
Etiology and Management of Pediatric Intestinal Failure: Focus on the Non-Digestive Causes

 , ,
, ,
Abstract
:1. Introduction
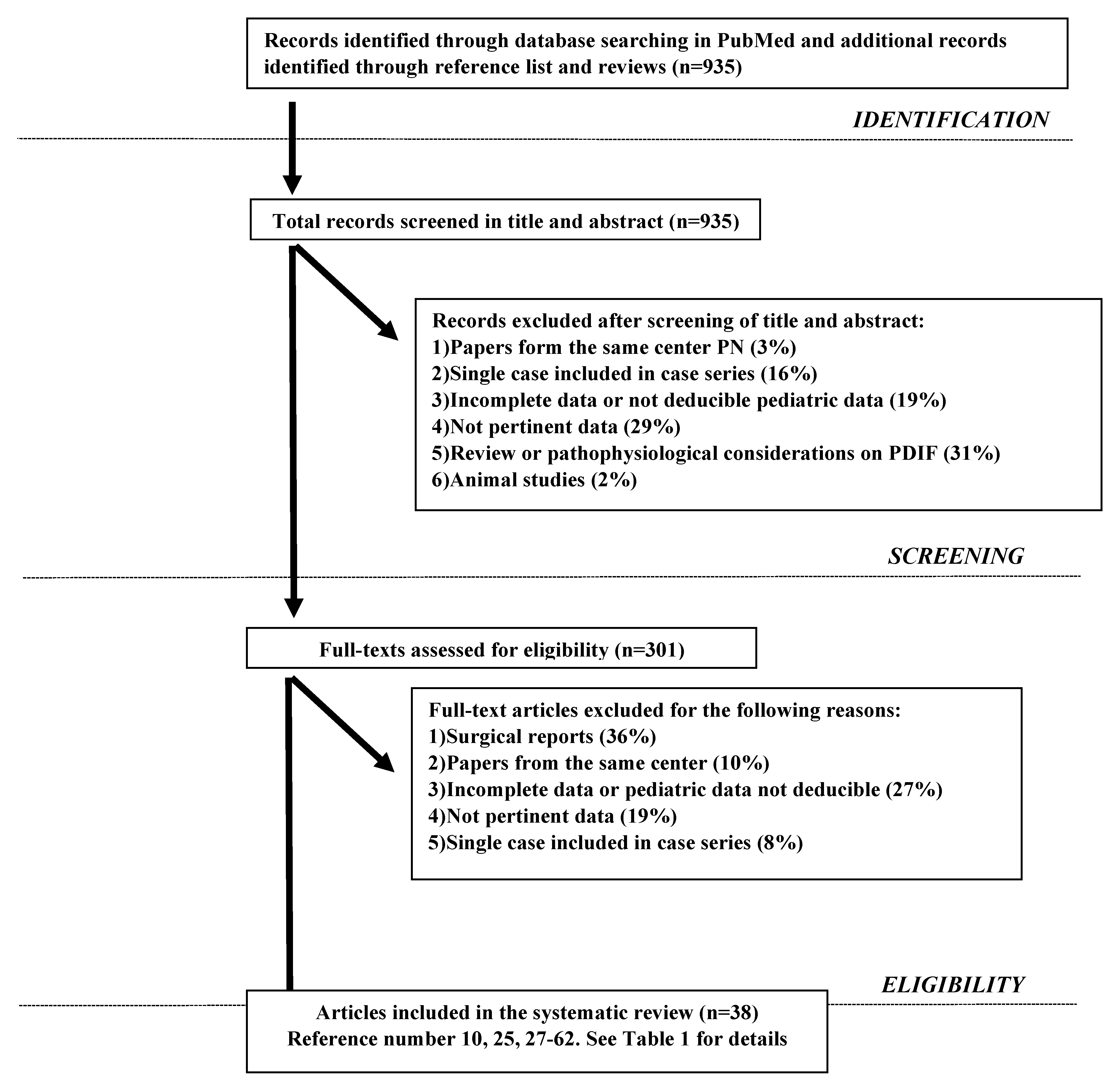
2. Materials and Methods
2.1. Search Strategy
2.2. Inclusion Criteria
2.3. Exclusion Criteria
2.4. Data Extraction, Synthesis and Analysis
2.5. Endpoints
3. Results
3.1. Pathophysiological Mechanisms of NDIF
3.1.1. Intestinal Fistulas
3.1.2. Short Bowel Syndrome (SBS)
3.1.3. Dysmotility
3.1.4. Malabsorption/Maldigestion
3.2. Management
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fleming, C.R.; Remington, M. Intestinal failure. In Nutrition and the Surgical Patient; Hill, G.L., Ed.; Churchill Livingstone: Edinburgh, UK, 1981; pp. 219–235. [Google Scholar]
- Pironi, L.; Arends, J.; Bozzetti, F.; Cuerda, C.; Gillanders, L.; Jeppesen, P.B.; Joly, F.; Kelly, D.; Lal, S.; Staun, M.; et al. ESPEN guidelines on chronic intestinal failure in adults. Clin. Nutr. 2016, 35, 247–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marco, G.; Barabino, A.; Gambarara, M.; Diamanti, A.; Martelossi, S.; Guarino, A. Network approach to the child with primary intestinal failure. J. Pediatr. Gastroenterol. Nutr. 2006, 43, S61–S67. [Google Scholar] [CrossRef] [PubMed]
- Colomb, V.; Dabbas-Tyan, M.; Taupin, P.; Talbotec, C.; Révillon, Y.; Jan, D.; De Potter, S.; Gorski-Colin, A.-M.; Lamor, M.; Herreman, K.; et al. Long-term Outcome of Children Receiving Home Parenteral Nutrition: A 20-year Single-center Experience in 302 Patients. J. Pediatr. Gastroenterol. Nutr. 2007, 44, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Terrin, G.; Tomaiuolo, R.; Passariello, A.; Elce, A.; Amato, F.; Di Costanzo, M.; Castaldo, G.; Canani, R.B. Congenital Diarrheal Disorders: An Updated Diagnostic Approach. Int. J. Mol. Sci. 2012, 13, 4168–4185. [Google Scholar] [CrossRef] [Green Version]
- Canani, R.B.; Pezzella, V.; Amoroso, A.; Cozzolino, T.; Di Scala, C.; Passariello, A. Diagnosing and treating intolerance to carbohydrates in children. Nutrients 2016, 8, 157. [Google Scholar] [CrossRef] [Green Version]
- Uhlig, H.H.; Schwerd, T.; Koletzko, S.; Shah, N.; Kammermeier, J.; Elkadri, A.; Ouahed, J.; Wilson, D.C.; Travis, S.P.; Turner, D.; et al. The Diagnostic Approach to Monogenic Very Early Onset Inflammatory Bowel Disease. Gastroenterology 2014, 147, 990–1007.e3. [Google Scholar] [CrossRef] [Green Version]
- Capriati, T.; Cardile, S.; Papadatou, B.; Romano, C.; Knafelz, D.; Bracci, F.; Diamanti, A. Pediatric inflammatory bowel disease: Specificity of very early onset. Expert Rev. Clin. Immunol. 2016, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pironi, L.; Hébuterne, X.; Van Gossum, A.; Messing, B.; Lyszkowska, M.; Colomb, V.; Forbes, A.; Micklewright, A.; Villares, J.M.M.; Thul, P.; et al. Candidates for Intestinal Transplantation: A Multicenter Survey in Europe. Am. J. Gastroenterol. 2006, 101, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Hizarcioglu-Gulsen, H.; Saltik-Temizel, I.N.; Demir, H.; Gurakan, F.; Ozen, H.; Yuce, A. Intractable diarrhea of infancy: 10 years of experience. J. Pediatr. Gastroenterol. Nutr. 2014, 59, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Walker-Smith, J.A. Intractable diarrhea of infancy. Saudi J. Gastroenterol. 1995, 1, 152–156. [Google Scholar] [PubMed]
- Catassi, C.; Fabiani, E.; Spagnuolo, M.I.; Barera, G.; Guarino, A. Severe and protracted diarrhea: Results of the 3-year SIGENP multicenter survey. Working Group of the Italian Society of Pediatric Gastroenterology and Hepatology (SIGENP). J. Pediatr. Gastroenterol. Nutr. 1999, 29, 63–68. [Google Scholar] [CrossRef]
- Guarino, A.; Spagnuolo, M.I.; Russo, S.; Albano, F.; Guandalini, S.; Capano, G.; Cucchiara, S.; Vairano, P.; Liguori, R.; Casola, A.; et al. Etiology and Risk Factors of Severe and Protracted Diarrhea. J. Pediatr. Gastroenterol. Nutr. 1995, 20, 173–178. [Google Scholar] [CrossRef]
- Beck, N.S.; Kang, I.S.; Suh, Y.L. Protracted Diarrhea: Results of the Five-year Survey in a Tertiary Hospital in Korea. J. Korean Med. Sci. 2001, 16, 736–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapar, N.; Saliakellis, E.; Benninga, M.A.; Borrelli, O.; Curry, J.; Faure, C.; De Giorgio, R.; Gupte, G.; Knowles, C.H.; Staiano, A.; et al. Pediatric intestinal pseudo-obstruction: Evidence and consensus-based rec-ommendations from an ESPGHAN-Led expert group. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 991–1019. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.H. Parenteral nutrition use in children with cancer. Pediatr. Blood Cancer 2019, 66, e28000. [Google Scholar] [CrossRef]
- Oya, Y.; Sugawara, Y.; Honda, M.; Yoshii, D.; Isono, K.; Hayashida, S.; Yamamoto, H.; Inomata, Y. Living Donor Liver Transplantation for Progressive Familial Intrahepatic Cholestasis Type 1: Two Reported Cases. Transpl. Proc. 2017, 49, 1123–1125. [Google Scholar] [CrossRef]
- Janecke, A.R.; Heinz-Erian, P.; Müller, T. Congenital Sodium Diarrhea: A Form of Intractable Diarrhea, With a Link to Inflammatory Bowel Disease. J. Pediatr. Gastroenterol. Nutr. 2016, 63, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Braamskamp, M.J.; Dolman, K.M.; Tabbers, M.M. Clinical practice. Protein-losing enteropathy in children. Eur. J. Pediatr. 2010, 169, 1179–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulou, A.; Lloyd, D.R.; Williams, M.D.; Darbyshire, P.J.; Booth, I.W. Gastrointestinal and nutritional sequelae of bone marrow transplantation. Arch. Dis. Child. 1996, 75, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.N.; Driscoll, D.J.; O’Leary, P.W. Protein-Losing Enteropathy and the Fontan Operation. Nutr. Clin. Pract. 2012, 27, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Mutanen, A.; Wales, P.W. Etiology and prognosis of pediatric short bowel syndrome. Semin. Pediatr. Surg. 2018, 27, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Posovszky, C. Congenital intestinal diarrheal diseases: A diagnostic and therapeutic challenge. Best Pract. Res. Clin. Gastroenterol. 2016, 30, 187–211. [Google Scholar] [CrossRef]
- Sherman, P.M.; Cutz, E.; Goulet, O. Immune and autoimmune enteropathies. Ann. Nestlé 2006, 64, 7–13. [Google Scholar] [CrossRef]
- Nader, E.A.; Lambe, C.; Talbotec, C.; Pigneur, B.; Lacaille, F.; Garnier-Lengliné, H.; Petit, L.-M.; Poisson, C.; Rocha, A.; Corriol, O.; et al. Outcome of home parenteral nutrition in 251 children over a 14-y period: Report of a single center. Am. J. Clin. Nutr. 2016, 103, 1327–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulet, O.; Ruemmele, F.; Lacaille, F.; Colomb, V. Irreversible Intestinal Failure. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 250–269. [Google Scholar] [CrossRef]
- Prasad, D.; Srivastava, A.; Tambe, A. Clinical profile, response to therapy, and outcome of children with primary intes-tinal lymphangiectasia. Dig. Dis. 2019, 37, 458–466. [Google Scholar] [CrossRef]
- LaRusso, K.; Schaack, G.; Fung, T.; McGregor, K.; Long, J.; Dumas, M.P.; Attari, Z.; Yousef, Y.; Girgis, H.; Raghunathan, R. Should you pick the PICC? Prolonged use of peripherally inserted central venous catheters in children with intestinal failure. J. Pediatr. Surg. 2019, 54, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Diamanti, A.; Fusaro, F.; Caldaro, T.; Capriati, T.; Candusso, M.; Nobili, V.; Borrelli, O. Pediatric Intestinal Pseudo-obstruction: Impact of Neonatal and Later Onset on Clinical and Nutritional Outcomes. J. Pediatr. Gastroenterol. Nutr. 2019, 69, 212–217. [Google Scholar] [CrossRef]
- Zemrani, B.; McLeod, E.; Rogers, E.; Lawrence, J.; Feldman, D.; Evans, V.; Shalley, H.; Bines, J. Vascular Ehlers-Danlos Syndrome: An Unusual Cause of Chronic Intestinal Failure in a Child. J. Pediatr. Gastroenterol. Nutr. 2019, 68, e14–e15. [Google Scholar] [CrossRef] [PubMed]
- Olivares, Y.Z.; Bunster, M.I.H.; Bayón, M.L.C.; Osiac, L.R.; Lorca, J.C.; Reus, G.A.; Antilef, R.M.; Balboa, P.; Cors, C.; De la Fuente, G. Home parenteral nutrition in pediatric patients with intestinal insufficiency. Rev. Chil. Pediatría 2019, 90, 60–68. [Google Scholar]
- Fayard, J.; Collardeau, S.; Bertrand, Y.; Cordier, M.-P.; Malcus, C.; Dubois, R.; Mure, P.-Y.; Basile, G.D.S.; Louazon, T.; Rohmer, B.; et al. TTC7A mutation must be considered in patients with repeated intestinal atresia associated with early inflammatory bowel disease: Two new case reports and a literature review. Archives de Pédiatrie 2018, 25, 334–339. [Google Scholar] [CrossRef]
- Raphael, B.P.; Hazekamp, C.; Samnaliev, M.; Ozonoff, A. Analysis of healthcare institutional costs of pediatric home parenteral nu-trition central line infections. J. Pediatr. Gastroenterol. Nutr. 2018, 67, e77–e81. [Google Scholar] [CrossRef]
- Gunnar, R.; Lumia, M.; Pakarinen, M.; Merras-Salmio, L. Children With Intestinal Failure Undergoing Intestinal Rehabilitation Are at Risk for Essential Fatty Acid Deficiency. J. Parenter. Enter. Nutr. 2018, 42, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Merras-Salmio, L.; Mutanen, A.; Ylinen, E.; Rintala, R.; Koivusalo, A.; Pakarinen, M.P. Pediatric Intestinal Failure: The Key Outcomes for the First 100 Patients Treated in a National Tertiary Referral Center During 1984-2017. J. Parenter. Enter. Nutr. 2018, 42, 1304–1313. [Google Scholar] [CrossRef]
- Diamanti, A.; Capriati, T.; Gandullia, P.; Di Leo, G.; Lezo, A.; Lacitignola, L.; Spagnuolo, M.I.; Gatti, S.; D’Antiga, L.; Verlato, G.; et al. Pediatric chronic intestinal failure in Italy: Report from the 2016 survey on be-half of Italian society for gastroenterology, hepatology and nutrition (SIGENP). Nutrients 2017, 9, 1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blotte, C.; Styers, J.; Zhu, H.; Channabasappa, N.; Piper, H.G. A comparison of Broviac® and peripherally inserted central catheters in children with in-testinal failure. J. Pediatr. Surg. 2017, 52, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Giabicani, E.; Lemale, J.; Dainese, L.; Boudjemaa, S.; Coulomb, A.; Tounian, P.; Dubern, B. Chronic intestinal pseudo-obstruction in a child with Treacher Collins syndrome. Arch. Pédiatrie 2017, 24, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Germán-Díaz, M.; Rodriguez-Gil, Y.; Cruz-Rojo, J.; Charbit-Henrion, F.; Cerf-Bensussan, N.; Manzanares, J.M.-L.; Moreno-Villares, J.M. A New Case of Congenital Malabsorptive Diarrhea and Diabetes Secondary to Mutant Neurogenin-3. Pediatr. 2017, 140, e20162210. [Google Scholar] [CrossRef]
- Gonzalez-Hernandez, J.; Daoud, Y.; Styers, J.; Journeycake, J.M.; Channabasappa, N.; Piper, H.G. Central venous thrombosis in children with intestinal failure on long-term parenteral nutrition. J. Pediatr. Surg. 2016, 51, 790–793. [Google Scholar] [CrossRef]
- Stýblová, J.; Kalousová, J.; Adamcová, M. Paediatric home parenteral nutrition in the Czech Republic and its devel-opment: Multicentre retrospective study 1995–2011. Ann. Nutr. Metab. 2017, 71, 99–106. [Google Scholar]
- Hashimura, Y.; Morioka, I.; Hisamatsu, C.; Yokoyama, N.; Taniguchi-Ikeda, M.; Yokozaki, H.; Murayama, K.; Ohtake, A.; Itoh, K.; Takeshima, Y.; et al. Mitochondrial respiratory chain complex IV deficiency complicated with chronic intestinal pseudo-obstruction in a neonate. Pediatr. Int. 2016, 58, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Furtado, S.; Ahmed, N.; Forget, S. Outcomes of patients with intestinal failure after the development and implementa-tion of a multidisciplinary team. Can. J. Gastroenterol. Hepatol. 2015, 16954. [Google Scholar] [CrossRef] [Green Version]
- Pichler, J.; Watson, T.; McHugh, K.; Hill, S. Prevalence of Gallstones Compared in Children With Different Intravenous Lipids. J. Pediatr. Gastroenterol. Nutr. 2015, 61, 253–259. [Google Scholar] [CrossRef]
- Mirabel-Chambaud, E.; N’Guyen, M.; Valdeyron, M.L. Dramatic increase of associated complications in children with intestinal failure. J. Parenter. Enter. Nutr. 2016, 40, 815–819. [Google Scholar]
- Mezoff, E.A.; Fei, L.; Troutt, M.; Klotz, K.; Kocoshis, S.A.; Cole, C.R. Ethanol Lock Efficacy and Associated Complications in Children With Intestinal Failure. J. Parenter. Enter. Nutr. 2016, 40, 815–819. [Google Scholar] [CrossRef] [Green Version]
- Neelis, E.G.; Roskott, A.M.; Dijkstra, G. Presentation of a nation wide multicenter registry of intestinal failure and intes-tinal transplantation. Clin. Nutr. 2016, 35, 225–229. [Google Scholar] [CrossRef]
- Pichler, J.; Simchowitz, V.; Macdonald, S. Comparison of liver function with two new/mixed intravenous lipid emul-sions in children with intestinal failure. Eur. J. Clin. Nutr. 2014, 68, 1161–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhi, A.D.; Goyal, A.; Davison, J.M.; Regueiro, M.D.; Roche, R.L.; Ranganathan, S. Pediatric autoimmune enteropathy: An entity frequently associated with immunodeficiency disorders. Mod. Pathol. 2013, 27, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Godart, F.; Bakhti, O.M.; Bonnevalle, M. Intestinal ischaemia as a severe presentation of Kawasaki disease leading to short-bowel syndrome. Cardiol. Young 2014, 24, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Duro, D.; Mitchell, P.D.; Mehta, N.M. Variability of resting energy expenditure in infants and young children with intes-tinal failure-associated liver disease. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 637–641. [Google Scholar] [CrossRef] [Green Version]
- Courtney-Martin, G.; Kosar, C.; Campbell, A.; Avitzur, Y.; Wales, P.W.; Steinberg, K.; Harrison, D.; Chambers, K. Plasma Aluminum Concentrations in Pediatric Patients Receiving Long-Term Parenteral Nutrition. J. Parenter. Enter. Nutr. 2015, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.G.; Lindberg, I.; Solorzano-Vargas, R.S. Congenital proprotein convertase 1/3 deficiency causes malabsorptive diarrhea and other endocrinopathies in a pediatric cohort. Gastroenterology 2013, 145, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, E.M.; Majewski, J.; Alirezaie, N.; Fernandez, I.; Casals, F.; Patey, N.; Decaluwe, H.; Gosselin, I.; Haddad, E.; Hodgkinson, A.; et al. Exome sequencing identifies mutations in the geneTTC7Ain French-Canadian cases with hereditary multiple intestinal atresia. J. Med. Genet. 2013, 50, 324–329. [Google Scholar] [CrossRef] [Green Version]
- Ubesie, A.C.; Heubi, J.E.; Kocoshis, S.A.; Henderson, C.J.; Mezoff, A.G.; Rao, M.B.; Cole, C.R. Vitamin D Deficiency and Low Bone Mineral Density in Pediatric and Young Adult Intestinal Failure. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 372–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derepas, C.; Kosar, C.; Avitzur, Y.; Wales, P.W.; Courtney-Martin, G. Decreased Bone Turnover Markers in Children on Long-Term Parenteral Nutrition (PN) for Intestinal Failure (IF). J. Parenter. Enter. Nutr. 2013, 39, 85–94. [Google Scholar] [CrossRef]
- Sadlier, C. Helping children who require long-term parenteral nutrition. Nurs. Child. Young People 2013, 25, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, K.P.; Nespor, C.; Ng, M. Evaluation of ethanol lock therapy in pediatric patients on long-term parenteral nutri-tion. Nutr. Clin. Pract. 2013, 28, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Javid, P.J.; Malone, F.R.; Bittner, R. The optimal timing of referral to an intestinal failure program: The relationship be-tween hyperbilirubinemia and mortality. J. Pediatr. Surg. 2011, 46, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Diamanti, A.; Bizzarri, C.; Basso, M.S. How does long-term parenteral nutrition impact the bone mineral status of chil-dren with intestinal failure? J. Bone Miner Metab. 2010, 28, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.; Moammarb, H.; Mitra, D.K. Visceral myopathy causing chronic intestinal pseudo obstruction and intestinal failure in a child with Sanjad-Sakati syndrome. J. Ped. Surg. 2010, 45, 430–434. [Google Scholar] [CrossRef]
- Gonzalez-Hernandez, J.; Prajapati, P.; Ogola, G.; Channabasappa, N.; Drews, B.; Piper, H.G. Predicting time to full enteral nutrition in children after significant bowel resection. J. Pediatr. Surg. 2017, 52, 764–767. [Google Scholar] [CrossRef]
- Corcos, O.; Nuzzo, A. Gastro-Intestinal Vascular Emergencies. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 709–725. [Google Scholar] [CrossRef]
- Boley, S.J.; Brandt, L.J.; Sammartano, R.J. History of mesenteric ischemia. The evolution of a diagnosis and management. Surg. Clin. N. Am. 1997, 77, 275–288. [Google Scholar] [CrossRef]
- Clair, D.G.; Beach, J.M. Mesenteric Ischemia. N. Engl. J. Med. 2016, 374, 959–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, I.A.; Swenson, O. Mesenteric Vascular Occlusion in Infancy and Childhood. N. Engl. J. Med. 1960, 263, 1122–1125. [Google Scholar] [CrossRef]
- Hebra, A.; Brown, M.F.; Hirschl, R.B.; McGeehin, K.; O’Neill, J.A.; Norwood, W.I.; Ross, A.J. Mesenteric ischemia in hypoplastic left heart syndrome. J. Pediatr. Surg. 1993, 28, 606–611. [Google Scholar] [CrossRef]
- Sathe, M.; Houwen, R. Meconium ileus in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16, S32–S39. [Google Scholar] [CrossRef] [Green Version]
- Carlyle, B.E.; Borowitz, D.S.; Glick, P.L. A review of pathophysiology and management of fetuses and neonates with meco-nium ileus for the pediatric surgeon. J. Pediatr. Surg. 2012, 47, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Yang, N.; Quinton, P.M. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regula-tor-dependent bicarbonate secretion. J. Clin. Investig. 2009, 119, 2613–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigorgne, A.E.; Farin, H.F.; Lemoine, R.; Mahlaoui, N.; Lambert, N.; Gil, M.; Schulz, A.; Philippet, P.; Schlesser, P.; Abrahamsen, T.G.; et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J. Clin. Investig. 2014, 124, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.; Patey, N.; Marchand, V.; Birlea, M.; Maranda, B.; Haddad, E.; Decaluwe, H.; Le Deist, F. Multiple Intestinal Atresia With Combined Immune Deficiency Related to TTC7A Defect Is a Multiorgan Pathology. Medicine 2014, 93, e327. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Giliani, S.; Lanzi, G. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) muta-tions for combined immunodeficiency with intestinal atresias. J. Allergy Clin. Immunol. 2013, 132, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kammermeier, J.; Dziubak, R.; Pescarin, M. Phenotypic and genotypic characterization of inflammatory bowel disease presenting before the age of 2 years. J. Crohns. Colitis 2017, 11, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, R.K.; Coccia, P.F.; Talmadge, J.E. Donor immune reconstitution after liver-small bowel transplantation for multi-ple intestinal atresia with immunodeficiency. Blood 2004, 103, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Sunkara, T.; Rawla, P.; Yarlagadda, K.S.; Gaduputi, V. Eosinophilic gastroenteritis: Diagnosis and clinical perspectives. Clin. Exp. Gastroenterol. 2019, 12, 239–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Mayer, L. Diagnosis and Treatment of Gastrointestinal Disorders in Patients With Primary Immunodeficiency. Clin. Gastroenterol. Hepatol. 2013, 11, 1050–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, D.; Travaglini, L.; Drouin, C.A. MEDNIK syndrome: A novel defect of copper metabolism treatable by zinc acetate therapy. Brain 2013, 136, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, D.; Dionisi-Vici, C. AP1S1 defect causing MEDNIK syndrome: A new adaptinopathy associated with defective copper metabolism. Ann. N. Y. Acad. Sci. 2014, 1314, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Sundar, U.; Lakkas, Y.; Asole, D. Gitelman’s syndrome presenting as recurrent paralytic ileus due to chronic renal tub-ular K+ wasting. J. Assoc. Physicians India 2010, 58, 322–324. [Google Scholar]
- Hyman, P.E.; Bursch, B.; Beck, D. Discriminating pediatric condition falsification from chronic intestinal pseu-do-obstruction in toddlers. Child Maltreat 2002, 7, 132–137. [Google Scholar] [CrossRef]
- Hall, J.G.; Aldinger, K.A.; Tanaka, K.I. Amyoplasia Revisited. Am. J. Med. Genet. A 2014, 164, 700–730. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kong, M.-S. Primary intestinal lymphangiectasia diagnosed by endoscopy following the intake of a high-fat meal. Eur. J. Nucl. Med. Mol. Imaging 2007, 167, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Freeman, H.J.; Nimmo, M. Intestinal lymphangectasia in adults. World J. Gastrointest Oncol. 2011, 3, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A.; Steinfeld, J.L.; Dutcher, T.F.; Davidson, J.D.; Gordon, R.S., Jr. The role of the gastrointestinal system in “idio-pathic hypoproteinemia”. Gastroenterology 1961, 41, 197–207. [Google Scholar] [CrossRef]
- Wen, J.; Tang, Q.; Wu, J.; Wang, Y.; Cai, W. Primary intestinal lymphangiectasia: Four case reports and a review of the litera-ture. Dig. Dis. Sci. 2010, 55, 3466–3472. [Google Scholar] [CrossRef] [PubMed]
- Siham, A.S.; Rawahi, Y.A.J.; Abdoon, H. Octreotide in Hennekam syndrome-associated intestinal lymphangiectasia. World J. Gastroenterol. 2012, 18, 6333–6337. [Google Scholar]
- Al Balushi, M.D.A.; Andrew, S. Mackie. Protein-Losing Enteropathy Following Fontan Palliation. Canadian. J. Cardiol. 2019, 35, 1857–1860. [Google Scholar]
- Itkin, M.; Piccoli, D.A.; Nadolski, G.; Rychik, J.; DeWitt, A.; Pinto, E.; Rome, J.; Dori, Y. Protein-Losing Enteropathy in Patients With Congenital Heart Disease. J. Am. Coll. Cardiol. 2017, 69, 2929–2937. [Google Scholar] [CrossRef]
- Matsumoto, T.; Yamagami, T.; Kato, T.; Hirota, T.; Yoshimatsu, R.; Masunami, T.; Nishimura, T. The effectiveness of lymphangiography as a treatment method for various chyle leakages. Br. J. Radiol. 2009, 82, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, J.L.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef]
- Gratwohl, A.; Baldomero, H.; Passweg, J.F.; Frassoni, F.; Niederwieser, D.; Schmitz, N.; Urbano-Ispizua, A. Hematopoietic stem cell transplantation for hematological malignancies in Europe. Leukemia 2003, 17, 941–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsohn, D.A.; Vogelsang, G.B. Acute graft versus host disease. Orphanet. J. Rare Dis. 2007, 2, 35. [Google Scholar] [CrossRef] [Green Version]
- Nassereddine, S.; Rafei, H.; Elbahesh, E.; Tabbara, I. Acute Graft Versus Host Disease: A Comprehensive Review. Anticancer. Res. 2017, 37, 1547–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquini, M.; Wang, Z.; Horowitz, M.M.; Gale, R.P. 2013 report from the Center for International Blood and Marrow Trans-plant Research (CIBMTR): Current uses and outcomes of hematopoietic cell transplants for blood and bone marrow disorders. Clin. Transpl. 2013, 8, 187–197. [Google Scholar]
- Martin, P.J.; McDonald, G.B.; Sanders, J.E. Increasingly frequent diagnosis of acute gastrointestinal graft-versus-host dis-ease after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2004, 10, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Fløisand, Y.; Lazarevic VLAertens, J. Safety and effectiveness of Vedolizumab in patients with steroid-refractory gas-trointestinal acute graft-versus-host disease: Retrospective Record Review. Biol. Blood Marrow Transplant. 2019, 25, 720–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeken, J.; Péanne, R. What is new in CDG? J. Inherit. Metab. Dis. 2017, 40, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Freeze, H.H.; Aebi, M. Altered glycan structures: The molecular basis of congenital disorders of glycosylation. Curr. Opin. Struct. Biol. 2005, 15, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Damen, G.; De Klerk, H.; Huijmans, J.; Hollander, J.D.; Sinaasappel, M. Gastrointestinal and Other Clinical Manifestations in 17 Children With Congenital Disorders of Glycosylation Type Ia, Ib, and Ic. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 282–287. [Google Scholar] [CrossRef]
- Liem, Y.S.; Bode, L.; Freeze, H.H.; Leebeek, F.W.; Zandbergen, A.A.; Wilson, J.P. Using heparin therapy to reverse protein-losing enteropathy in a patient with CDG-Ib. Nat. Clin. Pract. Gastroenterol. Hepatol. 2008, 5, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Zentilin Boyer, M.; de Lonlay, P.; Seta, N. Failure to thrive and intestinal diseases in congenital disorders of glycosyla-tion. Arch. Pediatr. 2003, 10, 590–595. [Google Scholar] [CrossRef]
- Jaeken, J.; Matthijs, G.; Saudubray, J.-M.; Dionisi-Vici, C.; Bertini, E.; De Lonlay, P.; Henri, H.; Carchon, H.; Schollen, E.; Van Schaftingen, E. Phosphomannose Isomerase Deficiency: A Carbohydrate-Deficient Glycoprotein Syndrome with Hepatic-Intestinal Presentation. Am. J. Hum. Genet. 1998, 62, 1535–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altassan, R.; Péanne, R.; Jaeken, J.; Barone, R.; Bidet, M.; Borgel, D.; Brasil, S.; Cassiman, D.; Cechova, A.; Coman, D.; et al. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up. J. Inherit. Metab. Dis. 2019, 42, 5–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čechová, A.; Altassan, R.; Borgel, D.; Bruneel, A.; Correia, J.; Girard, M.; Harroche, A.; Kiec-Wilk, B.; Mohnike, K.; Pascreau, T.; et al. Consensus guideline for the diagnosis and management of mannose phosphate isomerase-congenital disorder of glycosylation. J. Inherit. Metab. Dis. 2020, 43, 671–693. [Google Scholar] [CrossRef]
- Unsworth, D.J.; Walker-Smith, J.A. Autoimmunity in Diarrhoeal Disease. J. Pediatr. Gastroenterol. Nutr. 1985, 4, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Bacchetta, R.; Barzaghi, F.; Roncarolo, M.-G. From IPEX syndrome to FOXP3 mutation: A lesson on immune dysregulation. Ann. N. Y. Acad. Sci. 2018, 1417, 5–22. [Google Scholar] [CrossRef]
- Marciano, B.E.; Rosenzweig, S.D.; Kleiner, D.E.; Anderson, V.L.; Darnell, D.N.; Anaya-O’Brien, S.; Hilligoss, D.M.; Malech, H.L.; Gallin, J.I.; Holland, S.M. Gastrointestinal Involvement in Chronic Granulomatous Disease. Pediatrics 2004, 114, 462–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, B.H.; Leto, T.L.; Gallin, J.I.; Malech, H.L.; Holland, S.M. Genetic, biochemical, and clinical features of chronic granuloma-tous disease. Medicine 2000, 79, 170–200. [Google Scholar] [CrossRef]
- Johnston, R.B., Jr. Clinical aspects of chronic granulomatous disease. Curr. Opin. Hematol. 2001, 8, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, D.; Wright, V.M.; Shaw, D.G.; Raafat, F. Walker-Smith JA. Chronic granulomatous disease mimicking Crohn’s disease. J. Pediatr. Gastroenterol. Nutr. 1985, 4, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Mitomi, H.; Mikami, T.; Takahashi, H. Colitis in chronic granulomatous disease resembling Crohn’s disease: Compara-tive analysis of CD68- positive cells between two disease entities. Dig. Dis. Sci. 1999, 4, 452–456. [Google Scholar] [CrossRef]
- Schäppi, M.G.; Klein, N.J.; Lindley, K.J.; Rampling, D.; Smith, V.V.; Goldblatt, D.; Milla, P.J. The Nature of Colitis in Chronic Granulomatous Disease. J. Pediatr. Gastroenterol. Nutr. 2003, 36, 623–631. [Google Scholar] [CrossRef]
- Fisher, J.E.; Khan, A.R.; Heitlinger, L.; Allen, J.E.; Afshani, E. Chronic granulomatous disease of childhood with acute ulcerative colitis: A unique association. Pediatr. Pathol. 1987, 7, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Oil, C.Y.; Durie, P.R. Cystic fibrosis from the gastroenterologist’s perspective. Nat. Rev. 2016, 13, 175–185. [Google Scholar]
- Wilschanski, M.; Durie, P.R. Patterns of GI disease in adulthood associated with mutations in the CFTR gene. Gut 2007, 56, 1153–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werlin, S.L. Evidence of intestinal inflammation in patients with cystic fibrosis. J. Pediatr. Gastrtroenterol. Nutr. 2010, 51, 304–308. [Google Scholar] [CrossRef]
- Fine, K.D.; Schiller, L.R. AGA Technical Review on the Evaluation and Management of Chronic Diarrhea☆. Gastroenterol. 1999, 116, 1464–1486. [Google Scholar] [CrossRef]
- Lee, C.S.; Perreault, N.; Brestelli, J.E.; Kaestner, K.H. Neurogenin 3 is essential for the proper specification of gastric enteroen-docrine cells and the maintenance of gastric epithelial cell identity. Genes Dev. 2002, 16, 1488–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonhoff, S.E.; Giel-Moloney, M.; Leiter, A.B. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differen-tiate into both endocrine and non-endocrine cell types. Dev. Biol. 2004, 270, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. Neurogenin3 is required for the development of theour endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA 2000, 97, 1607–1611. [Google Scholar] [CrossRef] [Green Version]
- Capriati, T.; Mosca, A.; Alterio, T.; Spagnuolo, M.I.; Gandullia, P.; Lezo, A.; Lionetti, P.; D’Antiga, L.; Fusaro, F.; Diamanti, A. To Wean or Not to Wean: The Role of Autologous Reconstructive Surgery in the Natural History of Pediatric Short Bowel Syndrome on Behalf of Italian Society for Gastroenterology, Hepatology and Nutrition (SIGENP). Nutrients 2020, 12, 2136. [Google Scholar] [CrossRef] [PubMed]
- Kocoshis, S.A.; Merritt, R.J.; Hill, S.; Protheroe, S.; Carter, B.A.; Horslen, S.; Hu, S.; Kaufman, S.S.; Mercer, D.F.; Pakarinen, M.P.; et al. Safety and Efficacy of Teduglutide in Pediatric Patients With Intestinal Failure due to Short Bowel Syndrome: A 24-Week, Phase III Study. J. Parenter. Enter. Nutr. 2020, 44, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.A.; Cole, C.R. Ultra-short bowel syndrome during infancy: Improving outcomes and novel therapies. Cur. Opin. Ped. 2019, 31, 177–181. [Google Scholar] [CrossRef]
- Di Mauro, S.; Hirano, M. Pathogenesis and treatment of mitochondrial disorders. Adv. Exp. Med. Biol. 2009, 652, 139–170. [Google Scholar]
- Finsterer, J.; Frank, M. Gastrointestinal manifestations of mitochondrial disorders: A systematic review. Ther. Adv. Gastroenterol. 2017, 10, 142–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, I.; Spinazzola, A.; Papadimitriou, A.; Hammans, S.; Steiner, I.; Hahn, C.D.; Connolly, A.M.; Verloes, A.; Guimarães, J.; Maillard, I.; et al. Mitochondrial neurogastrointestinal encephalomyopathy: An autosomal recessive disorder due to thymidine phosphorylase mutations. Ann. Neurol. 2000, 47, 792–800. [Google Scholar] [CrossRef]
- Howard, L.; Ament, M.; Fleming, C.R. Current use and clinical outcome of home parenteral and enteral nutrition thera-pies in the United States. Gastroenterology 1995, 109, 355–365. [Google Scholar] [CrossRef]
- Howard, L.; Heaphey, L.; Fleming, C.R.; Lininger, L.; Steiger, E. Four Years of North American Registry Home Parenteral Nutrition Outcome Data and Their Implications for Patient Management. J. Parenter. Enter. Nutr. 1991, 15, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Beath, S.V.; Gowen, H.; Puntis, J.W.L. Trends in pediatric home parenteral nutrition and implications for service devel-opment. Clin. Nutr. 2011, 30, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Dalzell, A.M. Management of intestinal failure in children. Arch. Dis. Child. 2015, 100, 980–983. [Google Scholar] [CrossRef]
- D’Antiga, L.; Goulet, O. Intestinal failure in children: The European view. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Hill, S. Use of GLP-2 may herald a new era of iproved outcome of short bowel syndrome-associated intestinal failure. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.A.; Cohran, V.C.; Cole, C.R.; Corkins, M.R.; Dimmitt, R.A.; Duggan, C.; Hill, S.; Horslen, S.; Lim, J.D.; Mercer, D.F.; et al. Outcomes from a 12-Week, Open-Label, Multicenter Clinical Trial of Teduglutide in Pediatric Short Bowel Syndrome. J. Pediatr. 2017, 181, 102–111.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos Boluda, E.; Redecillas Ferreiro, S.; Manrique Moral, O. Experience with teduglutide in pediatric short bowel syndrome: First real-life data. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 734–739. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| REF. | Author (year) | Country | Study-Period | n° of Patients | NDIF (%) | DIF (%) |
|---|---|---|---|---|---|---|
| [27] | Prasad D (2019) | India | 2017–2017 | 6 | 6 (100) | 0 |
| [28] | LaRusso K (2019) | Canada | 2006–2018 | 37 | 1 (3) | 36 (97) |
| [29] | Diamanti A (2019) | Italy | 1988–2018 | 48 | 9 (19) | 39 (81) |
| [30] | Zemrani B (2019) | Australia | 2018 | 1 | 1 (100) | / |
| [31] | Zapata Olivares Y (2019) | Chile | 2016–2017 | 46 | 3 (7) | 43 (93) |
| [32] | Fayard J (2018) | France | 2000–2017 | 65 | 65 (100) | / |
| [33] | Raphael BP. (2018) | USA | 2001–2016 | 78 | 2 (3) | 76 (97) |
| [34] | Gunnar R. (2018) | Finland | 2012–2015 | 49 | / | 49 (100) |
| [35] | Merras-Salmio L. (2018) | Finland | 1984–2017 | 100 | 3 (3) | 97 (97) |
| [36] | Diamanti A. (2017) | Italy | 2016 | 145 | 23 (16) | 122 (84) |
| [37] | Blotte C. (2017) | USA | 2012–2016 | 36 | / | 36 (100) |
| [38] | Giabicani E. (2017) | France | 2016 | 1 | 1 (100) | / |
| [39] | German-Diaz M. (2017) | Spain | 2016 | 1 | 1 (100) | / |
| [40] | Gonzalez-Hernandez J. (2017) | USA | 1999–2012 | 71 | / | 71 (100) |
| [41] | Stýblová J. (2017) | Czech Republic | 1995–2011 | 66 | 3 (5) | 63 (95) |
| [42] | Hashimura Y. (2016) | Japan | 2016 | 1 | / | 1 (100) |
| [25] | AbiNader E. (2016) | France | 2000–2013 | 251 | 34 (14) | 217 (86) |
| [40] | Gonzalez-Hernandez J. (2016) | USA | 2010–2014 | 30 | / | 30 (100) |
| [43] | Furtado S. (2015) | CANADA | 2007–2012 | 55 | / | 55 (100) |
| [44] | Pichler J. (2015) | UK | 2002–2010 | 71 | 7 (10) | 64 (90 |
| [45] | Mirabel-Chambaud E (2015) | France | 2007–2014 | 183 | 19 (10) | 164 (90) |
| [46] | Mezoff EA (2015) | USA | 2010–2013 | 30 | 3 (10) | 27 (90) |
| [47] | Neelis EG (2015) | Netherland | 2013 | 37 | / | 37 (100) |
| [48] | Pichler J (2014) | UK | 2006–2010 | 127 | 46 (36) | 81 (64) |
| [49] | Singhi AD (2014) | USA | 1996–2013 | 14 | / | 14 (100) |
| [50] | Godart F. (2014) | France | 2014 | 1 | 1 (100) | / |
| [10] | Hizarcioglu-Gulsen H. (2014) | Turkey | 2000–2010 | 60 | 37 (62) | 23 (38) |
| [51] | Duro D. (2014) | US | 2013 | 28 | / | 28 (100) |
| [52] | Courtney-Martin G. (2014) | Canada | 27 | 3 (11) | 24 (89) | |
| [53] | Martin GM (2013) | USA | 2004–2012 | 14 | 14 (100) | / |
| [54] | Samuels ME. (2013) | Canada | 2013 | 5 | 5 (100) | / |
| [55] | Ubesie AC. (2013) | USA | 2007–2012 | 123 | / | 123 (100) |
| [56] | Derepas C. (2013) | Canada | 2012 | 13 | 2 (15) | 11 (85) |
| [57] | Sadlier C. (2013) | UK | 2007–2010 | 36 | 13 (36,1%) | 23 (63.8%) |
| [58] | Pieroni KP. (2013) | USA | 2007–2011 | 14 | 1 (7) | 13 (93) |
| [59] | Javid PJ. (2011) | USA | 2005–2009 | 62 | / | 62 (100) |
| [60] | Diamanti A. (2010) | Italy | 2005–2007 | 24 | 1 (4) | 23 (96) |
| [61] | Pal K. (2010) | Saudi Arabia | 2009 | 1 | 1 (100) | / |
| Ref. n, Etiology | n° (%) | |
|---|---|---|
| Digestive IF | 1419 | |
| Short Bowel Syndrome | [25,28,31,32,33,34,35,36,37,40,41,43,44,45,46,47,48,51,52,55,56,57,58,59,60,62] | 943 (66) |
| Necrotizing enterocolitis | 364 | |
| Volvulus | 201 | |
| Gastroschisis | 191 | |
| Atresias | 177 | |
| Double malformations | 4 | |
| Spontaneous perforation | 3 | |
| Omphalocele | 3 | |
| Dismotility | [10,25,28,29,31,33,34,35,36,40,41,44,45,46,47,48,52,55,56,57,58,59,60,61] | 285 (20) |
| Chronic intestinal pseudo-obstruction | 154 | |
| Hirshprung disease | 107 | |
| Mitocondrial diseases | 21 | |
| Celiac disease | 2 | |
| Gastroschisis | 1 | |
| Malabsorption/maldigestion | [10,28,31,33,35,37,40,41,42,45,46,48,51,54,55,57,58,59] | 191 (14) |
| Congenital enteropathy | 93 | |
| Not classified | 16 | |
| Tufting enteropathy | 41 | |
| Mycrovillous inclusion disease | 31 | |
| Trichoepatoentehric Syndrome | 5 | |
| IBD | 49 | |
| Autoimmune enteropathy | 30 | |
| Selective malabsorption | 19 | |
| Glucose-galactoses | 12 | |
| Sucrase-isomaltases | 1 | |
| Diacylglycerol acyltransferase (DGAT 1) deficiency | 2 | |
| Abetalipoproteinemia | 4 |
| Ref. n, Etiology | n (%) | |
|---|---|---|
| Non-Digestive IF | 237 | |
| Intestinal Fistulas | [30] | 1 (0.4) |
| Ehlers-Danlos Syndrome | 1 | |
| SBS | [25,36,41,46,50,57] | 9 (3,8) |
| Trauma | 2 | |
| Cardiovascular diseases | 5 | |
| Mesenteric ischemia | 4 | |
| Kawasaky Syndrome | 1 | |
| Pancreatic diseases | 2 | |
| Meconium ileus (Cystic Fibrois) | 2 | |
| Dismotility | [29,36,38,52,57,61] | 14 (5.9) |
| Neurodisabling diseases | 8 | |
| Neuroevolutive disorders | 7 | |
| Teacher Collins Syndrome | 1 | |
| Chronic electrolytes/minerals imbalances | 4 | |
| Gitelman Syndrome | 2 | |
| Sanjad-Sakati Syndrome | 2 | |
| Fabricated disease | 1 | |
| Conditions affecting GI smooth muscle | 1 | |
| Congenital amioplasia | 1 | |
| Malabsorption/maldigestion | [10,25,27,28,31,32,33,35,36,39,41,42,43,44,45,46,48,49,51,52,53,54,57] | 213 (89.9) |
| Primary immunodeficiency | 109 | |
| Not classified | 39 | |
| IPEX Syndrome. | 2 | |
| Chronic granulomatosis | 1 | |
| Di George Syndrome | 1 | |
| TTC7A mutations | 66 | |
| Cancer + HSCT | 37 | |
| Not classified | 35 | |
| GVHD | 2 | |
| Pancreatic diseases (Pseudocystis and CF) | 9 | |
| Metabolic diseases | 2 | |
| Protein Glycosilation deficiency | 1 | |
| Mednik Syndrome | 1 | |
| Entero-endocrine | 17 | |
| Enteric anendocrinosis (Mut. in Neurogenin 3 gene) | 2 | |
| Pro-protein convertase 1/3 (PC1/3) deficiency | 15 | |
| Allergic enteropthy | 13 | |
| Cardiovascular diseases | 26 | |
| Congenital hearth defects | 2 | |
| Intestinal lymphangectasia | 24 |
| SPECIFIC TREATMENTS FOR PRIMARY DISEASES | |
|---|---|
| Primary disease | Treatments |
| Immunodeficiency | HSCT |
| Cystic Fibrosis | CFTR modulators to correct the basic defect |
| GVHD | Steroids is the first choice; for patients who develop steroid refractory GVHD the following rescue therapies are available: pentostatin, antithymocyte globulin, alemtuzumab, infliximab, basiliximab/infliximab combination therapy, ruxolitinib, tocilizumab, brentuximab vedotin, vedolizumab |
| Eosinophilic gastroenteritis |
|
| NON-SPECIFIC TREATMENTS FOR SYMPTOMS OF IF | |
| Pathophysiological mechanism | Treatments |
| PLE | Standard treatments: Low-fat diet (<25% of the caloric intake from fat), MCT supplementation, high protein diet (>2 gr/Kg/die), human albumin transfusion, vitamins, electrolyte supplements, gamma globulin infusions (if recurrent infections, preceded by low serum IgG), steroids and octreotide. Specific treatment for heart disorders at increased central venous pressure:
|
| Inflammation and recurrent infections in immunodeficiency | Steroids; immunosuppression; antibiotic and antifungal prophylaxis; prophylactic use of IFN-γ, infliximab in patients with colitis; immunoglobulin substitution. |
| Inflammation in Cystic Fibrosis | Careful management of enzyme usage, dysmotility and bacterial overgrowth |
| NON-SPECIFIC TREATMENTS FOR CHRONIC IF | |
| Cause of chronic IF | Treatments |
| SBS |
|
| PIPO |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diamanti, A.; Calvitti, G.; Martinelli, D.; Santariga, E.; Capriati, T.; Bolasco, G.; Iughetti, L.; Pujia, A.; Knafelz, D.; Maggiore, G. Etiology and Management of Pediatric Intestinal Failure: Focus on the Non-Digestive Causes. Nutrients 2021, 13, 786. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13030786
Diamanti A, Calvitti G, Martinelli D, Santariga E, Capriati T, Bolasco G, Iughetti L, Pujia A, Knafelz D, Maggiore G. Etiology and Management of Pediatric Intestinal Failure: Focus on the Non-Digestive Causes. Nutrients. 2021; 13(3):786. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13030786
Chicago/Turabian StyleDiamanti, Antonella, Giacomo Calvitti, Diego Martinelli, Emma Santariga, Teresa Capriati, Giulia Bolasco, Lorenzo Iughetti, Arturo Pujia, Daniela Knafelz, and Giuseppe Maggiore. 2021. "Etiology and Management of Pediatric Intestinal Failure: Focus on the Non-Digestive Causes" Nutrients 13, no. 3: 786. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13030786