Mechanisms of Dietary Sodium-Induced Impairments in Endothelial Function and Potential Countermeasures


Department of Kinesiology and Applied Physiology, University of Delaware, 540 S College Ave., Newark, DE 19713, USA
*
Author to whom correspondence should be addressed.
Nutrients 2021, 13(1), 270; https://0-doi-org.brum.beds.ac.uk/10.3390/nu13010270
Submission received: 19 December 2020
/
Revised: 13 January 2021
/
Accepted: 16 January 2021
/
Published: 19 January 2021
(This article belongs to the Special Issue Salt in Health and Disease—a Delicate Balance)
Abstract
:Despite decades of efforts to reduce sodium intake, excess dietary sodium remains commonplace, and contributes to increased cardiovascular morbidity and mortality independent of its effects on blood pressure. An increasing amount of research suggests that high-sodium diets lead to reduced nitric oxide-mediated endothelial function, even in the absence of a change in blood pressure. As endothelial dysfunction is an early step in the progression of cardiovascular diseases, the endothelium presents a target for interventions aimed at reducing the impact of excess dietary sodium. In this review, we briefly define endothelial function and present the literature demonstrating that excess dietary sodium results in impaired endothelial function. We then discuss the mechanisms through which sodium impairs the endothelium, including increased reactive oxygen species, decreased intrinsic antioxidant defenses, endothelial cell stiffening, and damage to the endothelial glycocalyx. Finally, we present selected research findings suggesting that aerobic exercise or increased intake of dietary potassium may counteract the deleterious vascular effects of a high-sodium diet.
1. Introduction
Over 90% of adults in the United States consume high-salt diets, with an average daily sodium intake of 3600 mg, which far exceeds the 2300 mg/day as recommended in the Dietary Guidelines for Americans [1,2]. Globally, excess dietary sodium is a contributing factor to 1.65 million deaths annually, with 40% of these deaths occurring before the age of 70 [3]. As such, the consequences of high-sodium diets remain an important topic of study.
The deleterious effects of excess dietary sodium are commonly attributed to the effect of sodium on blood pressure (BP). Importantly, a positive linear association between BP and 24-h urinary sodium excretion, the gold standard marker of sodium intake, has been noted in numerous epidemiological studies [4,5]. Likewise, sodium restriction results in reduced BP in individuals with elevated BP [6,7]. However, among young adults, the magnitude of change in systolic and diastolic BP associated with changes in dietary sodium consumption is a rather modest 1.9 and 0.4 mmHg per 2300 mg/day change in sodium, respectively, after adjustment for BMI and alcohol consumption [5]. In fact, the majority of normotensive individuals are considered salt-resistant in that they do not experience a marked change in BP with large changes in sodium intake [8], especially those under the age of 45 [9].
Yet, even in the absence of elevated BP, high dietary sodium is associated with negative impacts on cardiovascular health. Increased sodium intake is related to increased risk of acute coronary events and cardiovascular mortality independent of BP [10]. Likewise, overweight individuals in the highest quartile of sodium intake have an increased risk of congestive heart failure compared to those in the lowest quartile, even after controlling for demographics, alcohol consumption, smoking, physical activity, and BP [11]. Additionally, greater sodium intake is related to greater left ventricular mass in both normotensive and hypertensive individuals [12,13], and left ventricular hypertrophy is reversed with sodium reduction [14]. Notably, endothelial dysfunction often precedes the development of cardiovascular disease (CVD) [15,16,17] and thus may be an early marker of the deleterious effects of excessive dietary sodium consumption.
As outlined in Figure 1, this review presents the current evidence for BP-independent effects of high dietary sodium consumption on endothelial function in humans, and summarizes the literature examining the potential mechanisms responsible for this phenomenon. Additionally, recent reports suggest that the vascular consequences of excess sodium may be mitigated by physical activity or increased dietary potassium consumption. As such, we will provide a brief review of how these behaviors may address specific mechanisms of dietary sodium-induced endothelial dysfunction.
2. Assessment of Endothelial Function
The endothelium is the monolayer of cells lining the lumen of the vasculature throughout the body. Beyond a simple barrier between the blood and the vascular wall, a healthy endothelium serves to prevent thrombosis and coagulation and control vascular tone [18]. In response to chemical or mechanical stimuli, the endothelium produces vasodilating or vasoconstricting substances that act on the adjacent vascular smooth muscle (VSM) cells [18] (Figure 2). One such substance is endothelium-derived nitric oxide (NO), which is frequently studied due to its anti-atherogenic effects, in addition to NO being a key mediator of vascular tone via its potent vasodilating effects [19].
In response to binding of agonists (e.g., acetylcholine or ACh) to endothelium receptors or an increase in shear stress on the endothelium, nitric oxide (NO) is produced when endothelial nitric oxide synthase (eNOS) catalyzes the conversion of oxygen and L-arginine into L-citrulline and NO (Figure 2) [19,20]. The gaseous NO then diffuses into the VSM, initiating an intracellular cascade and causing the removal of cytosolic calcium and/or cellular hyperpolarization, thus producing the relaxation of the VSM and the dilation of the vessel [19,20]. This response can be assessed in vitro in cultured cells and isolated vessels or in vivo in animals and humans. Intra-arterial infusions of ACh or methacholine (MCh) are utilized to test resistance artery endothelial function, and can be used in combination with an NOS inhibitor, such as L-NG-Nitro arginine methyl ester (L-NAME) and NG-Monomethyl-L-arginine (L-NMMA), to quantify the NO component of the dilation. Ultrasonic assessment of flow-mediated dilation (FMD) of a major conduit artery (i.e., brachial artery) has become commonplace due to its non-invasive nature and the availability of sophisticated analysis software. During the assessment of FMD, the artery in question is imaged via ultrasound before and after a 5-min period of circulatory arrest via a cuff applied to the limb distal to imaging and inflated to a suprasystolic pressure. The increases in blood flow that occur following cuff release increase shear stress on the arterial endothelium, resulting in dilation that is largely NO-mediated [21]. Importantly, impaired endothelium-dependent dilation of the peripheral vasculature is associated with increased risk of future cardiovascular events, and this relation exists whether endothelial function is assessed via FMD [22] or ACh infusion [23,24].
While the studies utilizing FMD demonstrate the effects of excess sodium on the macro-vascular endothelium, the cutaneous circulation can be used as a model of the microvasculature in humans [25]. For instance, a high-sodium diet reduces the NO-mediated increase in skin blood flow in response to local heating in young, salt-resistant normotensive individuals [26]. In agreement with this finding, a high-sodium diet blunts the cutaneous blood flow response to ACh delivered via intradermal infusion [27] or iontophoresis [28,29]. Likewise, cutaneous post-occlusive reactive hyperemia is blunted in the absence of a change in BP following excess sodium consumption [28,30]; however, cutaneous reactive hyperemia appears to be mediated by endothelial release of prostaglandins rather than NO [30,31], and is beyond the scope of this review.
3. Dietary Sodium and Impaired Endothelial Function in Humans
A deleterious vascular effect of high dietary sodium in the absence of a change in BP was first noted in the Dahl salt-resistant rat model in 1987 [32]. Subsequent studies, however, suggested that this effect might not occur in humans, as no effect of a short-term high-sodium diet was observed on endothelium-dependent forearm blood flow responses in a small group of young men [33] or middle-aged hypertensives [34,35]. In contrast, Tzemos and colleagues studied young, normotensive adults and found that a five-day high-sodium diet impaired forearm dilation to intra-arterial ACh infusions [36]. When on the high-sodium diet, subjects demonstrated less of an attenuation in ACh-induced dilation by L-NMMA, suggesting that high-sodium diets specifically inhibit NO-mediated dilation [36]. Notably, systolic BP increased on the high-sodium diet, suggesting that a significant proportion of the subjects were salt-sensitive [36], leaving the possibility that the change in BP was responsible for the impairment of endothelial function.
Later studies attempted to eliminate the potential confounding effects of BP. Jablonski et al. observed an improvement in both ACh-induced forearm blood flow and brachial artery FMD after statistically controlling for changes in BP following a four-week reduction in dietary sodium (~1500 vs. 3600 mg/day) amongst middle-aged and older adults with elevated BP [7]. Similarly, DuPont and colleagues observed a significantly blunted brachial artery FMD in young, normotensive, salt-resistant subjects on a high-sodium diet compared to a low-sodium condition [37]. In a subset of subjects, nitroglycerin induced similar dilation of the brachial artery across both diets, providing evidence that the impairment occurred at the endothelium [37] and not the VSM cells. High dietary sodium-induced reductions in FMD have been found in multiple [30,38] but not all [27] studies. There is some evidence that the impact of a high-sodium diet on FMD is greater in men than women [39]. Though these studies typically administer the excess sodium for five to eight days, one research team has demonstrated that FMD is attenuated acutely after a single high-sodium meal [40], highlighting the rapid effect of excess sodium on the vasculature, though this is not a universal finding [41,42]. Table 1 summarizes the details of relevant studies on the endothelial effects of high-sodium diets in humans.
4. Mechanisms Contributing to Dietary Sodium-Induced Impairment in Endothelial Function
4.1. Oxidative Stress
Excess oxidative stress is the most well-studied mechanistic explanation for dietary sodium-induced endothelial dysfunction. Oxidative stress occurs due to an imbalance between endogenous antioxidant activity and production of reactive oxygen species (ROS), particularly superoxide (O2−), which results in impaired endothelial function by decreasing NO bioavailability [45]. NO readily reacts with O2− to produce the free radical peroxynitrite (ONOO−). O2− and ONOO− can then oxidize the eNOS cofactor tetrahydrobiopterin (BH4), resulting in the uncoupling of eNOS and generation of O2− rather than NO [46,47]. Lenda and colleagues provided early evidence of ROS involvement in high-sodium-induced endothelial dysfunction with the observation that dilation of skeletal muscle arterioles in rats fed a high-sodium diet was restored in the presence of the O2− scavenger superoxide dismutase (SOD) [48]. Zhu and colleagues later demonstrated that aortas of normotensive rats fed a high-sodium diet for three days produced less NO and more O2− when stimulated by MCh, whereas Tempol, a SOD mimetic, decreased O2− production and increased NO [49].
Human studies support a role for oxidative stress in dietary sodium-induced impairments in endothelial function, as outlined in Figure 2. In middle-aged adults with elevated BP who habitually consume a high-sodium diet, ACh-induced forearm blood flow and brachial artery FMD were each augmented following ascorbic acid infusion [7]. Likewise, Greaney et al. demonstrated that local infusion of ascorbic acid restored the NO-mediated increase in skin blood flow to local heating in normotensive, salt-resistant individuals consuming excess sodium [26]. Oral supplementation with the antioxidant vitamins C and E during a high-sodium diet (5500 mg/day) similarly restored the skin blood flow response to cutaneous ACh iontophoresis while also preventing an increase in markers of oxidative stress in the plasma and urine [28].
Within endothelial cells, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase, uncoupled eNOS, and mitochondria can produce O2−. The origin of O2− induced by excess dietary sodium has been investigated by multiple laboratories and/or may vary by species and/or the vascular bed studied. For instance, significantly greater mRNA expression of NADPH oxidase subunits has been reported in the renal cortex of rats fed high-sodium for one week [50]. Likewise, NADPH oxidase and xanthine oxidase activity were increased in the skeletal muscle arterioles of rats fed high-sodium chow, however, inhibition of these enzymes did not restore endothelium-dependent relaxation [51]. Since O2− scavenging restores dilation in the skeletal muscle microvasculature [48], the authors speculated that uncoupled eNOS secondary to ONOO− oxidation of BH4 is the source of O2− [51]. Similarly, O2− production in the skeletal muscle arterioles of high sodium fed mice was decreased in the presence of both Tempol and L-NMMA [52]. The follow-up study by Nurkiewicz et al. confirmed that mice fed high-sodium were BH4 deficient relative to mice on a normal sodium diet, and that this difference and the resulting attenuation in NO-mediated dilation were restored by the addition of L-arginine to the drinking water [53]. Together, these findings suggest that uncoupled eNOS, due to a deficiency of BH4 or L-Arginine, is a source of sodium-induced increases in O2−.
Zhu and colleagues found that apocynin, an inhibitor of NADPH oxidase, restored dilation in mesenteric arteries of rats fed high-sodium [54]. Additionally, inhibition of eNOS did not decrease O2− production in the high-sodium rats, suggesting that uncoupled eNOS was not the source of O2− in the mesenteric artery [54]. Likewise, Guers et al. reported that dietary sodium markedly increased NAPDH oxidase expression in rat femoral arteries [55]. Two recent reports demonstrated that apocynin mitigates sodium-associated reductions in cutaneous microvascular dilation to local heating [44] and ACh infusion [27], suggesting that excess dietary sodium increases NADPH oxidase activity in normotensive, salt-resistant humans. Whether this occurs in other vascular beds in humans has not been determined.
In addition to increased production of O2−, it appears that high-sodium diets may decrease endogenous antioxidant defenses. During inhibition of the cytosolic isoform of SOD, copper-zinc SOD (CuZn SOD or SOD-1), the skeletal muscle microcirculation of rats on a normal sodium diet showed a greater increase in O2− than the microcirculation in rats fed a high-sodium diet, suggesting attenuated activity of SOD-1 in the high-sodium condition [56]. Notably, in this study, both groups of rats displayed similar SOD expression and similar responses to catalase inhibition, suggesting that activity of SOD-1 was decreased in the high-sodium condition. Likewise, sodium decreased SOD-1 expression in the femoral arteries of high-sodium fed versus normal chow fed rats [55]. Rats consuming a high-sodium diet also exhibited decreased expression of SOD-1 [57,58] and the mitochondrial isoform, manganese SOD (MnSOD or SOD-2) [57], in the middle cerebral artery; however, this effect may be vascular bed dependent, as the same laboratory did not observe an effect of sodium consumption on either isoform of SOD in the mesenteric arteries [59]. To date, whether vascular expression or activity of SOD are decreased due to dietary sodium has not been directly tested in humans; however, such an effect may partly explain the Tempol-induced increases in NO-mediated cutaneous microvascular function in participants on a high-sodium diet [27,44].
4.2. Endothelial Cell Stiffening
In addition to oxidative stress, there is evidence that high dietary sodium intake may decrease NO release via a change in the mechanical properties of endothelial cells. Using cultured endothelial cells, Oberleithner and colleagues report that an increase in the sodium content of the bath, within the physiological range for humans, results in stiffening of endothelial cells, as measured by atomic force microscopy, and attenuated NO release [60]. This only occurred when cells were treated with physiological concentrations of aldosterone, and the stiffening was prevented by amiloride, indicating that opening of epithelial sodium channels (ENaC) induces sodium entry and subsequent cell swelling. Importantly, changes in endothelial cell deformability precede changes in shear-induced NO release, whereas inhibition of NO does not stiffen the cell, suggesting that sodium-induced endothelial stiffening is likely not due to loss of NO bioavailability [61].
Finally, intriguing recent evidence suggests that sodium-induced damage of the endothelial glycocalyx (eGC) may initiate endothelial cell stiffening and subsequent endothelial dysfunction, as illustrated in Figure 3. The eGC is a 0.5 to 4.5 μm thick mesh-like layer of proteoglycans, glycoproteins, and glycosaminoglycans (i.e., heparin sulfate and hyaluronic acid) on the luminal surface of endothelial cells that serves as a protective barrier between the endothelium and red blood cells, and plays an important role in mechano-transduction of shear stress [62]. Deterioration of the eGC occurs with conditions that increase the risk of CVD, including aging [63], untreated hypertension [64], diabetes [65,66], and obesity [67], and may be a consequence of oxidative stress [68]. In the context of dietary sodium, the negatively charged eGC binds the positively charged sodium ions, thus buffering increases in plasma sodium levels and providing the initial endothelial cell barrier to sodium [69]. However, when endothelial cells are exposed to 150 mmol of sodium rather than 130 mmol ex vivo, the height of their eGC is reduced in a manner consistent with heparinase-induced degradation [69]. As a result, sodium has increased access to ENaC, and intracellular sodium increases rapidly, leading to augmented endothelial stiffness [70]. The sodium-induced decrease in eGC also increases monocyte adhesion and induces local inflammation [71], thus initiating further endothelial dysfunction.
5. Potential Countermeasures Against High Dietary Sodium-Induced Endothelial Dysfunction
Population-wide sodium reduction is proposed as the best means to mitigate the consequences of high-sodium consumption for both salt-resistant and salt-sensitive individuals [72]. For instance, by instituting policies aimed at decreasing sodium intake, Finland has reduced sodium consumption by ~40% since 1980, which contributed to an 80% reduction in the middle-aged death rate from strokes and coronary artery disease, despite increases in smoking and obesity [73]. Similarly, the sodium reduction plan instituted by the United Kingdom has decreased sodium intake by 15%, and is estimated to have prevented 9000 deaths per year [72]. However, as the sodium intake in the United States has been stable since the middle of the twentieth century [74] and a large portion of the population has no interest in reducing their sodium consumption [75], it is crucial that scientists identify non-sodium centered lifestyle changes that may help to counteract the consequences of excess sodium intake. Aerobic exercise and/or increased dietary potassium intake may diminish the vascular effects of excess sodium. If so, these behaviors could be targets for public health interventions, especially in populations that tend to resist sodium reduction.
5.1. Aerobic Exercise
Physical activity, or aerobic exercise in particular, is associated with reduced cardiovascular morbidity [76] and mortality [77] in adults. However, only approximately 60% of this reduction can be attributed to the effect of exercise on traditional CVD risk factors [78], suggesting non-traditional risk factors, such as endothelial function, may account for some of the remaining benefits of exercise [79]. Indeed, exercise appears to improve vascular function beyond vessels supplying the working muscles [80,81,82,83], though some controversy on this topic still exists [84]. Thus, regular physical activity may protect the vasculature from the effects of a high-sodium diet. Rebholz and colleagues report that the prevalence of salt sensitivity of BP is lowest among those in the highest quartile of physical activity [85]. While this study did not address vascular function directly, it suggests that habitual physical activity level mitigates the effects of sodium in humans; however, there is a paucity of research on this topic.
Our group recently published a report on the effects of voluntary wheel running on endothelial function in Sprague Dawley rats fed either a high or low-sodium diet for six weeks [55]. Consistent with previous reports, the high-sodium diet attenuated ACh-induced endothelium-dependent relaxation of the femoral artery, despite no group difference in BP. However, the effect of sodium on ACh-mediated relaxation was abolished in the rats who had access to a running wheel. Much of the systemic benefit of exercise on endothelial function is thought to be due to the effect of increased shear and circumferential stress on NO production (reviewed in [86]). In agreement with this hypothesis, Guers et al. showed that physical activity reversed high-sodium diet induced decreases in the ratio of phosphorylated eNOS relative to total eNOS expression. Additionally, voluntary wheel running abolished the high-sodium diet-associated increases in the expression of NADPH oxidase subunits and the decrease in expression of SOD-1. These findings support the hypothesis that regular aerobic exercise may protect against the adverse vascular effects of excess sodium consumption.
While the study by Guers et al. was not designed to determine the mechanism responsible for the beneficial effects of exercise, we can speculate on a few potential possibilities. Exercise elicits a marked increase in angiotensin II (Ang II) [87]. Ang II is a potent vasoconstrictor, and high concentrations are known to activate NADPH oxidase generation of O2− [88]. Yet, increased sodium intake significantly suppresses plasma Ang II levels. Interestingly, when Ang II is chronically infused in animals fed high-sodium diets to restore physiological levels, endothelium-dependent relaxation is restored [57,58,89,90], O2− production is decreased [89], and the sodium-induced decrease in SOD is abolished [57,58]. Together, this suggests that exercise may exert its vascular benefits via increases in circulating Ang II. However, this explanation is unlikely, as the half-life of Ang II in the blood is approximately 30 s [91], whereas the positive effect of subpressor doses of Ang II in animals occurs with chronic infusions.
Another possible mechanism for the endothelium-protective effects of exercise in high salt fed rats is the activation of nuclear factor erythroid 2-related factor 2 (NRF2). NRF2 is a transcription factor activated by exercise-induced ROS that, in turn, regulates expression of antioxidants and other cyto-protective genes [92,93,94]. Recently, Priestly and colleagues demonstrated that the herbal supplement Protandim, previously shown to activate NRF2 [95] and upregulate endogenous antioxidant enzymes [96], corrected sodium-induced reductions in ACh-mediated relaxation in both rat cerebral and mesenteric arteries, as well as in hamster cheek pouch arterioles [97]. Importantly, these animals exhibited increased SOD and decreased mitochondrial ROS, yet there was no effect in the NRF2(−/−) knockout rats, signifying that Protandim worked specifically on NRF2 [97]. Further work is needed to determine if NRF2 activation via exercise is beneficial to rats and humans consuming a high-sodium diet.
Aerobic exercise may also protect the endothelium from excessive sodium intake by maintaining the eGC. While not studied in the context of sodium consumption, exposure to fluid shear stress in cultured endothelial cells results in a thicker eGC [98], consistent with a beneficial effect of increased blood flow, like that seen in exercise. Indeed, 20 weeks of moderate intensity aerobic training decreases markers of eGC shedding in young men [99]. Likewise, a four-week high-intensity interval training program resulted in increased eGC thickness in the sublingual microvasculature of healthy young adults and an increase in circulating microRNAs associated with eGC thickness [100]. Theoretically, such protection could prevent dietary sodium-induced eGC damage and subsequent endothelial dysfunction. A recent modelling study suggests that greater blood flow, like that occurring during exercise, reduces the number of sodium ions that bind to the eGC, thus allowing the eGC to maintain its sodium buffering capacity and mitigate the effect of sodium on endothelial function [101]. As yet, however, the potential for aerobic exercise to protect the eGC from high-sodium diets has not been experimentally tested in humans.
5.2. Dietary Potassium Intake
Increased dietary intake of potassium is associated with reduced risk of cardiovascular morbidity and mortality [102]. Some of the beneficial effects of potassium may come from its positive influence on endothelial function. For example, the blunted ACh-induced dilation in stroke-prone spontaneously hypertensive rats is completely prevented by eight weeks of a high-potassium diet, despite no effect on BP [103]. Similarly, 2500 mg/day of potassium supplementation in hypertensive humans who typically consumed ~3000 mg/day increased brachial artery FMD without a clinically significant reduction in BP [104]. This effect also occurred when increased potassium (5690 vs. 3110 mg/day) was consumed via food sources [105], although this benefit to endothelial function was not observed with a more moderate increase in dietary potassium (780–1560 mg/day) in individuals who typically consume ~2300 mg/day [106].
Raij and colleagues demonstrated that the addition of 3.6% potassium citrate to a high-sodium diet (8% sodium chloride) mitigated sodium-induced endothelial dysfunction in Dahl salt-sensitive rats, independent of any effect on BP, providing support for the use of potassium to combat the vascular consequences of excess dietary sodium [107]. Sundhir and colleagues also observed a protective effect of potassium supplementation (2.1% potassium chloride or 2.1% potassium bicarbonate) against the vascular insult of sodium in Dahl rats; however, these findings appeared to be related to a direct effect of potassium on systolic BP [108]. Notably, in young, salt-resistant humans, a high-potassium diet (4700 mg/day) abolished the reduction in brachial artery FMD caused by a seven-day high-sodium diet with moderate potassium intake (2500 mg/day), despite no change in either laboratory-measured or 24-hr ambulatory BP [38]. Furthermore, added dietary potassium counteracts an acute sodium insult without changing BP, as the postprandial reduction in FMD following a single high-sodium meal containing only 117 mg of potassium was completely abolished when the high-sodium meal contained 1482 mg of potassium [43]. Taken together, these findings suggest that increased consumption of potassium may be a relatively simple means to protect the vascular endothelium from a high-sodium diet.
The mechanisms through which high-potassium diets may confer vascular protection against excess dietary sodium have not been fully elucidated, but it appears potassium plays a role in modulating oxidative stress. For instance, McCabe et al. demonstrated that cultured endothelial cells and monocytes produced less ROS as the potassium content of the culture media increased [109]. Likewise, rabbits fed a low-potassium diet exhibited increased O2− production and decreased endothelium-dependent dilation [110]. Furthermore, high-potassium feeding in Dahl salt-sensitive rats abolished high sodium induced increases in vascular O2− and NADPH oxidase mRNA expression [111]. In addition to its effects on oxidative stress, potassium appears to regulate the deformability of endothelial cells, thus influencing how they respond to shear stress. Oberleithner and colleagues demonstrated that an increase in extracellular potassium concentration softens endothelial cells and increases NO release in response to shear stress [112]; however, more research is needed to determine the precise mechanisms behind this effect.
6. Conclusions
Consumption of excess dietary sodium increases the risk for CVD-related morbidity and mortality. While the processes through which dietary sodium promotes CVD are likely multifactorial, mounting evidences suggests that endothelial dysfunction may be an early contributor to this risk, as it is regularly observed as a consequence of high-sodium diets. Importantly, sodium impairs endothelial functioning in healthy individuals who do not have salt-sensitive BP, in addition to those for whom sodium increases BP. Extensive work in cell culture and animal models has been performed to examine the mechanisms through which dietary sodium harms the endothelium. This preclinical evidence suggests roles for increased oxidative stress and damage to the endothelial glycocalyx. Translation of these mechanistic studies to humans is ongoing, and will inform public health and clinical strategies to mitigate the effects of dietary sodium.
Ideally, all adults should meet the current recommendation of 2300 mg or less of sodium per day to decrease risk of CVD. However, excess sodium consumption remains ubiquitous, despite decades of attempts at reducing sodium intake. As such, additional approaches to lessen the impact of dietary sodium consumption must be explored. Aerobic exercise and increased dietary potassium consumption, for example, each directly benefit the vascular endothelium, and may protect it in the face of a high-sodium diet. More research is needed to determine if these non-sodium focused interventions are effective at mitigating the deleterious effects of dietary sodium on the vasculature, and whether these effects translate into a decrease in the burden of CVD.
Author Contributions
Conceptualization, J.C.P., S.L.L., W.B.F., and D.G.E.; writing—original draft preparation, J.C.P.; writing—review and editing, J.C.P., S.L.L., W.B.F., and D.G.E.; funding acquisition, J.C.P., S.L.L., W.B.F., and D.G.E. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the American Heart Association, grant number 20POST35080171, and the National Institutes of Health, grant numbers P20GM113125, R01HL145055, R01HL104106.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not applicable.
Acknowledgments
All figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cogswell, M.E.; Loria, C.M.; Terry, A.L.; Zhao, L.; Wang, C.Y.; Chen, T.C.; Wright, J.D.; Pfeiffer, C.M.; Merritt, R.; Moy, C.S.; et al. Estimated 24-Hour Urinary Sodium and Potassium Excretion in US Adults. JAMA 2018, 319, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Dietary Guidelines for Americans 2020–2025, 9th ed.; U.S. Department of Agriculture and U.S. Department of Health and Human Services: Washington, DC, USA, 2020. Available online: Dietaryguidelines.gov (accessed on 5 January 2021).
- Mozaffarian, D.; Fahimi, S.; Singh, G.M.; Micha, R.; Khatibzadeh, S.; Engell, R.E.; Lim, S.; Danaei, G.; Ezzati, M.; Powles, J.; et al. Global sodium consumption and death from cardiovascular causes. N. Engl. J. Med. 2014, 371, 624–634. [Google Scholar] [CrossRef] [Green Version]
- Intersalt Cooperative Research Group. Intersalt: An International Study of Electrolyte Excretion and Blood Pressure. Results for 24 Hour Urinary Sodium and Potassium Excretion. BMJ Br. Med. J. 1988, 297, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, P.; Stamler, J.; Nichols, R.; Dyer, A.R.; Stamler, R.; Kesteloot, H.; Marmot, M. Intersalt revisited: Further analyses of 24 hour sodium excretion and blood pressure within and across populations. Intersalt Cooperative Research Group. BMJ 1996, 312, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, G.A.; Markandu, N.D.; Best, F.E.; Elder, D.M.; Cam, J.M.; Sagnella, G.A.; Squires, M. Double-blind randomised crossover trial of moderate sodium restriction in essential hypertension. Lancet 1982, 1, 351–355. [Google Scholar] [CrossRef]
- Jablonski, K.L.; Racine, M.L.; Geolfos, C.J.; Gates, P.E.; Chonchol, M.; McQueen, M.B.; Seals, D.R. Dietary sodium restriction reverses vascular endothelial dysfunction in middle-aged/older adults with moderately elevated systolic blood pressure. J. Am. Coll. Cardiol. 2013, 61, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, M.H.; Miller, J.Z.; Luft, F.C.; Grim, C.E.; Fineberg, N.S. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension 1986, 8, II127. [Google Scholar] [CrossRef] [Green Version]
- Overlack, A.; Ruppert, M.; Kolloch, R.; Kraft, K.; Stumpe, K.O. Age is a major determinant of the divergent blood pressure responses to varying salt intake in essential hypertension. Am. J. Hypertens. 1995, 8, 829–836. [Google Scholar] [CrossRef]
- Tuomilehto, J.; Jousilahti, P.; Rastenyte, D.; Moltchanov, V.; Tanskanen, A.; Pietinen, P.; Nissinen, A. Urinary sodium excretion and cardiovascular mortality in Finland: A prospective study. Lancet 2001, 357, 848–851. [Google Scholar] [CrossRef]
- He, J.; Ogden, L.G.; Bazzano, L.A.; Vupputuri, S.; Loria, C.; Whelton, P.K. Dietary sodium intake and incidence of congestive heart failure in overweight US men and women: First National Health and Nutrition Examination Survey Epidemiologic Follow-up Study. Arch. Intern. Med. 2002, 162, 1619–1624. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, C.J.; Bibbins-Domingo, K.; Jin, Z.; Daviglus, M.L.; Goff, D.C., Jr.; Jacobs, D.R., Jr. Association of sodium and potassium intake with left ventricular mass: Coronary artery risk development in young adults. Hypertension 2011, 58, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Kuznetsova, T.; Maillard, M.; Richart, T.; Thijs, L.; Bochud, M.; Herregods, M.C.; Burnier, M.; Fagard, R.; Staessen, J.A. Independent relations of left ventricular structure with the 24-hour urinary excretion of sodium and aldosterone. Hypertension 2009, 54, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, L.A.; de Simone, G.; Pasanisi, F.; Mancini, M.; Mancini, M. Left ventricular mass reduction during salt depletion in arterial hypertension. Hypertension 1984, 6, 755–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, T.J.; Charbonneau, F.; Buithieu, J.; Rose, M.S.; Conradson, H.; Hildebrand, K.; Fung, M.; Verma, S.; Lonn, E.M. Microvascular Function Predicts Cardiovascular Events in Primary Prevention Long-Term Results From the Firefighters and Their Endothelium (FATE) Study. Circulation 2011, 123, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokce, N.; Keaney, J.F., Jr.; Hunter, L.M.; Watkins, M.T.; Menzoian, J.O.; Vita, J.A. Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function: A prospective study. Circulation 2002, 105, 1567–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perticone, F.; Ceravolo, R.; Pujia, A.; Ventura, G.; Iacopino, S.; Scozzafava, A.; Ferraro, A.; Chello, M.; Mastroroberto, P.; Verdecchia, P. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation 2001, 104, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F., Jr.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Fulton, D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front. Physiol. 2013, 4, 347. [Google Scholar] [CrossRef] [Green Version]
- Ignarro, L.J. Biological actions and properties of endothelium-derived nitric oxide formed and released from artery and vein. Circ. Res. 1989, 65, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Thijssen, D.H.J.; Black, M.A.; Pyke, K.E.; Padilla, J.; Atkinson, G.; Harris, R.A.; Parker, B.; Widlansky, M.E.; Tschakovsky, M.E.; Green, D.J. Assessment of flow-mediated dilation in humans: A methodological and physiological guideline. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2–H12. [Google Scholar] [CrossRef] [Green Version]
- Inaba, Y.; Chen, J.A.; Bergmann, S.R. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: A meta-analysis. Int. J. Cardiovasc. Imaging 2010, 26, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Takase, B.; Hamabe, A.; Satomura, K.; Akima, T.; Uehata, A.; Matsui, T.; Ohsuzu, F.; Ishihara, M.; Kurita, A. Comparable Prognostic Value of Vasodilator Response to Acetylcholine in Brachial and Coronary Arteries for Predicting Long-Term Cardiovascular Events in Suspected Coronary Artery Disease. Circ. J. 2006, 70, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, L.; Berglund, L.; Larsson, A.; Sundstrom, J. Endothelial function in resistance and conduit arteries and 5-year risk of cardiovascular disease. Circulation 2011, 123, 1545–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holowatz, L.A.; Thompson-Torgerson, C.S.; Kenney, W.L. The human cutaneous circulation as a model of generalized microvascular function. J. Appl. Physiol. 2008, 105, 370–372. [Google Scholar] [CrossRef] [Green Version]
- Greaney, J.L.; DuPont, J.J.; Lennon-Edwards, S.L.; Sanders, P.W.; Edwards, D.G.; Farquhar, W.B. Dietary sodium loading impairs microvascular function independent of blood pressure in humans: Role of oxidative stress. J. Physiol. 2012, 590, 5519–5528. [Google Scholar] [CrossRef]
- Alba, B.K.; Stanhewicz, A.E.; Dey, P.; Bruno, R.S.; Kenney, W.L.; Alexander, L.M. Controlled Feeding of an 8-d, High-Dairy Cheese Diet Prevents Sodium-Induced Endothelial Dysfunction in the Cutaneous Microcirculation of Healthy, Older Adults through Reductions in Superoxide. J. Nutr. 2020, 150, 55–63. [Google Scholar] [CrossRef]
- Barić, L.; Drenjančević, I.; Mihalj, M.; Matić, A.; Stupin, M.; Kolar, L.; Mihaljević, Z.; Mrakovčić-Šutić, I.; Šerić, V.; Stupin, A. Enhanced Antioxidative Defense by Vitamins C and E Consumption Prevents 7-Day High-Salt Diet-Induced Microvascular Endothelial Function Impairment in Young Healthy Individuals. J. Clin. Med. 2020, 9, 843. [Google Scholar] [CrossRef] [Green Version]
- Baric, L.; Drenjancevic, I.; Matic, A.; Stupin, M.; Kolar, L.; Mihaljevic, Z.; Lenasi, H.; Seric, V.; Stupin, A. Seven-Day Salt Loading Impairs Microvascular Endothelium-Dependent Vasodilation without Changes in Blood Pressure, Body Composition and Fluid Status in Healthy Young Humans. Kidney Blood Press. Res. 2019, 44, 835–847. [Google Scholar] [CrossRef] [Green Version]
- Cavka, A.; Jukic, I.; Ali, M.; Goslawski, M.; Bian, J.T.; Wang, E.; Drenjancevic, I.; Phillips, S.A. Short-term high salt intake reduces brachial artery and microvascular function in the absence of changes in blood pressure. J. Hypertens. 2016, 34, 676–684. [Google Scholar] [CrossRef]
- Wong, B.J.; Wilkins, B.W.; Holowatz, L.A.; Minson, C.T. Nitric oxide synthase inhibition does not alter the reactive hyperemic response in the cutaneous circulation. J. Appl. Physiol. 2003, 95, 504–510. [Google Scholar] [CrossRef] [Green Version]
- Lüscher, T.; Raij, L.; Vanhoutte, P.M. Endothelium-dependent vascular responses in normotensive and hypertensive Dahl rats. Hypertension 1987, 9, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.M.; Nelson, R.; Brown, M.; Wood, M.; Wood, A.J. Dietary sodium intake modulates vasodilation mediated by nitroprusside but not by methacholine in the human forearm. Hypertension 1995, 25, 1220–1223. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, A.; Suzuki, H.; Fujiwara, M.; Masai, M.; Iwasaki, T. Impairment of endothelial function in salt-sensitive hypertension in humans. Am. J. Hypertens. 1997, 10, 1083–1090. [Google Scholar] [CrossRef]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Kimura, M.; Noma, K.; Sasaki, S.; Hara, K.; Matsuura, H.; Chayama, K.; Oshima, T. Sodium chloride loading does not alter endothelium-dependent vasodilation of forearm vasculature in either salt-sensitive or salt-resistant patients with essential hypertension. Hypertens. Res. 2001, 24, 711–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzemos, N.; Lim, P.O.; Wong, S.; Struthers, A.D.; MacDonald, T.M. Adverse cardiovascular effects of acute salt loading in young normotensive individuals. Hypertension 2008, 51, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuPont, J.J.; Greaney, J.L.; Wenner, M.M.; Lennon-Edwards, S.L.; Sanders, P.W.; Farquhar, W.B.; Edwards, D.G. High dietary sodium intake impairs endothelium-dependent dilation in healthy salt-resistant humans. J. Hypertens. 2013, 31, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Smiljanec, K.; Mbakwe, A.; Ramos Gonzalez, M.; Farquhar, W.B.; Lennon, S.L. Dietary Potassium Attenuates the Effects of Dietary Sodium on Vascular Function in Salt-Resistant Adults. Nutrients 2020, 12, 1206. [Google Scholar] [CrossRef]
- Lennon-Edwards, S.; Ramick, M.G.; Matthews, E.L.; Brian, M.S.; Farquhar, W.B.; Edwards, D.G. Salt loading has a more deleterious effect on flow-mediated dilation in salt-resistant men than women. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, K.M.; Clifton, P.M.; Keogh, J.B. Endothelial function is impaired after a high-salt meal in healthy subjects. Am. J. Clin. Nutr. 2011, 93, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Migdal, K.U.; Robinson, A.T.; Watso, J.C.; Babcock, M.C.; Lennon, S.L.; Martens, C.R.; Serrador, J.M.; Farquhar, W.B. A high salt meal does not impair cerebrovascular reactivity in healthy young adults. Physiol. Rep. 2020, 8, e14585. [Google Scholar] [CrossRef]
- Smiljanec, K.; Mbakwe, A.U.; Ramos-Gonzalez, M.; Pohlig, R.T.; Lennon, S.L. Antioxidant cocktail following a high-sodium meal does not affect vascular function in young, healthy adult humans: A randomized controlled crossover trial. Nutr. Res. 2020, 79, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Blanch, N.; Clifton, P.M.; Petersen, K.S.; Keogh, J.B. Effect of sodium and potassium supplementation on vascular and endothelial function: A randomized controlled trial. Am. J. Clin. Nutr. 2015, 101, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramick, M.G.; Brian, M.S.; Matthews, E.L.; Patik, J.C.; Seals, D.R.; Lennon, S.L.; Farquhar, W.B.; Edwards, D.G. Apocynin and Tempol ameliorate dietary sodium-induced declines in cutaneous microvascular function in salt-resistant humans. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H97–H103. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Harrison, D.G.; Drexler, H. Oxidant stress—A major cause of reduced endothelial nitric oxide availability in cardiovascular disease. Eur. J. Clin. Pharmacol. 2005, 62, 13–19. [Google Scholar] [CrossRef]
- Milstien, S.; Katusic, Z. Oxidation of tetrahydrobiopterin by peroxynitrite: Implications for vascular endothelial function. Biochem. Biophys. Res. Commun. 1999, 263, 681–684. [Google Scholar] [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Lenda, D.M.; Sauls, B.A.; Boegehold, M.A. Reactive oxygen species may contribute to reduced endothelium-dependent dilation in rats fed high salt. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H7–H14. [Google Scholar] [CrossRef]
- Zhu, J.; Mori, T.; Huang, T.; Lombard, J.H. Effect of high-salt diet on NO release and superoxide production in rat aorta. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H575–H583. [Google Scholar] [CrossRef]
- Kitiyakara, C.; Chabrashvili, T.; Chen, Y.; Blau, J.; Karber, A.; Aslam, S.; Welch, W.J.; Wilcox, C.S. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J. Am. Soc. Nephrol. JASN 2003, 14, 2775–2782. [Google Scholar] [CrossRef] [Green Version]
- Lenda, D.M.; Boegehold, M.A. Effect of a high-salt diet on oxidant enzyme activity in skeletal muscle microcirculation. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H395–H402. [Google Scholar] [CrossRef] [Green Version]
- Nurkiewicz, T.R.; Boegehold, M.A. High salt intake reduces endothelium-dependent dilation of mouse arterioles via superoxide anion generated from nitric oxide synthase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1550–R1556. [Google Scholar] [CrossRef] [PubMed]
- Nurkiewicz, T.R.; Wu, G.; Li, P.; Boegehold, M.A. Decreased arteriolar tetrahydrobiopterin is linked to superoxide generation from nitric oxide synthase in mice fed high salt. Microcirculation 2010, 17, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, T.; Lombard, J.H. Effect of high-salt diet on vascular relaxation and oxidative stress in mesenteric resistance arteries. J. Vasc. Res. 2007, 44, 382–390. [Google Scholar] [CrossRef]
- Guers, J.J.; Kasecky-Lardner, L.; Farquhar, W.B.; Edwards, D.G.; Lennon, S.L. Voluntary wheel running prevents salt-induced endothelial dysfunction: Role of oxidative stress. J. Appl. Physiol. 2019, 126, 502–510. [Google Scholar] [CrossRef]
- Lenda, D.M.; Boegehold, M.A. Effect of a high salt diet on microvascular antioxidant enzymes. J. Vasc. Res. 2002, 39, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Durand, M.J.; Lombard, J.H. Low-dose angiotensin II infusion restores vascular function in cerebral arteries of high salt-fed rats by increasing copper/zinc superoxide dimutase expression. Am. J. Hypertens. 2013, 26, 739–747. [Google Scholar] [CrossRef] [PubMed]
- McEwen, S.T.; Schmidt, J.R.; Somberg, L.; Cruz Lde, L.; Lombard, J.H. Time-course and mechanisms of restored vascular relaxation by reduced salt intake and angiotensin II infusion in rats fed a high-salt diet. Microcirculation 2009, 16, 220–234. [Google Scholar] [CrossRef]
- Raffai, G.; Durand, M.J.; Lombard, J.H. Acute and chronic angiotensin-(1-7) restores vasodilation and reduces oxidative stress in mesenteric arteries of salt-fed rats. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1341–H1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberleithner, H.; Riethmuller, C.; Schillers, H.; MacGregor, G.A.; de Wardener, H.E.; Hausberg, M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc. Natl. Acad. Sci. USA 2007, 104, 16281–16286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fels, J.; Callies, C.; Kusche-Vihrog, K.; Oberleithner, H. Nitric oxide release follows endothelial nanomechanics and not vice versa. Pflug. Arch. 2010, 460, 915–923. [Google Scholar] [CrossRef]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.M.J.; oude Egbrink, M.G.A. The endothelial glycocalyx: Composition, functions, and visualization. Pflüg. Arch. Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machin, D.R.; Bloom, S.I.; Campbell, R.A.; Phuong, T.T.T.; Gates, P.E.; Lesniewski, L.A.; Rondina, M.T.; Donato, A.J. Advanced age results in a diminished endothelial glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H531–H539. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Voumvourakis, A.; Makavos, G.; Triantafyllidi, H.; Pavlidis, G.; Katogiannis, K.; Benas, D.; Vlastos, D.; Trivilou, P.; Varoudi, M.; et al. Association of impaired endothelial glycocalyx with arterial stiffness, coronary microcirculatory dysfunction, and abnormal myocardial deformation in untreated hypertensives. J. Clin. Hypertens. (Greenwich) 2018, 20, 672–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwdorp, M.; Mooij, H.L.; Kroon, J.; Atasever, B.; Spaan, J.A.; Ince, C.; Holleman, F.; Diamant, M.; Heine, R.J.; Hoekstra, J.B.; et al. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes 2006, 55, 1127–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekhuizen, L.N.; Lemkes, B.A.; Mooij, H.L.; Meuwese, M.C.; Verberne, H.; Holleman, F.; Schlingemann, R.O.; Nieuwdorp, M.; Stroes, E.S.; Vink, H. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 2010, 53, 2646–2655. [Google Scholar] [CrossRef] [Green Version]
- Fancher, I.S.; Le Master, E.; Ahn, S.J.; Adamos, C.; Lee, J.C.; Berdyshev, E.; Dull, R.O.; Phillips, S.A.; Levitan, I. Impairment of Flow-Sensitive Inwardly Rectifying K(+) Channels via Disruption of Glycocalyx Mediates Obesity-Induced Endothelial Dysfunction. Arter. Thromb. Vasc. Biol. 2020, 40, e240–e255. [Google Scholar] [CrossRef]
- Lipowsky, H.H.; Lescanic, A. The effect of doxycycline on shedding of the glycocalyx due to reactive oxygen species. Microvasc. Res. 2013, 90, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Oberleithner, H.; Peters, W.; Kusche-Vihrog, K.; Korte, S.; Schillers, H.; Kliche, K.; Oberleithner, K. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflug. Arch. 2011, 462, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Korte, S.; Wiesinger, A.; Straeter, A.S.; Peters, W.; Oberleithner, H.; Kusche-Vihrog, K. Firewall function of the endothelial glycocalyx in the regulation of sodium homeostasis. Pflug. Arch. 2012, 463, 269–278. [Google Scholar] [CrossRef]
- Schierke, F.; Wyrwoll, M.J.; Wisdorf, M.; Niedzielski, L.; Maase, M.; Ruck, T.; Meuth, S.G.; Kusche-Vihrog, K. Nanomechanics of the endothelial glycocalyx contribute to Na+-induced vascular inflammation. Sci. Rep. 2017, 7, 46476. [Google Scholar] [CrossRef]
- He, F.J.; Tan, M.; Ma, Y.; MacGregor, G.A. Salt Reduction to Prevent Hypertension and Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Karppanen, H.; Mervaala, E. Sodium intake and hypertension. Prog. Cardiovasc. Dis. 2006, 49, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, A.M.; Willett, W.C. Trends in 24-h urinary sodium excretion in the United States, 1957–2003: A systematic review. Am. J. Clin. Nutr. 2010, 92, 1172–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newson, R.S.; Elmadfa, I.; Biro, G.; Cheng, Y.; Prakash, V.; Rust, P.; Barna, M.; Lion, R.; Meijer, G.W.; Neufingerl, N.; et al. Barriers for progress in salt reduction in the general population. An international study. Appetite 2013, 71, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maessen, M.F.; Verbeek, A.L.; Bakker, E.A.; Thompson, P.D.; Hopman, M.T.; Eijsvogels, T.M. Lifelong Exercise Patterns and Cardiovascular Health. Mayo Clin. Proc. 2016, 91, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Hamer, M.; O’Donovan, G.; Stamatakis, E. Association between physical activity and sub-types of cardiovascular disease death causes in a general population cohort. Eur. J. Epidemiol. 2019, 34, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Mora, S.; Cook, N.; Buring, J.E.; Ridker, P.M.; Lee, I.M. Physical activity and reduced risk of cardiovascular events: Potential mediating mechanisms. Circulation 2007, 116, 2110–2118. [Google Scholar] [CrossRef] [Green Version]
- Green, D.J.; O’Driscoll, G.; Joyner, M.J.; Cable, N.T. Exercise and cardiovascular risk reduction: Time to update the rationale for exercise? J. Appl. Physiol. 2008, 105, 766–768. [Google Scholar] [CrossRef] [Green Version]
- Edwards, D.G.; Schofield, R.S.; Lennon, S.L.; Pierce, G.L.; Nichols, W.W.; Braith, R.W. Effect of exercise training on endothelial function in men with coronary artery disease. Am. J. Cardiol. 2004, 93, 617–620. [Google Scholar] [CrossRef]
- Green, D.J.; Walsh, J.H.; Maiorana, A.; Best, M.J.; Taylor, R.R.; O’Driscoll, J.G. Exercise-induced improvement in endothelial dysfunction is not mediated by changes in CV risk factors: Pooled analysis of diverse patient populations. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2679–H2687. [Google Scholar] [CrossRef] [Green Version]
- Clarkson, P.; Montgomery, H.E.; Mullen, M.J.; Donald, A.E.; Powe, A.J.; Bull, T.; Jubb, M.; World, M.; Deanfield, J.E. Exercise training enhances endothelial function in young men. J. Am. Coll. Cardiol. 1999, 33, 1379–1385. [Google Scholar] [CrossRef] [Green Version]
- Hodges, G.J.; Sharp, L.; Stephenson, C.; Patwala, A.Y.; George, K.P.; Goldspink, D.F.; Tim Cable, N. The effect of 48 weeks of aerobic exercise training on cutaneous vasodilator function in post-menopausal females. Eur. J. Appl. Physiol. 2010, 108, 1259–1267. [Google Scholar] [CrossRef]
- Thijssen, D.H.; Hopman, M.T. Counterpoint: Exercise training does not induce vascular adaptations beyond the active muscle beds. J. Appl. Physiol. 2008, 105, 1004–1006; discussion 1006–1007. [Google Scholar] [CrossRef] [PubMed]
- Rebholz, C.M.; Gu, D.; Chen, J.; Huang, J.F.; Cao, J.; Chen, J.C.; Li, J.; Lu, F.; Mu, J.; Ma, J.; et al. Physical activity reduces salt sensitivity of blood pressure: The Genetic Epidemiology Network of Salt Sensitivity Study. Am. J. Epidemiol. 2012, 176 (Suppl. 7), S106–S113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padilla, J.; Simmons, G.H.; Bender, S.B.; Arce-Esquivel, A.A.; Whyte, J.J.; Laughlin, M.H. Vascular effects of exercise: Endothelial adaptations beyond active muscle beds. Physiology 2011, 26, 132–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staessen, J.; Fagard, R.; Hespel, P.; Lijnen, P.; Vanhees, L.; Amery, A. Plasma renin system during exercise in normal men. J. Appl. Physiol. 1987, 63, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Priestley, J.R.; Buelow, M.W.; McEwen, S.T.; Weinberg, B.D.; Delaney, M.; Balus, S.F.; Hoeppner, C.; Dondlinger, L.; Lombard, J.H. Reduced angiotensin II levels cause generalized vascular dysfunction via oxidant stress in hamster cheek pouch arterioles. Microvasc. Res. 2013, 89, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Drenjancevic-Peric, I.; McEwen, S.; Friesema, J.; Schulta, D.; Yu, M.; Roman, R.J.; Lombard, J.H. Role of superoxide and angiotensin II suppression in salt-induced changes in endothelial Ca2+ signaling and NO production in rat aorta. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H929–H938. [Google Scholar] [CrossRef] [Green Version]
- van Kats, J.P.; de Lannoy, L.M.; Jan Danser, A.H.; van Meegen, J.R.; Verdouw, P.D.; Schalekamp, M.A. Angiotensin II type 1 (AT1) receptor-mediated accumulation of angiotensin II in tissues and its intracellular half-life in vivo. Hypertension 1997, 30, 42–49. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuland, D.J.; McCord, J.M.; Hamilton, K.L. The role of Nrf2 in the attenuation of cardiovascular disease. Exerc. Sport Sci. Rev. 2013, 41, 162–168. [Google Scholar] [CrossRef]
- Done, A.J.; Traustadottir, T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016, 10, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Velmurugan, K.; Alam, J.; McCord, J.M.; Pugazhenthi, S. Synergistic induction of heme oxygenase-1 by the components of the antioxidant supplement Protandim. Free Radic. Biol. Med. 2009, 46, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.K.; Bose, S.K.; Grunwald, G.K.; Myhill, P.; McCord, J.M. The induction of human superoxide dismutase and catalase in vivo: A fundamentally new approach to antioxidant therapy. Free Radic. Biol. Med. 2006, 40, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Priestley, J.R.; Fink, K.E.; McCord, J.M.; Lombard, J.H. NRF 2 activation with Protandim attenuates salt-induced vascular dysfunction and microvascular rarefaction. Microcirculation 2019, 26, e12575. [Google Scholar] [CrossRef]
- Gouverneur, M.; Spaan, J.A.; Pannekoek, H.; Fontijn, R.D.; Vink, H. Fluid shear stress stimulates incorporation of hyaluronan into endothelial cell glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H458-H452. [Google Scholar] [CrossRef] [Green Version]
- Majerczak, J.; Grandys, M.; Duda, K.; Zakrzewska, A.; Balcerczyk, A.; Kolodziejski, L.; Szymoniak-Chochol, D.; Smolenski, R.T.; Bartosz, G.; Chlopicki, S.; et al. Moderate-intensity endurance training improves endothelial glycocalyx layer integrity in healthy young men. Exp. Physiol. 2017, 102, 70–85. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, B.; Niehues, H.; Lenders, M.; Thorwesten, L.; Klose, A.; Kruger, M.; Brand, E.; Brand, S.M. Effects of high-intensity interval training on microvascular glycocalyx and associated microRNAs. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1538–H1551. [Google Scholar] [CrossRef]
- Jiang, X.Z.; Luo, K.H.; Ventikos, Y. Reducing Salt Intake and Exercising Regularly: Implications from Molecular Dynamics Simulations of Endothelial Glycocalyx. Front. Physiol. 2018, 9, 1667. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.; Mente, A.; Rangarajan, S.; McQueen, M.J.; Wang, X.; Liu, L.; Yan, H.; Lee, S.F.; Mony, P.; Devanath, A.; et al. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N. Engl. J. Med. 2014, 371, 612–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, T.; Tobian, L.; Ganguli, M.C. High potassium diets protect against dysfunction of endothelial cells in stroke-prone spontaneously hypertensive rats. Hypertension 1988, 11, 579–585. [Google Scholar] [CrossRef] [Green Version]
- He, F.J.; Marciniak, M.; Carney, C.; Markandu, N.D.; Anand, V.; Fraser, W.D.; Dalton, R.N.; Kaski, J.C.; MacGregor, G.A. Effects of potassium chloride and potassium bicarbonate on endothelial function, cardiovascular risk factors, and bone turnover in mild hypertensives. Hypertension 2010, 55, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Blanch, N.; Clifton, P.M.; Petersen, K.S.; Willoughby, S.R.; Keogh, J.B. Effect of high potassium diet on endothelial function. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Berry, S.E.; Mulla, U.Z.; Chowienczyk, P.J.; Sanders, T.A. Increased potassium intake from fruit and vegetables or supplements does not lower blood pressure or improve vascular function in UK men and women with early hypertension: A randomised controlled trial. Br. J. Nutr. 2010, 104, 1839–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raij, L.; Luscher, T.F.; Vanhoutte, P.M. High potassium diet augments endothelium-dependent relaxations in the Dahl rat. Hypertension 1988, 12, 562–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhir, K.; Kurtz, T.W.; Yock, P.G.; Connolly, A.J.; Morris, R.C., Jr. Potassium preserves endothelial function and enhances aortic compliance in Dahl rats. Hypertension 1993, 22, 315–322. [Google Scholar] [CrossRef] [Green Version]
- McCabe, R.D.; Bakarich, M.A.; Srivastava, K.; Young, D.B. Potassium inhibits free radical formation. Hypertension 1994, 24, 77–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.C.; Li, D.Y.; Weng, Y.F.; Lynch, J.; Wingo, C.S.; Mehta, J.L. Increased superoxide anion generation and altered vasoreactivity in rabbits on low-potassium diet. Am. J. Physiol. 1998, 274, H1955–H1961. [Google Scholar] [CrossRef]
- Kido, M.; Ando, K.; Onozato, M.L.; Tojo, A.; Yoshikawa, M.; Ogita, T.; Fujita, T. Protective effect of dietary potassium against vascular injury in salt-sensitive hypertension. Hypertension 2008, 51, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Oberleithner, H.; Callies, C.; Kusche-Vihrog, K.; Schillers, H.; Shahin, V.; Riethmuller, C.; Macgregor, G.A.; de Wardener, H.E. Potassium softens vascular endothelium and increases nitric oxide release. Proc. Natl. Acad. Sci. USA 2009, 106, 2829–2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Excess dietary sodium negatively (−) influences nitric oxide (NO)-mediated endothelial function via increases in oxidative stress, an increase in endothelial cell (EC) stiffness, and damage to the endothelial glycocalyx (eGC). Each of these factors results in decreased bioavailability of NO derived from endothelial nitric oxide synthase (eNOS). Evidence suggests that aerobic exercise and/or increased intake of dietary potassium positively (+) influence these factors and thus may be effective strategies to counteract the impact of excess dietary sodium.
Figure 1.
Excess dietary sodium negatively (−) influences nitric oxide (NO)-mediated endothelial function via increases in oxidative stress, an increase in endothelial cell (EC) stiffness, and damage to the endothelial glycocalyx (eGC). Each of these factors results in decreased bioavailability of NO derived from endothelial nitric oxide synthase (eNOS). Evidence suggests that aerobic exercise and/or increased intake of dietary potassium positively (+) influence these factors and thus may be effective strategies to counteract the impact of excess dietary sodium.

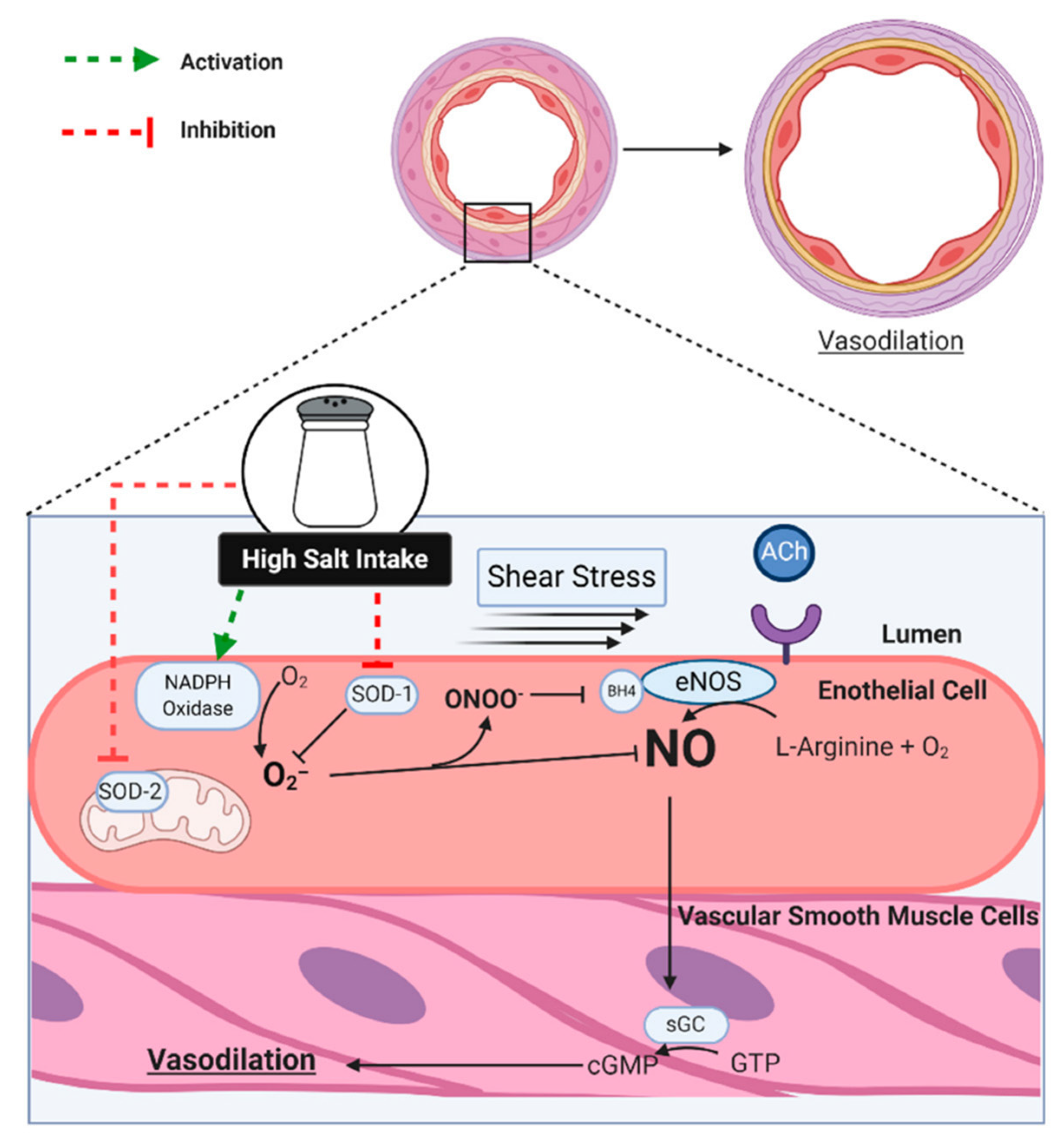
Figure 2.
In response to various physiological and pharmacological stimuli, such as shear stress and acetylcholine (ACh), the endothelium produces nitric oxide (NO) via endothelial nitric oxide synthase (eNOS) that diffuses into the vascular smooth muscle cells, where it activates soluble guanylate cyclase (sGC), which then converts guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP), ultimately leading to vasodilation. Excess dietary sodium increases superoxide (O2−) via activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, while also inhibiting cytosolic superoxide dismutase (SOD-1) and mitochondrial SOD (SOD-2). NO readily reacts with O2−, thus rendering it unable to diffuse into the VSM. The resulting peroxynitrite (ONOO−) oxidizes tetrahydrobiopterin (BH4), which leads to uncoupling of eNOS, leading to further reductions in NO and increases in O2− (not shown).
Figure 2.
In response to various physiological and pharmacological stimuli, such as shear stress and acetylcholine (ACh), the endothelium produces nitric oxide (NO) via endothelial nitric oxide synthase (eNOS) that diffuses into the vascular smooth muscle cells, where it activates soluble guanylate cyclase (sGC), which then converts guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP), ultimately leading to vasodilation. Excess dietary sodium increases superoxide (O2−) via activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, while also inhibiting cytosolic superoxide dismutase (SOD-1) and mitochondrial SOD (SOD-2). NO readily reacts with O2−, thus rendering it unable to diffuse into the VSM. The resulting peroxynitrite (ONOO−) oxidizes tetrahydrobiopterin (BH4), which leads to uncoupling of eNOS, leading to further reductions in NO and increases in O2− (not shown).

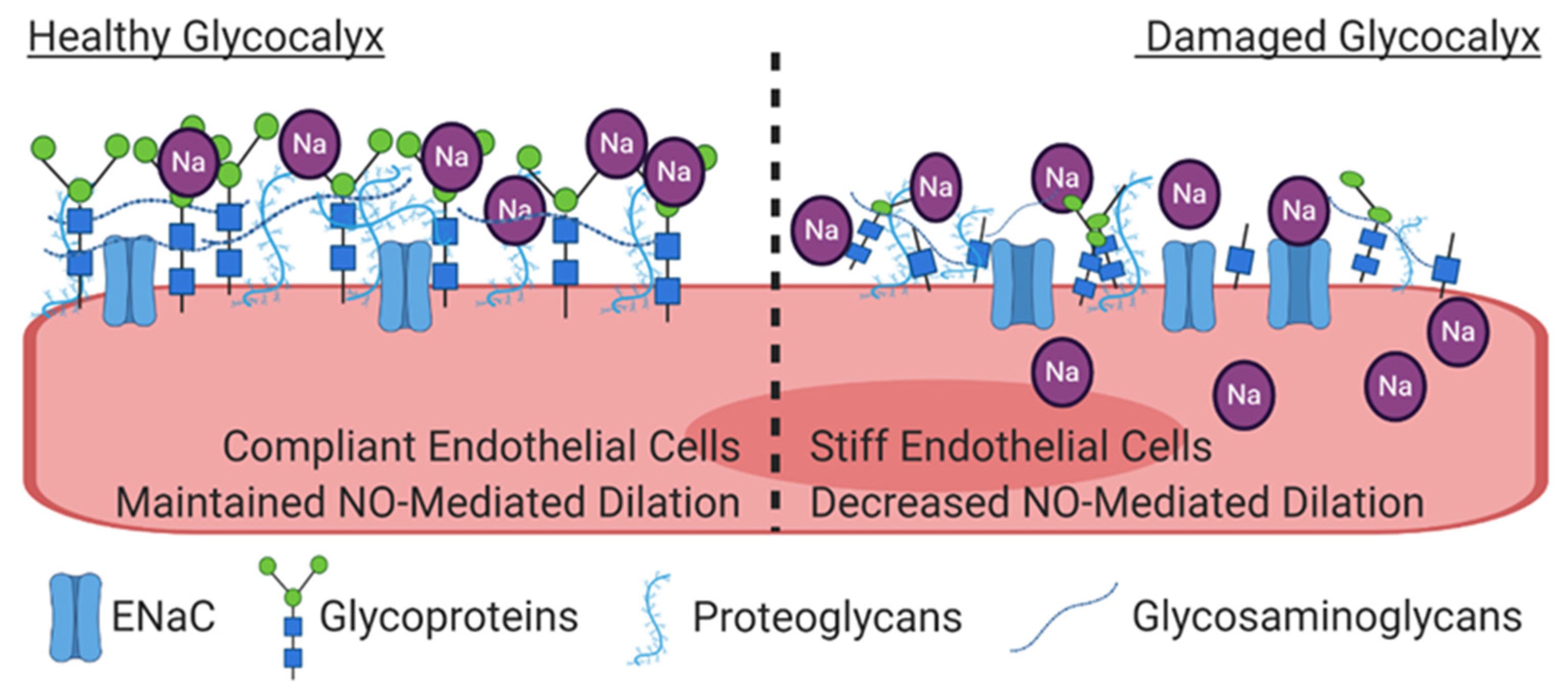
Figure 3.
On the left, the healthy glycocalyx functions as a barrier between the plasma and the endothelial cell. The negatively charged glycocalyx buffers the positively charged sodium ions. On the right, increases in plasma sodium cause damage to the glycocalyx, thus increasing the access of sodium to epithelial sodium channels (ENaC) on the surface of the endothelium, leading to increased intracellular sodium concentrations. Under these conditions, the cell becomes stiffer, and the transduction of shear stress into NO release is decreased.
Figure 3.
On the left, the healthy glycocalyx functions as a barrier between the plasma and the endothelial cell. The negatively charged glycocalyx buffers the positively charged sodium ions. On the right, increases in plasma sodium cause damage to the glycocalyx, thus increasing the access of sodium to epithelial sodium channels (ENaC) on the surface of the endothelium, leading to increased intracellular sodium concentrations. Under these conditions, the cell becomes stiffer, and the transduction of shear stress into NO release is decreased.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary table of studies that have investigated the effects of dietary sodium on endothelial function.
Table 1.
Summary table of studies that have investigated the effects of dietary sodium on endothelial function.
| Sodium Intervention | ||||||
|---|---|---|---|---|---|---|
| Study | Subject Characteristics | LS Content | HS Content | Duration | Endothelial Assessment | Relevant Findings |
| Alba et al., 2020 [27] | 65 ± 7 yr n = 5 M/6 W Pre-Hypertensive Salt Resistant BP | 1500 mg/day | 5500 mg/day | Eight days | Conduit artery: FMD Microvascular: Cutaneous ACh microdialysis | HS impaired ACh-induced cutaneous dilation during a low dairy diet compared to a high dairy diet. ACh-induced vasodialtion was restored with infusion of ascorbic acid, apocynin, and Tempol. There was no effect of HS on FMD. |
| Baric et al., 2019 [29] | 21 ± 2 yr n= 23M/25W Normotensive Salt Resistant BP | <1380 mg/day | ~5800 mg/day (4610 mg added to normal diet) | Seven days | Microvascular: Cutaneous ACh iontophoresis | HS blunted ACh-induced cutaneous dilation. |
| Baric et al., 2020 [28] | 20 ± 2 yr n = 25 M/26 W Normotensive Salt Resistant BP | <1380 mg/day | ~5800 mg/day (4610 mg added to normal diet) | Seven Days | Microvascular: Cutaneous ACh iontophoresis | ACh-induced dilation was reduced by HS compared to LS in the control group, but this decrement did not occur in the group that also supplemented with vitamin C and E. |
| Blanch et al., 2015 [43] | 37 ± 15 yr n = 21 M/18 W Normotensive | 138 mg | 1495 mg | Single Meal | Conduit Artery: FMD | FMD was reduced following the HS + low potassium meal, but not after the HS + high-potassium condition. |
| Cavka et al., 2016 [30] | 28 ± 7 yr n = 12 W Normotensive | N/A | ~4300 mg/day (2364 mg added to normal diet) | Seven days | Conduit Artery: FMD Microvascular: Ex-vivo Gluteal Fat Arteriole ACh and FMD | HS diet reduced brachial artery FMD from baseline. ACh- and flow-induced dilation of gluteal fat arterioles was maintained during HS diet. |
| Dickinson et al., 2011 [40] | 37 ± 18 yr n = 6 M/10 W Normotensive | 115 mg | 1495 mg | Single Meal | Conduit Artery: FMD | FMD decreased for at least 120 min after both LS and HS meals. The reduction in FMD in the first hour after the meal was augmented with HS. Reactive hyperemia index was unchanged following either meal. |
| DuPont et al., 2013 [37] | 33 ± 7 yr n = 9 M/5 W Normotensive Salt Resistant BP | 460 mg/day | ~7500 mg/day | Seven days | Conduit artery: FMD | Brachial artery FMD was significantly reduced following HS. |
| Greaney et al., 2012 [26] | 31 ± 7 yr n = 5 M/7 W Normotensive Salt Resistant BP | 460 mg/day | ~7500 mg/day | Seven days | Microvascular: Cutaneous local heating | Local heating-induced cutaneous dilation was reduced following HS. Local ascorbic acid treatment augmented dilation in the HS condition. |
| Higashi et al., 2001 [35] | 52 ± 16 yr n = 17 M/12 W Hypertensive Salt-sensitive and Salt Resistant BP | 1150 mg/day | 7820 mg/day | Seven days | Microvascular: Forearm blood flow w/ACh | ACh-induced dilation was not unaffected by dietary sodium in participants with both salt-sensitive and salt resistant BP. |
| Jablonski et al., 2013 [7] | 62 ± 7 yr n = 11 M/6 W Prehypertensive or Stage 1 hypertension | ~1300 mg/day | ~3100 mg/day (habitual intake) | Four weeks | Conduit Artery: FMD Microvascular: Forearm blood flow w/ACh | Compared to normal sodium intake (HS), LS improved both FMD and forearm blood flow responses to ACh. FMD was improved in HS with ascorbic acid and BH4 treatment. Ascorbic acid also restored the microvascular response to ACh. Sodium-induced changes in BP were accounted for statistically. |
| Lennon-Edwards et al., 2014 [39] | 30 ± 8 yr n = 16 M/14 W Normotensive, Salt Resistant | 460 mg/day | ~7500 mg/day | Seven days | Conduit Artery: FMD | FMD was decreased by HS in both men and women, but the decrement was greater in men. |
| Migdal et al., 2020 [41] | 25 ± 5 yr, n = 17 M/20 W Normotensive | 138 mg | 1495 mg | Single Meal | Conduit Artery: FMD | FMD, assessed 50 min postprandial, was unchanged after either meal. |
| Miyoshi et al., 1997 [34] | 46 ± 12 yr n = 15 M Hypertensive Salt-sensitive and Salt Resistant BP | 1970 mg/day | 7880 mg/day | Seven days | Microvascular: Forearm blood flow w/ACh | ACh-induced dilation was blunted in subjects with salt-sensitive BP versus salt resistant subjects. There was no effect of HS on ACh responses in the salt resistant group, but an increase in ACh-induced dilation occurred in the salt-sensitive group during HS. |
| Ramick et al., 2019 [44] | 34 ± 11 yr n = 18 M/11 F Normotensive Salt Resistant BP | 460 mg/day | 6900 mg/day | Seven days | Microvascular: Cutaneous local heating | Local heating-induced dilation was impaired in the HS condition compared to LS. Local infusion of ascorbic acid, tempol, and apocynin restored microvasculature function during HS. |
| Smiljanec et al., 2020 [38] | 27 ± 6 yr n = 16 M/17 W Normotensive Salt Resistant BP | 1150 mg/day | 6900 mg/day | Seven days | Conduit Artery: FMD | When HS was accompanied by moderate intake of potassium, FMD was reduced relative to the LS condition. High-potassium intake abolished the effects of HS on FMD. |
| Smiljanec et al., 2020 [42] | 24 ± 6 yr n = 20 M/21 W Normotensive | N/A | 1445 mg | Single Meal | Conduit Artery: FMD | FMD was unchanged from baseline at 60 and 120 min post-meal in both the oral antioxidant cocktail and placebo conditions. |
| Stein et al., 1995 [33] | 34 ± 7 yr n = 7 M Normotensive Salt Resistant BP | 230 mg/day | 5750 mg/day | Five days | Microvascular: Forearm blood flow w/MCh | MCh-induced dilation was similar in HS and LS. |
| Tzemos, et al., 2008 [36] | 25 ± 8 yr n = 16 M Normotensive | <920 mg/day | ~5500 mg/day (4620 mg added to LS) | Five days | Microvascular: Forearm blood flow w/ACh | Forearm blood flow responses to ACh were blunted in the HS condition relative to LS. Notably, systolic BP was increased in HS, suggesting that some subjects had salt-sensitive BP. |
Subject age is presented as mean ± standard deviation (SD) (calculated from published n and standard error where necessary). ACh: acetylcholine, BP: blood pressure, FMD: flow-mediated dilation, HS: high sodium intervention, LS: low sodium intervention, M: men, MCh: methacholine, W: women.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Patik, J.C.; Lennon, S.L.; Farquhar, W.B.; Edwards, D.G. Mechanisms of Dietary Sodium-Induced Impairments in Endothelial Function and Potential Countermeasures. Nutrients 2021, 13, 270. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13010270
AMA Style
Patik JC, Lennon SL, Farquhar WB, Edwards DG. Mechanisms of Dietary Sodium-Induced Impairments in Endothelial Function and Potential Countermeasures. Nutrients. 2021; 13(1):270. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13010270
Chicago/Turabian StylePatik, Jordan C., Shannon L. Lennon, William B. Farquhar, and David G. Edwards. 2021. "Mechanisms of Dietary Sodium-Induced Impairments in Endothelial Function and Potential Countermeasures" Nutrients 13, no. 1: 270. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13010270
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.