenosine nucleoside inosine, guanosine and uridine in Wistar Albino Glaxo Rijswijk rats. Neuroscience 2015, 300, 593–608. [Google Scholar] [CrossRef]
- Dobolyi, A.; Juhász, G.; Kovács, Z.; Kardos, J. Uridine function in the central nervous system. Curr. Top. Med. Chem. 2011, 11, 1058–1067. [Google Scholar] [CrossRef]
- Kardos, J.; Kovács, I.; Szárics, E.; Kovács, R.; Skuban, N.; Nyitrai, G.; Dobolyi, A.; Juhász, G. Uridine activates fast transmembrane Ca2+ ion fluxes in rat brain homogenates. Neuroreport 1999, 10, 1577–1582. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Ho, I.K.; Yamamoto, I. Uridine receptor: Discovery and its involvement in sleep mechanism. Sleep 2001, 24, 251–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Guo, S.; Xie, C.; Fang, J. Uridine metabolism and its role in glucose, lipid, and amino acid homeostasis. Biomed. Res. Int. 2020, 2020, 7091718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourget, P.A.; Tremblay, G.C. Pyrimidine biosynthesis in rat brain. J. Neurochem. 1972, 19, 1617–1624. [Google Scholar] [CrossRef]
- Kovács, Z.; Dobolyi, Á.; Kékesi, A.; Juhász, G. 5′-nucleotidases, nucleosides and their distribution in the brain: Pathological and therapeutic implications. Curr. Med. Chem. 2013, 20, 4217–4240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ipata, P.L.; Camici, M.; Micheli, V.; Tozz, M.G. Metabolic network of nucleosides in the brain. Curr. Top. Med. Chem. 2011, 11, 909–922. [Google Scholar] [CrossRef]
- Pooler, A.M.; Guez, D.H.; Benedictus, R.; Wurtman, R.J. Uridine enhances neurite outgrowth in nerve growth factor-differentiated pheochromocytoma cells. Neuroscience 2005, 134, 207–214. [Google Scholar] [CrossRef]
- De Campo, D.M.; Kossoff, E.H. Ketogenic dietary therapies for epilepsy and beyond. Curr. Opin. Clin. Nutr. Metab. Care. 2019, 22, 264–268. [Google Scholar] [CrossRef]
- Freeman, J.M.; Vining, E.P.; Pillas, D.J.; Pyzik, P.L.; Casey, J.C.; Kelly, L.M. The efficacy of the ketogenic diet-1998: A prospective evaluation of intervention in 150 children. Pediatrics. 1998, 102, 1358–1363. [Google Scholar] [CrossRef]
- Martin-McGill, K.J.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2020, 6, CD001903. [Google Scholar] [CrossRef]
- Vining, E.P.; Freeman, J.M.; Ballaban-Gil, K.; Camfield, C.S.; Camfield, P.R.; Holmes, G.L.; Shinnar, S.; Shuman, R.; Trevathan, E.; Wheless, J.W. A multicenter study of the efficacy of the ketogenic diet. Arch. Neurol. 1998, 55, 1433–1437. [Google Scholar] [CrossRef] [Green Version]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Ketogenic diet and epilepsy. Nutrients 2019, 11, 2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Li, X.J.; Jiang, W.L.; Sun, H.B.; Liu, J. Efficacy of and patient compliance with a ketogenic diet in adults with intractable epilepsy: A meta-analysis. J. Clin. Neurol. 2015, 11, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Hashim, S.A.; VanItallie, T.B. Ketone body therapy: From the ketogenic diet to the oral administration of ketone ester. J. Lipid Res. 2014, 55, 1818–1826. [Google Scholar] [CrossRef] [Green Version]
- Kovács, Z.; D’Agostino, D.P.; Dobolyi, A.; Ari, C. Adenosine A1 Receptor Antagonism Abolished the Anti-seizure Effects of Exogenous Ketone Supplementation in Wistar Albino Glaxo Rijswijk Rats. Front. Mol. Neurosci. 2017, 10, 235. [Google Scholar] [CrossRef] [Green Version]
- Poff, A.M.; Rho, J.M.; D’Agostino, D.P. Ketone administration for seizure disorders: History and rationale for ketone esters and metabolic alternatives. Front. Neurosci. 2019, 13, 1041. [Google Scholar] [CrossRef] [Green Version]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the brain: One molecule, multiple mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic control of vesicular glutamate transport and release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef] [Green Version]
- Lund, T.M.; Ploug, K.B.; Iversen, A.; Jensen, A.A.; Jansen-Olesen, I. The metabolic impact of β-hydroxybutyrate on neurotransmission: Reduced glycolysis mediates changes in calcium responses and KATP channel receptor sensitivity. J. Neurochem. 2015, 132, 520–531. [Google Scholar] [CrossRef] [Green Version]
- McNally, M.A.; Hartman, A.L. Ketone bodies in epilepsy. J. Neurochem. 2012, 121, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.K.; Rani, E.; Waheed, A.; Rajput, S.K. Pharmacoresistant epilepsy: A current update on non-conventional pharmacological and non-pharmacological interventions. J. Epilepsy Res. 2015, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sada, N.; Inoue, T. Electrical control in neurons by the ketogenic diet. Front. Cell Neurosci. 2018, 12, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, H.L.; Greene, R.W. Adenosine enhances afterhyperpolarization and accommodation in hippocampal pyramidal cells. Pflugers Arch. 1984, 402, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Simeone, T.A.; Simeone, K.A.; Rho, J.M. Ketone bodies as anti-seizure agents. Neurochem. Res. 2017, 42, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, D.; Pilla, R.; Held, H.; Landon, C.; Puchowicz, M.; Brunengraber, H.; Ari, C.; Arnold, P.; Dean, J.B. Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats. Am. J. Phys. Reg. Integr. Comp. Phys. 2013, 304, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Simeone, T.A.; Simeone, K.A.; Stafstrom, C.E.; Rho, J.M. Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology 2018, 133, 233–241. [Google Scholar] [CrossRef]
- Masino, S.A.; Kawamura, M.; Wasser, C.D.; Pomeroy, L.T.; Ruskin, D.N. Adenosine, ketogenic diet and epilepsy: The emerging therapeutic relationship between metabolism and brain activity. Curr. Neuropharmacol. 2009, 7, 257–268. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Rogawski, M.A.; Löscher, W.; Rho, J.M. Mechanisms of Action of Antiseizure Drugs and the Ketogenic Diet. Cold Spring Harb. Perspect. Med. 2016, 6, a022780. [Google Scholar] [CrossRef]
- Ari, C.; Kovács, Z.; Juhasz, G.; Murdun, C.; Goldhagen, C.R.; Koutnik, A.M.; Poff, A.M.; Kesl, S.L.; D’Agostino, D.P. Exogenous ketone supplements reduce anxiety-related behavior in Sprague-Dawley and Wistar Albino Glaxo/Rijswijk rats. Front. Mol. Neurosci. 2016, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Kovács, Z.; D’Agostino, D.P.; Diamond, D.M.; Ari, C. Exogenous Ketone Supplementation Decreased the Lipopolysaccharide-Induced Increase in Absence Epileptic Activity in Wistar Albino Glaxo Rijswijk Rats. Front. Mol. Neurosci. 2019, 12, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, Z.; D’Agostino, D.P.; Diamond, D.; Kindy, M.S.; Rogers, C.; Ari, C. Therapeutic Potential of Exogenous Ketone Supplement Induced Ketosis in the Treatment of Psychiatric Disorders: Review of Current Literature. Front. Psychiatry 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Coenen, A.M.; Van Luijtelaar, E.L. Genetic animal models for absence epilepsy: A review of the WAG/Rij strain of rats. Behav. Genet. 2003, 33, 635–655. [Google Scholar] [CrossRef] [PubMed]
- Meeren, H.K.; Pijn, J.P.; Van Luijtelaar, E.L.; Coenen, A.M.; Lopes da Silva, F.H. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J. Neurosci. 2002, 22, 1480–1495. [Google Scholar] [CrossRef] [PubMed]
- D’Alimonte, I.; D’Auro, M.; Citraro, R.; Biagioni, F.; Jiang, S.; Nargi, E.; Buccella, S.; Di Iorio, P.; Giuliani, P.; Ballerini, P.; et al. Altered distribution and function of A2A adenosine receptors in the brain of WAG/Rij rats with genetic absence epilepsy, before and after appearance of the disease. Eur. J. Neurosci. 2009, 30, 1023–1035. [Google Scholar] [CrossRef]
- Lakatos, R.K.; Dobolyi, Á.; Todorov, M.I.; Kékesi, K.A.; Juhász, G.; Aleksza, M.; Kovács, Z. Guanosine may increase absence epileptic activity by means of A2A adenosine receptors in Wistar Albino Glaxo Rijswijk rats. Brain Res. Bull. 2016, 124, 172–181. [Google Scholar] [CrossRef]
- Ari, C.; Murdun, C.; Koutnik, A.P.; Goldhagen, C.R.; Rogers, C.; Park, C.; Bharwani, S.; Diamond, D.M.; Kindy, M.S.; D’Agostino, D.P.; et al. Exogenous ketones lower blood glucose level in rested and exercised rodent models. Nutrients 2019, 11, 2330. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Watson, C. The Rat Brain Stereotaxic Coordinates; Academic Press: Orlando, FL, USA, 1998; ISBN 978-0125476171. [Google Scholar]
- Sitnikova, E.; van Luijtelaar, G. Electroencephalographic precursors of spike-wave discharges in a genetic rat model of absence epilepsy: Power spectrum and coherence EEG analyses. Epilepsy Res. 2009, 84, 159–171. [Google Scholar] [CrossRef]
- Depaulis, A.; Van Luijtelaar, G. Genetic models of absence epilepsy in the rat. In Models of Seizures and Epilepsy, 1st ed.; Pitkänen, A., Schwartzkroin, P.A., Moshé, S.L., Eds.; Academic Press: San Diego, CA, USA, 2005; Chapter 18; pp. 233–248. [Google Scholar]
- Kovács, Z.; Kékesi, K.A.; Szilágyi, N.; Ábrahám, I.; Székács, D.; Király, N.; Papp, E.; Császár, I.; Szego, E.; Barabás, K.; et al. Facilitation of spike-wave discharge activity by lipopolysaccharides in Wistar Albino Glaxo/Rijswijk rats. Neuroscience 2006, 140, 731–742. [Google Scholar] [CrossRef]
- Kovács, Z.; Czurkó, A.; Kékesi, K.A.; Juhász, G. The effect of intraperitoneally administered dimethyl sulfoxide on absence-like epileptic activity of freely moving WAG/RIJ rats. J. Neurosci. Methods 2011, 197, 133–136. [Google Scholar] [CrossRef]
- Koch, J.; Mayr, J.A.; Alhaddad, B.; Rauscher, C.; Bierau, J.; Kovacs-Nagy, R.; Coene, K.L.; Bader, I.; Holzhacker, M.; Prokisch, H.; et al. CAD mutations and uridine-responsive epileptic encephalopathy. Brain 2017, 140, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Miki, M.; Ikeda, M.; Yonemoto, S.; Watanabe, K.; Kondo, S.; Ho, I.K.; Yamamoto, I. Possible existence of a novel receptor for uridine analogues in the central nervous system using two isomers, N3-(S)-(+)- and N3-(R)-(−)-alpha-hydroxy-beta-phenethyluridines. Biol. Pharm. Bull. 2001, 24, 729–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarlone, S.L.; Grieco, J.C.; D’Agostino, D.P.; Weeber, E.J. Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol. Dis. 2016, 96, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Todd King, M.; Musa-Veloso, K.; Ho, M.; Roberts, A.; Robertson, J.; Vanitallie, T.B.; et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul. Toxicol. Pharmacol. 2012, 63, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yudkoff, M.; Daikhin, Y.; Melø, T.M.; Nissim, I.; Sonnewald, U.; Nissim, I. The ketogenic diet and brain metabolism of amino acids: Relationship to the anticonvulsant effect. Annu. Rev. Nutr. 2007, 27, 415–430. [Google Scholar] [CrossRef] [Green Version]
- Masino, S.A.; Li, T.; Theofilas, P.; Sandau, U.S.; Ruskin, D.N.; Fredholm, B.B.; Geiger, J.D.; Aronica, E.; Boison, D. A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J. Clin. Investig. 2011, 121, 2679–2683. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Che, X.; Yu, L.; Yang, X.; An, N.; Song, W.; Wu, C.; Yang, J. Uridine attenuates morphine-induced conditioned place preference and regulates glutamate/GABA levels in mPFC of mice. Pharmacol. Biochem. Behav. 2017, 163, 74–82. [Google Scholar] [CrossRef]
- Cunha, R.A. Neuroprotection by adenosine in the brain: From A(1) receptor activation to A (2A) receptor blockade. Purinergic Signal. 2005, 1, 111–134. [Google Scholar] [CrossRef] [Green Version]
- Sperlágh, B.; Szabó, G.; Erdélyi, F.; Baranyi, M.; Vizi, E.S. Homo- and heteroexchange of adenine nucleotides and nucleosides in rat hippocampal slices by the nucleoside transport system. Br. J. Pharmacol. 2003, 139, 623–633. [Google Scholar] [CrossRef]
- Ruskin, D.N.; Kawamura, M.; Masino, S.A. Adenosine and Ketogenic Treatments. J. Caffeine Adenosine Res. 2020, 10, 104–109. [Google Scholar] [CrossRef]
- Cremer, C.M.; Palomero-Gallagher, N.; Bidmon, H.J.; Schleicher, A.; Speckmann, E.J.; Zilles, K. Pentylenetetrazole-induced seizures affect binding site densities for GABA, glutamate and adenosine receptors in the rat brain. Neuroscience 2009, 163, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Lusardi, T.A.; Akula, K.K.; Coffman, S.Q.; Ruskin, D.N.; Masino, S.A.; Boison, D. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology 2015, 99, 500–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, T.; Cansev, M.; Wurtman, R.J. Oral supplementation with docosahexaenoic acid and uridine-5′-monophosphate increases dendritic spine density in adult gerbil hippocampus. Brain Res. 2007, 1182, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfers, M.; Sorkin, L. Effect of cytokines on neuronal excitability. Neurosci. Lett. 2008, 437, 188–193. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| % Cal from Fat | 10.0 |
| % Cal from Protein | 23.0 |
| % Cal from Carbohydrates | 67.0 |
| kcal/g | 3.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunner, B.; Rauch, E.; Ari, C.; D’Agostino, D.P.; Kovács, Z. Enhancement of Ketone Supplements-Evoked Effect on Absence Epileptic Activity by Co-Administration of Uridine in Wistar Albino Glaxo Rijswijk Rats. Nutrients 2021, 13, 234. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13010234
Brunner B, Rauch E, Ari C, D’Agostino DP, Kovács Z. Enhancement of Ketone Supplements-Evoked Effect on Absence Epileptic Activity by Co-Administration of Uridine in Wistar Albino Glaxo Rijswijk Rats. Nutrients. 2021; 13(1):234. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13010234
Chicago/Turabian StyleBrunner, Brigitta, Enikő Rauch, Csilla Ari, Dominic P. D’Agostino, and Zsolt Kovács. 2021. "Enhancement of Ketone Supplements-Evoked Effect on Absence Epileptic Activity by Co-Administration of Uridine in Wistar Albino Glaxo Rijswijk Rats" Nutrients 13, no. 1: 234. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13010234