
Examining the Interaction of the Gut Microbiome with Host Metabolism and Cardiometabolic Health in Metabolic Syndrome

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
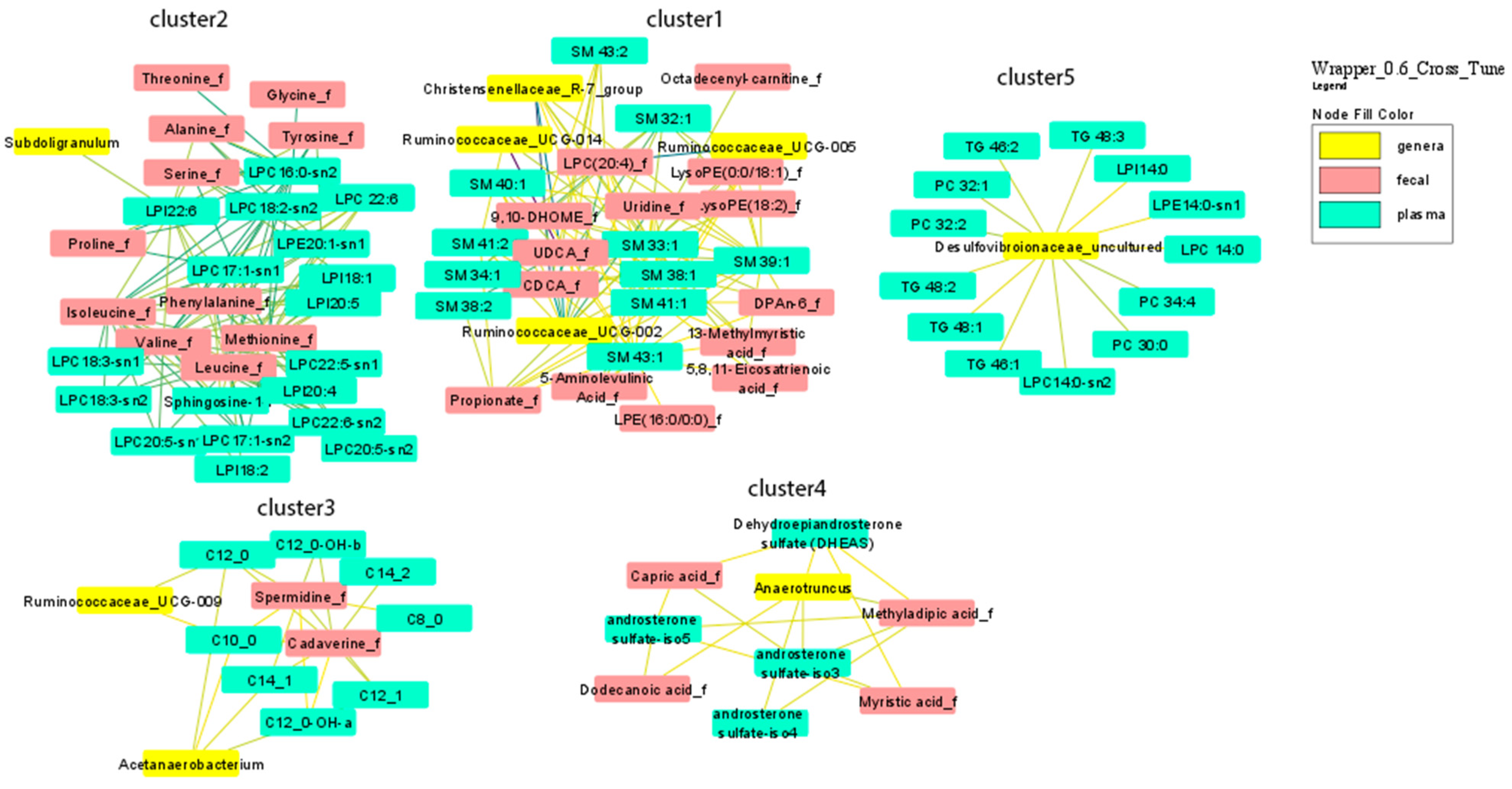
3.1. Multi-Omics Network of Correlations between Gut Microbiota, Fecal and Plasma Metabolites
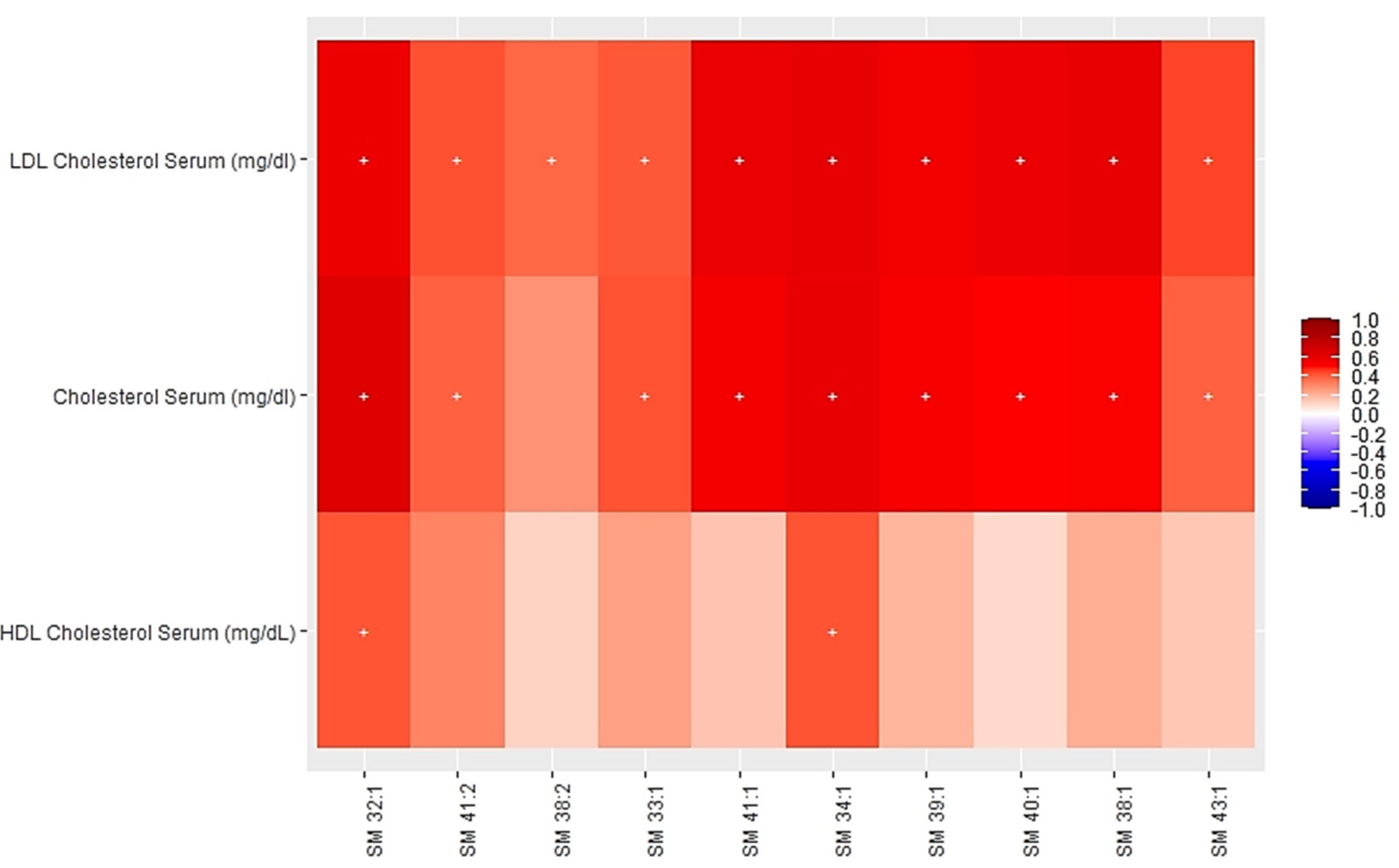
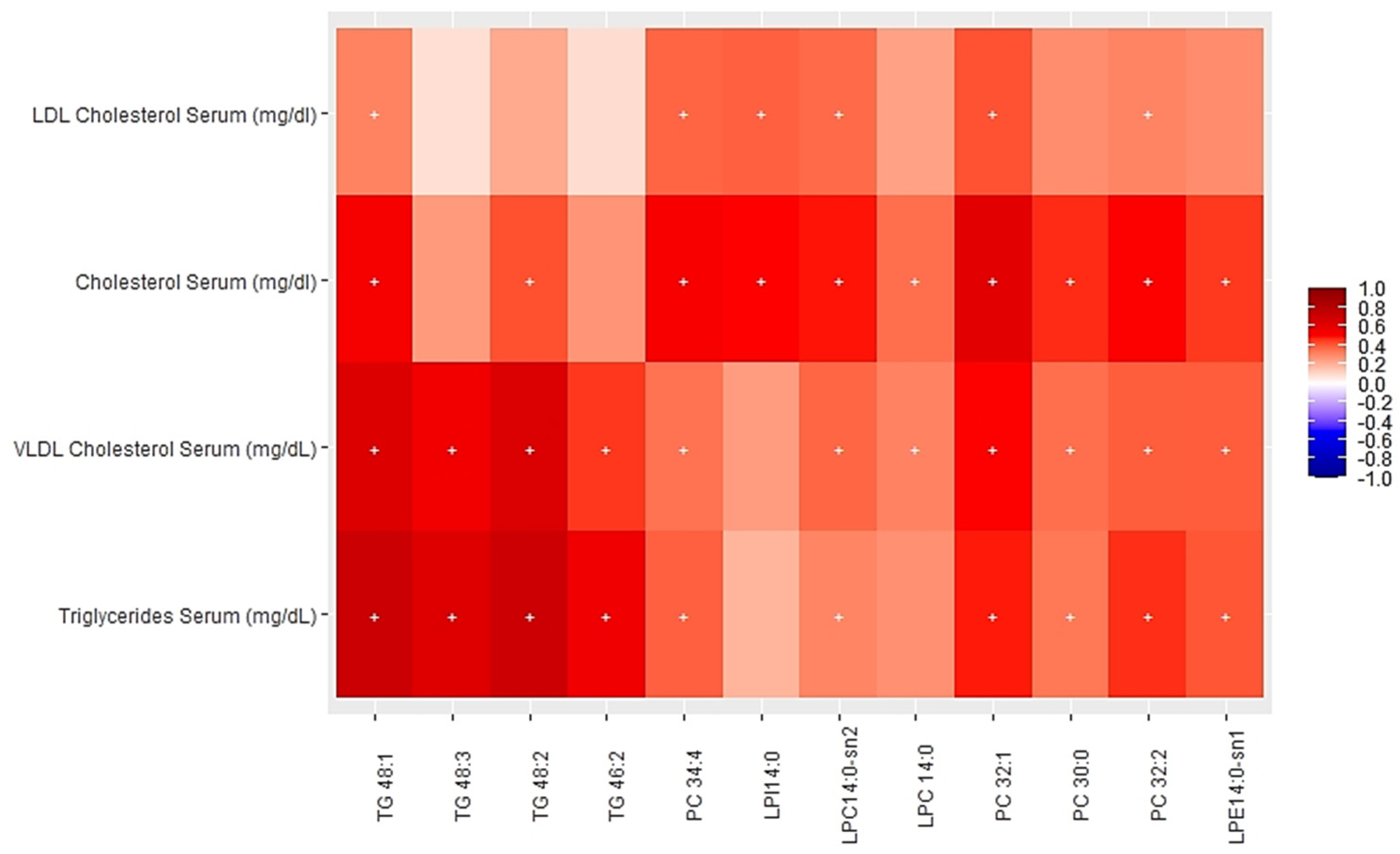
3.2. Associations between Multi-Omics Scores and Cardiometabolic Risk Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xie, H.; Guo, R.; Zhong, H.; Feng, Q.; Lan, Z.; Qin, B.; Ward, K.J.; Jackson, M.; Xia, Y.; Chen, X.; et al. Shotgun Metagenomics of 250 Adult Twins Reveals Genetic and Environmental Impacts on the Gut Microbiome. Cell Syst. 2016, 3, 572–584.e3. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.; Gordon, J.I. An Invitation to the Marriage of Metagenomics and Metabolomics. Cell 2008, 134, 708–713. [Google Scholar] [CrossRef] [Green Version]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Bäckhed, F. Diet–microbiota interactions as moderators of human metabolism. Nat. Cell Biol. 2016, 535, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Monnerie, S.; Comte, B.; Ziegler, D.; Morais, J.A.; Pujos-Guillot, E.; Gaudreau, P. Metabolomic and Lipidomic Signatures of Metabolic Syndrome and its Physiological Components in Adults: A Systematic Review. Sci. Rep. 2020, 10, 669. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.X.; Wang, S.-Y.; Kuo, C.-H.; Tsai, I.-L. Metabolome analysis for investigating host-gut microbiota interactions. J. Formos. Med. Assoc. 2019, 118, S10–S22. [Google Scholar] [CrossRef]
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.; Steves, C.; et al. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; de Rinaldis, E.; Bell, J.T.; Venter, J.C.; et al. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 2019, 10, 4505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festi, D.; Schiumerini, R.; Eusebi, L.H.; Marasco, G.; Taddia, M.; Colecchia, A. Gut microbiota and metabolic syndrome. World J. Gastroenterol. 2014, 20, 16079–16094. [Google Scholar] [CrossRef] [PubMed]
- Galié, S.; García-Gavilán, J.; Papandreou, C.; Camacho-Barcía, L.; Arcelin, P.; Palau-Galindo, A.; Rabassa, A.; Bulló, M. Effects of Mediterranean Diet on plasma metabolites and their relationship with insulin resistance and gut microbiota composition in a crossover randomized clinical trial. Clin. Nutr. 2021, 40, 3798–3806. [Google Scholar] [CrossRef]
- Antuna-Puente, B.; Disse, E.; Rabasa-Lhoret, R.; Laville, M.; Capeau, J.; Bastard, J.-P. How can we measure insulin sensitivity/resistance? Diabetes Metab. 2011, 37, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Galiè, S. Supplementary Data, Mendeley Data, V3, 2021. Available online: https://data.mendeley.com/datasets/nhh5cpsz4d/3 (accessed on 20 November 2021). [CrossRef]
- Mas-Lloret, J.; Obón-Santacana, M.; Ibáñez-Sanz, G.; Guinó, E.; Pato, M.L.; Rodriguez-Moranta, F.; Mata, A.; García-Rodríguez, A.; Moreno, V.; Pimenoff, V.N. Gut microbiome diversity detected by high-coverage 16S and shotgun sequencing of paired stool and colon sample. Sci. Data 2020, 7, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenenhaus, A.; Philippe, C.; Guillemot, V.; Le Cao, K.-A.; Grill, J.; Frouin, V. Variable selection for generalized canonical correlation analysis. Biostatistics 2014, 15, 569–583. [Google Scholar] [CrossRef] [Green Version]
- Benedetti, E.; Pučić-Baković, M.; Keser, T.; Gerstner, N.; Büyüközkan, M.; Štambuk, T.; Selman, M.H.J.; Rudan, I.; Polašek, O.; Hayward, C.; et al. A strategy to incorporate prior knowledge into correlation network cutoff selection. Nat. Commun. 2020, 11, 5153. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Amar, J. Microbiota–Host Crosstalk: A Bridge between Cardiovascular Risk Factors, Diet, and Cardiovascular Disease. Am. J. Hypertens. 2018, 31, 941–944. [Google Scholar] [CrossRef]
- Jiang, X.-C.; Paultre, F.; Pearson, T.A.; Reed, R.G.; Francis, C.K.; Lin, M.; Berglund, L.; Tall, A.R. Plasma Sphingomyelin Level as a Risk Factor for Coronary Artery Disease. Arter. Thromb. Vasc. Biol. 2000, 20, 2614–2618. [Google Scholar] [CrossRef] [Green Version]
- Zalloua, P.; Kadar, H.; Hariri, E.; Farraj, L.A.; Brial, F.; Hedjazi, L.; Le Lay, A.; Colleu, A.; Dubus, J.; Touboul, D.; et al. Untargeted Mass Spectrometry Lipidomics identifies correlation between serum sphingomyelins and plasma cholesterol. Lipids Health Dis. 2019, 18, 38. [Google Scholar] [CrossRef]
- Vojinovic, D.; Radjabzadeh, D.; Kurilshikov, A.; Amin, N.; Wijmenga, C.; Franke, L.; Ikram, M.A.; Uitterlinden, A.G.; Zhernakova, A.; Fu, J.; et al. Relationship between Gut Microbiota and Circulating Metabolites in Population-Based Cohorts. Nat. Commun. 2019, 10, 5813. [Google Scholar] [CrossRef] [Green Version]
- López-Contreras, B.E.; Morán-Ramos, S.; Villarruel-Vázquez, R.; Macías-Kauffer, L.; Villamil-Ramírez, H.; León-Mimila, P.; Vega-Badillo, J.; Sánchez-Muñoz, F.; Llanos-Moreno, L.E.; Canizalez-Román, A.; et al. Composition of gut microbiota in obese and normal-weight Mexican school-age children and its association with metabolic traits. Pediatr. Obes. 2018, 13, 381–388. [Google Scholar] [CrossRef]
- Russell, D.W. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavella, T.; Rampelli, S.; Guidarelli, G.; Bazzocchi, A.; Gasperini, C.; Pujos-Guillot, E.; Comte, B.; Barone, M.; Biagi, E.; Candela, M.; et al. Elevated gut microbiome abundance of Christensenellaceae, Porphyromonadaceae and Rikenellaceae is associated with reduced visceral adipose tissue and healthier metabolic profile in Italian elderly. Gut Microbes 2021, 13, 1–19. [Google Scholar] [CrossRef]
- Porez, G.; Prawitt, J.; Gross, B.; Staels, B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J. Lipid Res. 2012, 53, 1723–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simental-Mendía, L.E.; Simental-Mendía, M.; Sánchez-García, A.; Banach, M.; Serban, M.-C.; Cicero, A.F.G.; Sahebkar, A. Impact of ursodeoxycholic acid on circulating lipid concentrations: A systematic review and meta-analysis of randomized placebo-controlled trials. Lipids Health Dis. 2019, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.A.; Halliday, D.; Pittet, P.G. The use of 13-methyltetradecanoic acid as an indicator of adipose tissue turnover. Lipids 1980, 15, 572–579. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Waters, J.L.; Ley, R.E. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019, 17, 83. [Google Scholar] [CrossRef]
- Van Hul, M.; Le Roy, T.; Prifti, E.; Dao, M.C.; Paquot, A.; Zucker, J.-D.; Delzenne, N.M.; Muccioli, G.G.; Clément, K.; Cani, P.D. From correlation to causality: The case of Subdoligranulum. Gut Microbes 2020, 12, 1–13. [Google Scholar] [CrossRef]
- Ruiz-Canela, M.; Toledo, E.; Clish, C.; Hruby, A.; Liang, L.; Salas-Salvadó, J.; Razquin, C.; Corella, D.; Estruch, R.; Ros, E.; et al. Plasma Branched-Chain Amino Acids and Incident Cardiovascular Disease in the PREDIMED Trial. Clin. Chem. 2016, 62, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Canela, M.; Guasch-Ferré, M.; Toledo, E.; Clish, C.B.; Razquin, C.; Liang, L.; Wang, D.D.; Corella, D.; Estruch, R.; Hernáez, Á.; et al. Plasma branched chain/aromatic amino acids, enriched Mediterranean diet and risk of type 2 diabetes: Case-cohort study within the PREDIMED Trial. Diabetologia 2018, 61, 1560–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomino-Schätzlein, M.; Mayneris-Perxachs, J.; Caballano-Infantes, E.; Rodríguez, M.A.; Palomo-Buitrago, M.-E.; Xiao, X.; Mares, R.; Ricart, W.; Simó, R.; Herance, J.R.; et al. Combining metabolic profiling of plasma and faeces as a fingerprint of insulin resistance in obesity. Clin. Nutr. 2020, 39, 2292–2300. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Y.; Ong, C.-N.; Subramaniam, T.; Choi, H.W.; Yuan, J.-M.; Koh, W.-P.; Pan, A. Metabolic signatures and risk of type 2 diabetes in a Chinese population: An untargeted metabolomics study using both LC-MS and GC-MS. Diabetologia 2016, 59, 2349–2359. [Google Scholar] [CrossRef] [Green Version]
- Gannon, N.P.; Schnuck, J.K.; Vaughan, R.A. BCAA Metabolism and Insulin Sensitivity-Dysregulated by Metabolic Status? Mol. Nutr. Food Res. 2018, 62, e1700756. [Google Scholar] [CrossRef]
- Loftfield, E.; Herzig, K.-H.; Caporaso, J.G.; Derkach, A.; Wan, Y.; Byrd, D.A.; Vogtmann, E.; Männikkö, M.; Karhunen, V.; Knight, R.; et al. Association of Body Mass Index with Fecal Microbial Diversity and Metabolites in the Northern Finland Birth Cohort. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2289–2299. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, M.A.; Hoffmann, C.; Sherrill–Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.-Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High-Fat Diet Determines the Composition of the Murine Gut Microbiome Independently of Obesity. Gastroenterology 2009, 137, 1716–1724.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djekic, D.; Shi, L.; Brolin, H.; Carlsson, F.; Särnqvist, C.; Savolainen, O.; Cao, Y.; Bäckhed, F.; Tremaroli, V.; Landberg, R.; et al. Effects of a Vegetarian Diet on Cardiometabolic Risk Factors, Gut Microbiota, and Plasma Metabolome in Subjects with Ischemic Heart Disease: A Randomized, Crossover Study. J. Am. Hear. Assoc. 2020, 9, e016518. [Google Scholar] [CrossRef]
- Roeder, J.; Schink, B. Syntrophic Degradation of Cadaverine by a Defined Methanogenic Coculture. Appl. Environ. Microbiol. 2009, 75, 4821–4828. [Google Scholar] [CrossRef] [Green Version]
- Loke, M.F.; Chua, E.G.; Gan, H.M.; Thulasi, K.; Wanyiri, J.W.; Thevambiga, I.; Goh, K.L.; Wong, W.F.; Vadivelu, J. Metabolomics and 16S rRNA sequencing of human colorectal cancers and adjacent mucosa. PLoS ONE 2018, 13, e0208584. [Google Scholar] [CrossRef]
- Ma, J.; Yue, J.; Huang, R.; Liao, Y.; Li, S.; Liu, W. Reversion of aging-related DHEAS decline in mouse plasma alleviates aging-related glucose tolerance impairment by potentiation of glucose-stimulated insulin secretion of acute phase. Biochem. Biophys. Res. Commun. 2018, 500, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Baseline n = 47 |
|---|---|
| Age (years) | 50.6 (48.6, 52.6) |
| Women n (%) | 30 (63.8%) |
| BMI (Kg/m2) | 30.5 (29.9, 31.2) |
| Waist Circumference (cm) | 102.1 (99.3, 104.8) |
| SBP (mmHg) | 135.1 (131.7, 138.6) |
| DBP (mmHg) | 85.0 (82.3, 87.7) |
| Total Cholesterol (mg/dL) | 215.3 (206.1, 224.5) |
| LDLc (mg/dL) | 135.8 (128.1, 143.6) |
| HDLc (mg/dL) | 50.5 (47.6, 53.5) |
| VLDLc (mg/dL) | 28.1 (24.7, 31.5) |
| Triglycerides (mg/dL) | 147.7 (126.3, 169.2) |
| Glucose (mg/dL) | 100.2 (96.5, 103.9) |
| Insulin (mcUI/mL) | 13.4 (11.3, 15.5) |
| HOMA-IR | 3.3 (2.8, 3.9) |
| Multi-Omic Score 1 | Multi-Omic Score 2 | Multi-Omic Score 3 | Multi-Omic Score 4 | Multi-Omic Score 5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Factor | Mean ± SE | p 1 | Mean ± SE | p 1 | Mean ± SE | p 1 | Mean ± SE | p 1 | Mean ± SE | p 1 |
| Cholesterol (mg/dL) | 1.666 ± 0.435 | 0.001 | 0.249 ± 0.087 | 0.070 | 0.099 ± 0.215 | 0.983 | 0.121 ± 0.311 | 0.743 | 0.621 ± 0.148 | 0.0003 |
| LDLc (mg/dL) | 1.269 ± 0.336 | 0.005 | 0.161 ± 0.0692 | 0.083 | 0.062 ± 0.166 | 0.983 | 0.079 ± 0.239 | 0.743 | 0.327 ± 0.126 | 0.032 |
| HDLc (mg/dL) | 0.374 ± 0.149 | 0.053 | 0.0452 ± 0.029 | 0.330 | −0.001 ± 0.068 | 0.983 | 0.089 ± 0.098 | 0.743 | 0.0326 ± 0.055 | 0.803 |
| VLDLc (mg/dL) | −0.068 ± 0.179 | 0.755 | 0.036 ± 0.033 | 0.495 | 0.030 ± 0.076 | 0.983 | 0.050 ± 0.110 | 0.743 | 0.224 ± 0.052 | 0.0003 |
| Triglycerides (mg/dL) | 0.373 ± 1.186 | 0.755 | 0.234 ± 0.221 | 0.495 | 0.222 ± 0.505 | 0.983 | −0.498 ± 0.728 | 0.743 | 1.420 ± 0.351 | 0.0003 |
| Glucose (mg/dL) | 0.358 ± 0.191 | 0.170 | 0.087 ± 0.035 | 0.083 | 0.132 ± 0.0824 | 0.983 | 0.116 ± 0.121 | 0.743 | 0.043 ± 0.069 | 0.803 |
| Insulin (mcUI/mL) | 0.088 ± 0.114 | 0.738 | 0.007 ± 0.021 | 0.838 | −0.028 ± 0.0486 | 0.983 | −0.062 ± 0.070 | 0.743 | −0.010 ± 0.040 | 0.803 |
| HOMA IR | 0.034 ± 0.029 | 0.472 | 0.004 ± 0.005 | 0.656 | −0.0005 ± 0.012 | 0.983 | −0.008 ± 0.018 | 0.743 | −0.003 ± 0.010 | 0.803 |
| SBP (mmHg) | 0.077 ± 0.191 | 0.755 | 0.019 ± 0.036 | 0.749 | 0.105 ± 0.080 | 0.983 | −0.117 ± 0.117 | 0.743 | 0.019 ± 0.067 | 0.803 |
| DBP (mmHg) | 0.065 ± 0.148 | 0.755 | 0.002 ± 0.028 | 0.951 | 0.006 ± 0.063 | 0.983 | 0.045 ± 0.091 | 0.743 | 0.028 ± 0.052 | 0.803 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galié, S.; Papandreou, C.; Arcelin, P.; Garcia, D.; Palau-Galindo, A.; Gutiérrez-Tordera, L.; Folch, À.; Bulló, M. Examining the Interaction of the Gut Microbiome with Host Metabolism and Cardiometabolic Health in Metabolic Syndrome. Nutrients 2021, 13, 4318. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13124318
Galié S, Papandreou C, Arcelin P, Garcia D, Palau-Galindo A, Gutiérrez-Tordera L, Folch À, Bulló M. Examining the Interaction of the Gut Microbiome with Host Metabolism and Cardiometabolic Health in Metabolic Syndrome. Nutrients. 2021; 13(12):4318. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13124318
Chicago/Turabian StyleGalié, Serena, Christopher Papandreou, Pierre Arcelin, David Garcia, Antoni Palau-Galindo, Laia Gutiérrez-Tordera, Àlex Folch, and Mònica Bulló. 2021. "Examining the Interaction of the Gut Microbiome with Host Metabolism and Cardiometabolic Health in Metabolic Syndrome" Nutrients 13, no. 12: 4318. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13124318