
Colonic In Vitro Model Assessment of the Prebiotic Potential of Bread Fortified with Polyphenols Rich Olive Fiber

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fecal Donors
2.2. Materials
2.3. Experimental Bread and Controls
2.4. In vitro Gastric and Duodenal Digestion
2.5. Fecal Batch-Culture Fermentation and Samples Collection
2.6. Pipeline of Analytical Activities
2.6.1. DNA Extraction, Amplification and Sequencing
2.6.2. Sequence Data Analysis
2.6.3. Enumeration of Bacterial Groups
2.6.4. Volatilome Analysis
2.6.5. Statistical Analysis
3. Results and Discussion
3.1. Quality Controls for the Validation of MICODE
3.2. Changes in Fecal Bacterial Alpha and Beta Diversities
3.3. Fecal Bacterial Relative Abundance at the Phylum Level
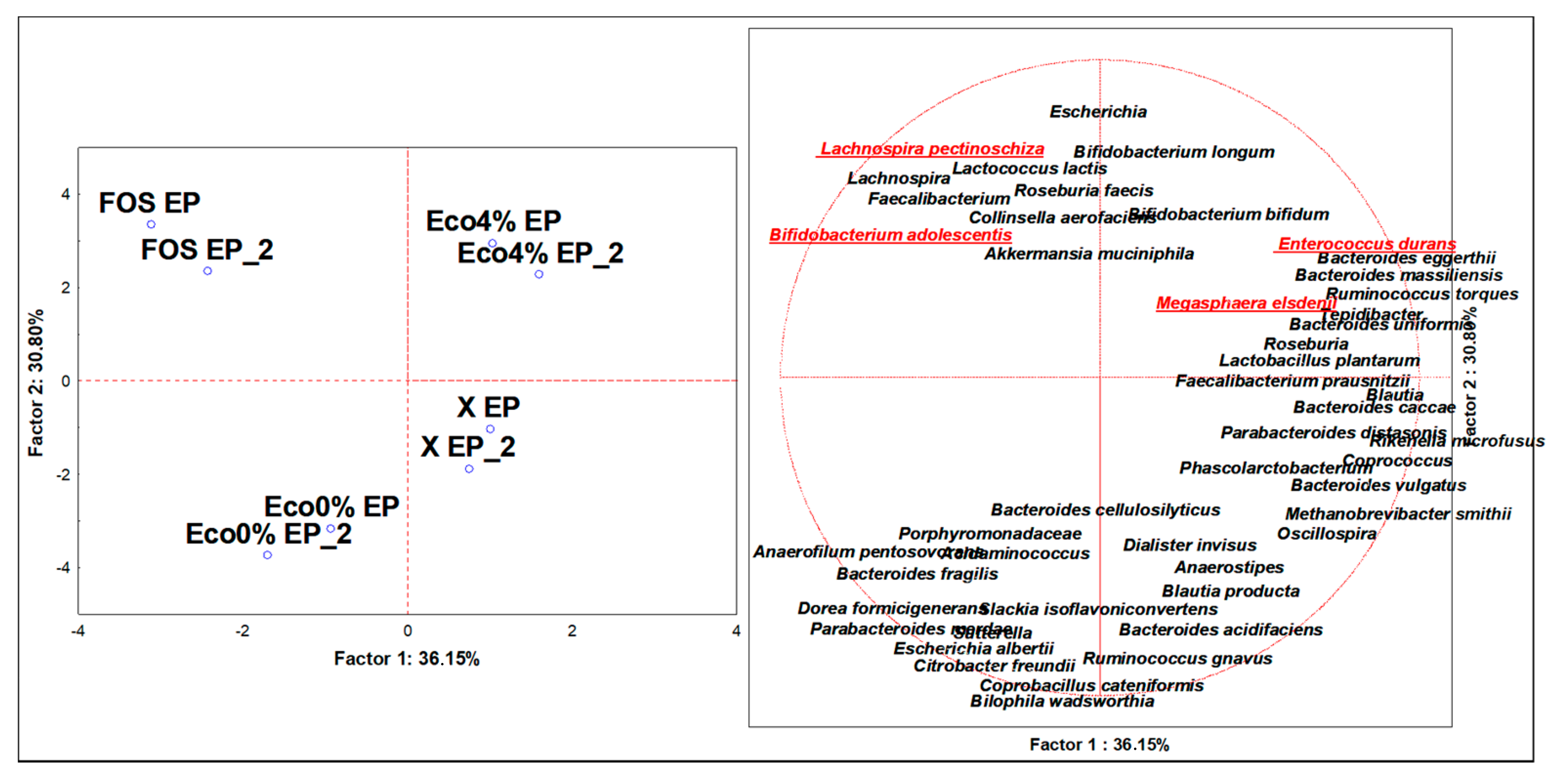
3.4. Discriminant Microbiota Populations at the Species Level
3.5. Changes in Selected Fecal Bacterial Populations Measured with qPCR
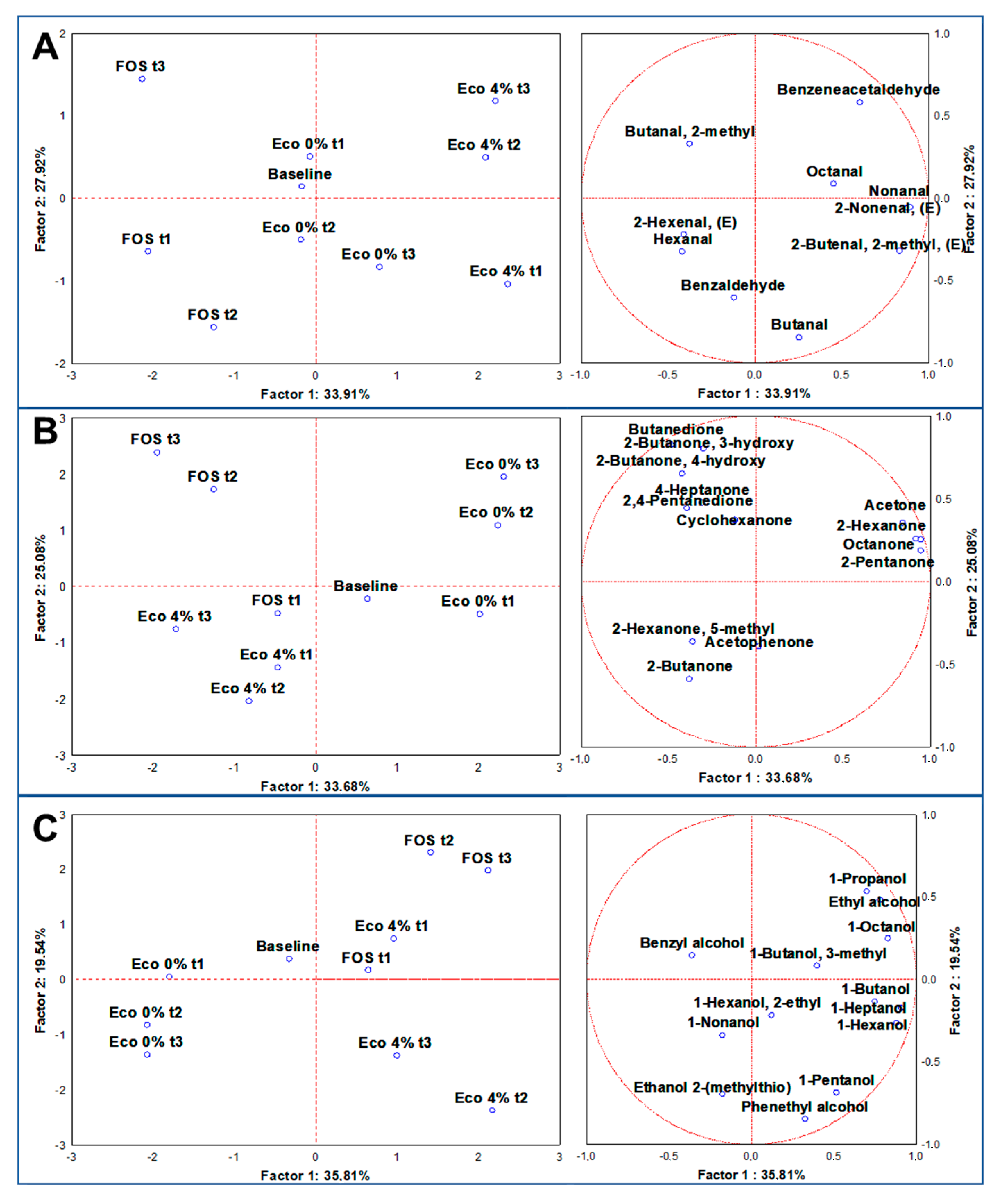
3.6. Volatilome Analysis through SPME GC/MS
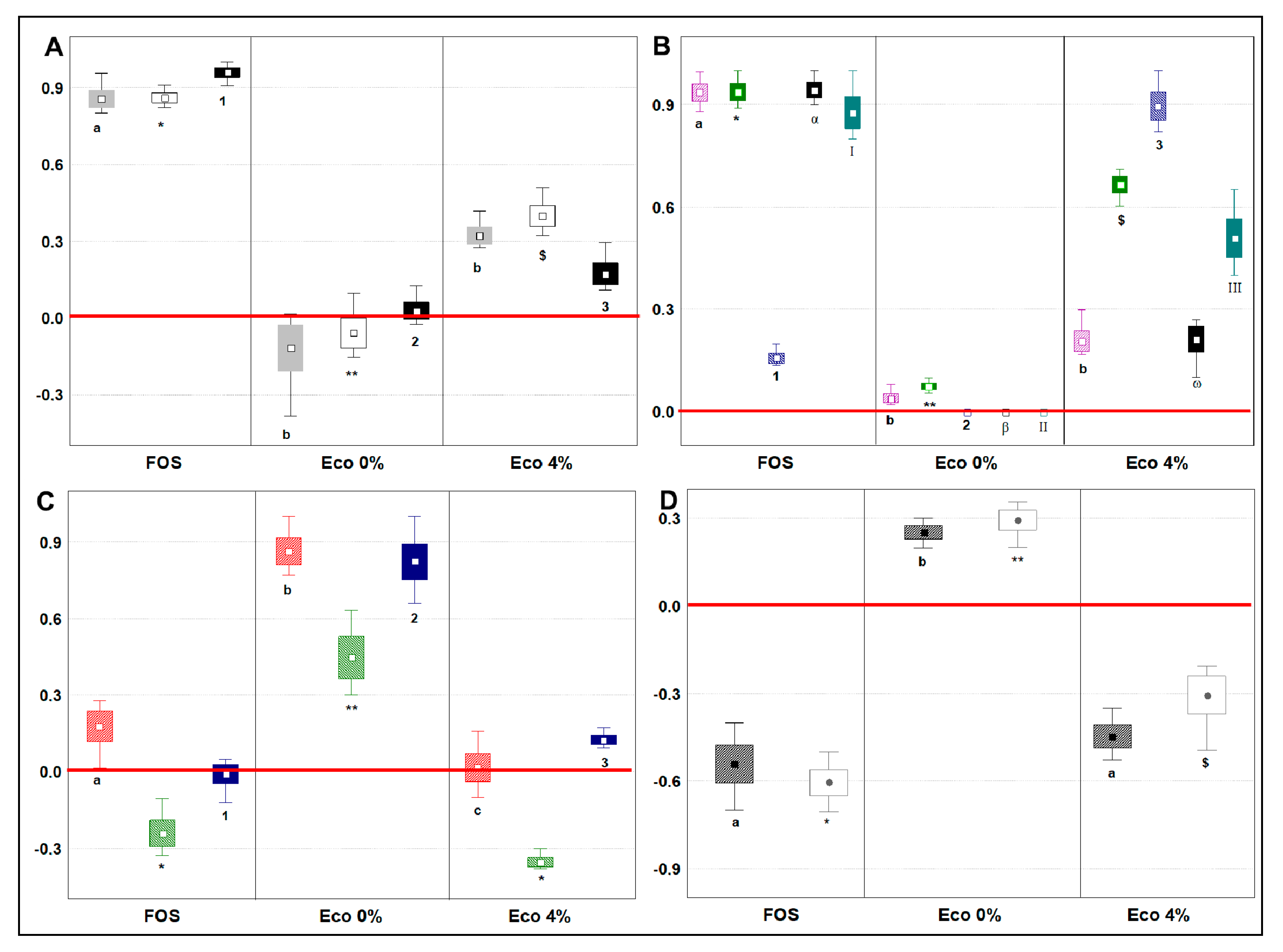
3.7. Changes in Main Microbial Metabolites Related to Prebiotic Potential
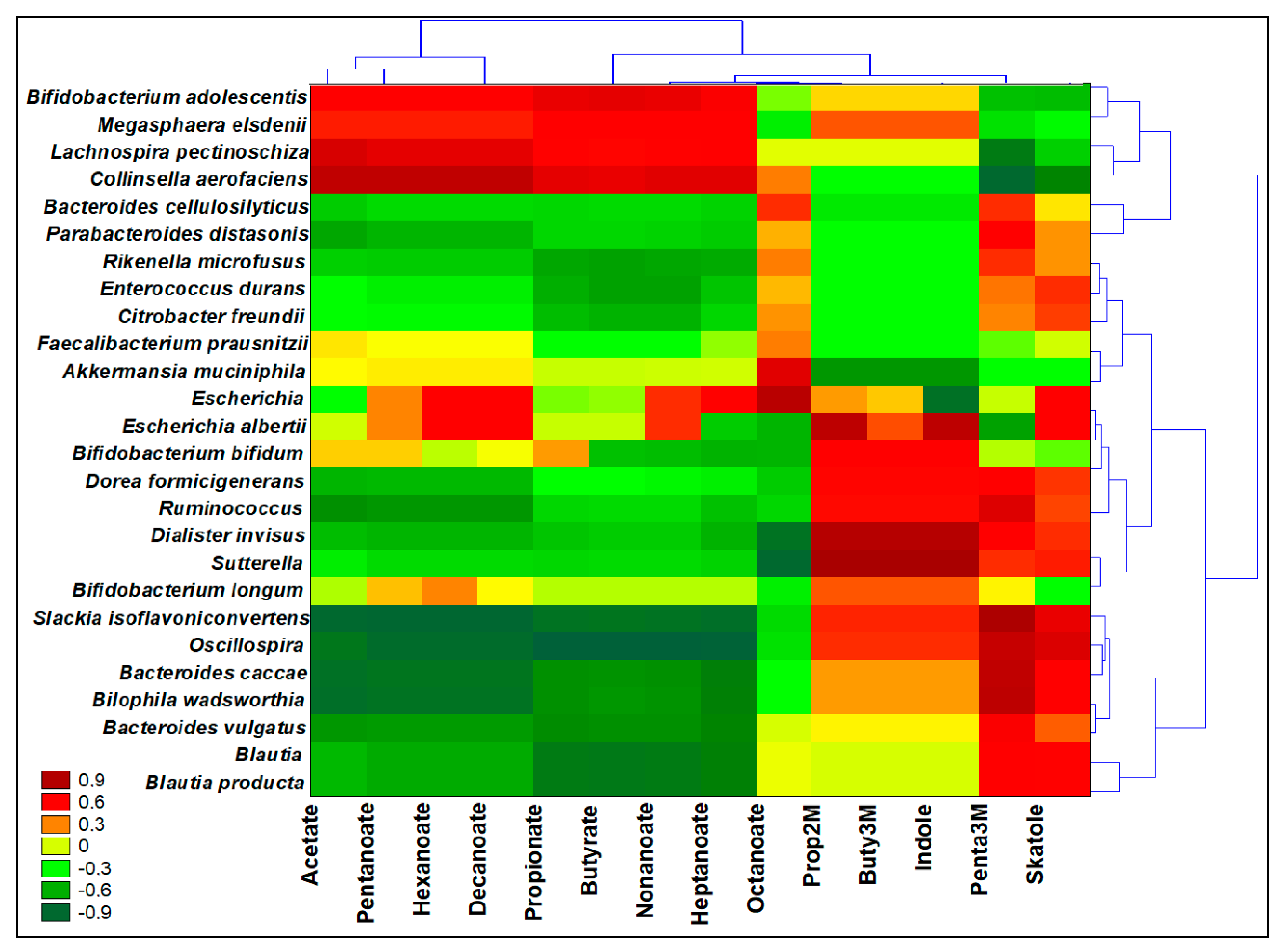
3.8. Interomics Correlations among Metabolites Related to Prebiotic Potential and the Microbiota
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boubaker, M.; El Omri, A.; Blecker, C.; Bouzouita, N. Fibre concentrate from artichoke (Cynara scolymus L.) stem by-products: Characterization and application as a bakery product ingredient. Food Sci. Technol. Int. 2016, 22, 759–768. [Google Scholar] [CrossRef]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, M.E.; Merenstein, D.J.; Reid, G.; Gibson, G.R.; Rastall, R.A. Probiotics and prebiotics in intestinal health and disease: From biology to the clinic. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, J.G.; Chain, F.; Martín, R.; Bermúdez-Humarán, L.G.; Courau, S.; Langella, P. Beneficial effects on host energy metabolism of short-chain fatty acids and vitamins produced by commensal and probiotic bacteria. Microb. Cell Fact. 2017, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Wu, W.; Chen, L.; Yang, W.; Huang, X.; Ma, C.; Chen, F.; Xiao, Y.; Zhao, Y.; Ma, C.; et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rial, S.A.; Karelis, A.D.; Bergeron, K.-F.; Mounier, C. Gut Microbiota and Metabolic Health: The Potential Beneficial Effects of a Medium Chain Triglyceride Diet in Obese Individuals. Nutrients 2016, 8, 281. [Google Scholar] [CrossRef] [Green Version]
- Oba, S.; Sunagawa, T.; Tanihiro, R.; Awashima, K.; Sugiyama, H.; Odani, T.; Nakamura, Y.; Kondo, A.; Sasaki, D.; Sasaki, K. Prebiotic effects of yeast mannan, which selectively promotes Bacteroides thetaiotaomicron and Bacteroides ovatus in a human colonic microbiota model. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Manor, O.; Dai, C.L.; Kornilov, S.A.; Smith, B.; Price, N.D.; Lovejoy, J.C.; Gibbons, S.M.; Magis, A.T. Health and disease markers correlate with gut microbiome composition across thousands of people. Nat. Commun. 2020, 11, 5206. [Google Scholar] [CrossRef]
- Wang, X.; Gibson, G.R.; Sailer, M.; Theis, S.; Rastall, R.A. Prebiotics Inhibit Proteolysis by Gut Bacteria in a Host Diet-Dependent Manner: A Three-Stage Continuous In vitro Gut Model Experiment. Appl. Environ. Microbiol. 2020, 86, 02730-19. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, L.; Casciano, F.; Gianotti, A. Intestinal fermentation in vitro models to study food-induced gut microbiota shift: An updated review. FEMS Microbiol. Lett. 2020, 367, 12. [Google Scholar] [CrossRef]
- Di Nunzio, M.; Picone, G.; Pasini, F.; Caboni, M.F.; Gianotti, A.; Bordoni, A.; Capozzi, F. Olive oil industry by-products. Effects of a polyphenol-rich extract on the metabolome and response to inflammation in cultured intestinal cell. Food Res. Int. 2018, 113, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nunzio, M.; Picone, G.; Pasini, F.; Chiarello, E.; Caboni, M.F.; Capozzi, F.; Gianotti, A.; Bordoni, A. Olive oil by-product as functional ingredient in bakery products. Influence of processing and evaluation of biological effects. Food Res. Int. 2020, 131, 108940. [Google Scholar] [CrossRef] [Green Version]
- Minekus, M.; Alminger, M.; Alvito, P.; Ballance, S.; Bohn, T.; Bourlieu, C.; Carrière, F.; Boutrou, R.; Corredig, M.; Dupont, D.; et al. A standardised static in vitro digestion method suitable for food—An international consensus. Food Funct. 2014, 5, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Connolly, M.L.; Tuohy, K.M.; Lovegrove, J.A. Wholegrain oat-based cereals have prebiotic potential and low glycaemic index. Br. J. Nutr. 2012, 108, 2198–2206. [Google Scholar] [CrossRef] [Green Version]
- Koutsos, A.; Lima, M.; Conterno, L.; Gasperotti, M.; Bianchi, M.; Fava, F.; Vrhovsek, U.; Lovegrove, J.A.; Tuohy, K.M. Effects of Commercial Apple Varieties on Human Gut Microbiota Composition and Metabolic Output Using an In vitro Colonic Model. Nutrients 2017, 9, 533. [Google Scholar] [CrossRef] [Green Version]
- Marino, M.; De Wittenau, G.D.; Saccà, E.; Cattonaro, F.; Spadotto, A.; Innocente, N.; Radovic, S.; Piasentier, E.; Marroni, F. Metagenomic profiles of different types of Italian high-moisture Mozzarella cheese. Food Microbiol. 2019, 79, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naìve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.K.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.J.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2011, 6, 610–618. [Google Scholar] [CrossRef]
- Tanner, S.A.; Berner, A.Z.; Rigozzi, E.; Grattepanche, F.; Chassard, C.; Lacroix, C. In vitro Continuous Fermentation Model (PolyFermS) of the Swine Proximal Colon for Simultaneous Testing on the Same Gut Microbiota. PLoS ONE 2014, 9, e94123. [Google Scholar] [CrossRef] [Green Version]
- Nissen, L.; Di Carlo, E.; Gianotti, A. Prebiotic potential of hemp blended drinks fermented by probiotics. Food Res. Int. 2020, 131, 109029. [Google Scholar] [CrossRef]
- Saa, D.T.; Turroni, S.; Serrazanetti, D.I.; Rampelli, S.; Maccaferri, S.; Candela, M.; Severgnini, M.; Simonetti, E.; Brigidi, P.; Gianotti, A. Impact of Kamut® Khorasan on gut microbiota and metabolome in healthy volunteers. Food Res. Int. 2014, 63, 227–232. [Google Scholar] [CrossRef]
- Koliada, A.; Syzenko, G.; Moseiko, V.; Budovska, L.; Puchkov, K.; Perederiy, V.; Gavalko, Y.; Dorofeyev, A.; Romanenko, M.; Tkach, S.; et al. Association between body mass index and Firmicutes/Bacteroidetes ratio in an adult Ukrainian population. BMC Microbiol. 2017, 17, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, B.S.; Hansen, E.E.; Manchester, J.K.; Coutinho, P.M.; Henrissat, B.; Fulton, R.; Latreille, P.; Kim, K.; Wilson, R.K.; Gordon, J.I. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc. Natl. Acad. Sci. USA 2007, 104, 10643–10648. [Google Scholar] [CrossRef] [Green Version]
- Takagi, R.; Sasaki, K.; Sasaki, D.; Fukuda, I.; Tanaka, K.; Yoshida, K.-I.; Kondo, A.; Osawa, R. A Single-Batch Fermentation System to Simulate Human Colonic Microbiota for High-Throughput Evaluation of Prebiotics. PLoS ONE 2016, 11, e0160533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Wang, Y.; Jacoby, J.J.; Jiang, Y.; Zhang, Y.; Yu, L.L. Effects of Medium- and Long-Chain Triacylglycerols on Lipid Metabolism and Gut Microbiota Composition in C57BL/6J Mice. J. Agric. Food Chem. 2017, 65, 6599–6607. [Google Scholar] [CrossRef]
- Farràs, M.; Martinez-Gili, L.; Portune, K.; Arranz, S.; Frost, G.; Tondo, M.; Blanco-Vaca, F. Modulation of the Gut Microbiota by Olive Oil Phenolic Compounds: Implications for Lipid Metabolism, Immune System, and Obesity. Nutrients 2020, 12, 2200. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Genet. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Hiippala, K.; Kainulainen, V.; Suutarinen, M.; Heini, T.; Bowers, J.R.; Jasso-Selles, D.; Lemmer, D.; Valentine, M.; Barnes, R.; Engelthaler, D.M.; et al. Isolation of Anti-Inflammatory and Epithelium Reinforcing Bacteroides and Parabacteroides Spp. from A Healthy Fecal Donor. Nutrients 2020, 12, 935. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Liao, M.; Zhou, N.; Bao, L.; Ma, K.; Zheng, Z.; Wang, Y.; Liu, C.; Wang, W.; Wang, J.; et al. Parabacteroides distasonis Alleviates Obesity and Metabolic Dysfunctions via Production of Succinate and Secondary Bile Acids. Cell Rep. 2019, 26, 222–235.e5. [Google Scholar] [CrossRef] [Green Version]
- Callaway, T.R.; Edrington, T.S.; Anderson, R.C.; Harvey, R.B.; Genovese, K.J.; Kennedy, C.N.; Venn, D.W.; Nisbet, D.J. Probiotics, prebiotics and competitive exclusion for prophylaxis against bacterial disease. Anim. Heal. Res. Rev. 2008, 9, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeney, D.D.; Zhai, Z.; Bendiks, Z.; Barouei, J.; Martinic, A.; Slupsky, C.; Marco, M.L. Lactobacillus plantarum bacteriocin is associated with intestinal and systemic improvements in diet-induced obese mice and maintains epithelial barrier integrity in vitro. Gut Microbes 2019, 10, 382–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltani, S.; Hammami, R.; Cotter, P.D.; Rebuffat, S.; Ben Said, L.; Gaudreau, H.; Bédard, F.; Biron, E.; Drider, D.; Fliss, I. Bacteriocins as a new generation of antimicrobials: Toxicity aspects and regulations. FEMS Microbiol. Rev. 2021, 45, 039. [Google Scholar] [CrossRef]
- Dewulf, E.M.; Cani, P.; Claus, S.P.; Fuentes, S.; Puylaert, P.G.B.; Neyrinck, A.M.; Bindels, L.B.; De Vos, W.M.; Gibson, G.R.; Thissen, J.-P.; et al. Insight into the prebiotic concept: Lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut 2012, 62, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Sasaki, T.; Itoh, K.; Kitahara, T.; Takema, Y.; Hiramatsu, K.; Ishikawa, D.; Shibuya, T.; Kobayashi, O.; Osada, T.; et al. A Soluble Fiber Diet Increases Bacteroides fragilis Group Abundance and Immunoglobulin A Production in the Gut. Appl. Environ. Microbiol. 2020, 86, 00405–00420. [Google Scholar] [CrossRef] [PubMed]
- Ganji, L.; Alebouyeh, M.; Shirazi, M.H.; Eshraghi, S.S.; Mirshafiey, A.; Daryani, E.N.; Zali, M.R. Dysbiosis of fecal microbiota and high frequency of Citrobacter, Klebsiella spp., and Actinomycetes in patients with irritable bowel syndrome and gastroen-teritis. Gastroenterol. Hepatol. Bed Bench 2016, 9, 325. [Google Scholar]
- Ooka, T.; Seto, K.; Kawano, K.; Kobayashi, H.; Etoh, Y.; Ichihara, S.; Kaneko, A.; Isobe, J.; Yamaguchi, K.; Horikawa, K.; et al. Clinical Significance ofEscherichia albertii. Emerg. Infect. Dis. 2012, 18, 488–492. [Google Scholar] [CrossRef] [Green Version]
- Henke, M.T.; Kenny, D.J.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. USA 2019, 116, 12672–12677. [Google Scholar] [CrossRef] [Green Version]
- Natividad, J.M.; Lamas, B.; Pham, H.P.; Michel, M.-L.; Rainteau, D.; Bridonneau, C.; Da Costa, G.; Vlieg, J.V.H.; Sovran, B.; Chamignon, C.; et al. Bilophila wadsworthia aggravates high fat diet induced metabolic dysfunctions in mice. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, M.; Prieto, I.; Abriouel, H.; Villarejo, A.B.; Ramírez-Sánchez, M.; Cobo, A.; Benomar, N.; Gálvez, A.; Martínez-Cañamero, M. Changes in Gut Microbiota Linked to a Reduction in Systolic Blood Pressure in Spontaneously Hypertensive Rats Fed an Extra Virgin Olive Oil-Enriched Diet. Plant Foods Hum. Nutr. 2017, 73, 1–6. [Google Scholar] [CrossRef]
- Nissen, L.; Rollini, M.; Picozzi, C.; Musatti, A.; Foschino, R.; Gianotti, A. Yeast-Free Doughs by Zymomonas mobilis: Evaluation of Technological and Fermentation Performances by Using a Metabolomic Approach. Microorganisms 2020, 8, 792. [Google Scholar] [CrossRef]
- Malaguarnera, G.; Giordano, M.; Nunnari, G.; Bertino, G.; Malaguarnera, M. Gut microbiota in alcoholic liver disease: Pathogenetic role and therapeutic perspectives. World J. Gastroenterol. 2014, 20, 16639–16648. [Google Scholar] [CrossRef] [PubMed]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, H.-K.; Lee, J.Y. Molecular Basis of Alcohol-Related Gastric and Colon Cancer. Int. J. Mol. Sci. 2017, 18, 1116. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.J.; Buescher, R.W.; Johnson, M.; Janes, M. Inactivation of Pathogenic Bacteria by Cucumber Volatiles (E,Z)-2,6-Nonadienal and (E)-2-Nonenal. J. Food Prot. 2004, 67, 1014–1016. [Google Scholar] [CrossRef] [PubMed]
- Mitro, S.; Gordon, A.R.; Olsson, M.J.; Lundström, J.N. The Smell of Age: Perception and Discrimination of Body Odors of Different Ages. PLoS ONE 2012, 7, e38110. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Mulero, A.; Tinahones, A.; Bandera, B.; Moreno-Indias, I.; Macías-González, M.; Tinahones, F.J. Keto microbiota: A powerful contributor to host disease recovery. Rev. Endocr. Metab. Disord. 2019, 20, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Bradberry, S. Acetone. Medicine 2007, 35, 581. [Google Scholar] [CrossRef]
- Tran, T.D.; Olsson, M.A.; McMillan, D.J.; Cullen, J.K.; Parsons, P.G.; Reddell, P.W.; Ogbourne, S.M. Potent Antibacterial Prenylated Acetophenones from the Australian Endemic Plant Acronychia crassipetala. Antibiotics 2020, 9, 487. [Google Scholar] [CrossRef] [PubMed]
- Taslimi, P.; Sujayev, A.; Karaman, M.; Maharramova, G.; Sadeghian, N.; Osmanova, S.; Sardarova, S.; Majdi, N.; Ozel, H.U.; Gulcin, I. N -Substituted pyrimidinethione and acetophenone derivatives as a new therapeutic approach in diabetes. Arch. Pharm. 2020, 353, e2000075. [Google Scholar] [CrossRef]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Moens, F.; Abbeele, P.V.D.; Basit, A.W.; Dodoo, C.; Chatterjee, R.; Smith, B.; Gaisford, S. A four-strain probiotic exerts positive immunomodulatory effects by enhancing colonic butyrate production in vitro. Int. J. Pharm. 2019, 555, 1–10. [Google Scholar] [CrossRef]
- Santos, M.M.; Piccirillo, C.; Castro, P.M.L.; Kalogerakis, N.; Pintado, M.E. Bioconversion of oleuropein to hydroxytyrosol by lactic acid bacteria. World J. Microbiol. Biotechnol. 2012, 28, 2435–2440. [Google Scholar] [CrossRef] [PubMed]
- Lundsgaard, A.-M.; Fritzen, A.M.; Sjøberg, K.A.; Kleinert, M.; Richter, E.A.; Kiens, B. Small Amounts of Dietary Medium-Chain Fatty Acids Protect Against Insulin Resistance During Caloric Excess in Humans. Diabetes 2021, 70, 91–98. [Google Scholar] [CrossRef]
- De Preter, V.; Machiels, K.; Joossens, M.; Arijs, I.; Matthys, C.; Vermeire, S.; Rutgeerts, P.; Verbeke, K. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut 2015, 64, 447–458. [Google Scholar] [CrossRef]
- Scarborough, M.J.; Myers, K.S.; Donohue, T.J.; Noguera, D.R. Medium-Chain Fatty Acid Synthesis by “Candidatus Weimeria bifida” gen. nov., sp. nov., and “Candidatus Pseudoramibacter fermentans” sp. nov. Appl. Environ. Microbiol. 2019, 86, 02242-19. [Google Scholar] [CrossRef] [Green Version]
- Rivière, A.; Selak, M.; Geirnaert, A.; Abbeele, P.V.D.; De Vuyst, L. Complementary Mechanisms for Degradation of Inulin-Type Fructans and Arabinoxylan Oligosaccharides among Bifidobacterial Strains Suggest Bacterial Cooperation. Appl. Environ. Microbiol. 2018, 84, 02893-17. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, M.; Venema, K. Challenges in simulating the human gut for understanding the role of the microbiota in obesity. Benef. Microbes 2017, 8, 31–53. [Google Scholar] [CrossRef]
- Yao, C.K.; Muir, J.G.; Gibson, P.R. Review article: Insights into colonic protein fermentation, its modulation and potential health implications. Aliment. Pharmacol. Ther. 2016, 43, 181–196. [Google Scholar] [CrossRef] [Green Version]
- Diether, N.E.; Willing, B.P. Microbial Fermentation of Dietary Protein: An Important Factor in Diet–Microbe–Host Interaction. Microorganisms 2019, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Hendrikx, T.; Schnabl, B. Indoles: Metabolites produced by intestinal bacteria capable of controlling liver disease manifestation. J. Intern. Med. 2019, 286, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2009, 107, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.J.; Harrison, A.P.; Stahl, D.; Pace, B.; Giovannoni, S.J.; Olsen, G.J.; Pace, N.R. Evolutionary relationships among sulfur- and iron-oxidizing eubacteria. J. Bacteriol. 1992, 174, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartosch, S.; Fite, A.; Macfarlane, G.T.; McMurdo, M.E.T. Characterization of Bacterial Communities in Feces from Healthy Elderly Volunteers and Hospitalized Elderly Patients by Using Real-Time PCR and Effects of Antibiotic Treatment on the Fecal Microbiota. Appl. Environ. Microbiol. 2004, 70, 3575–3581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, J.; Hertel, C.; Tannock, G.W.; Lis, C.M.; Munro, K.; Hammes, W.P. Detection of Lactobacillus, Pediococcus, Leuconostoc, and Weissella Species in Human Feces by Using Group-Specific PCR Primers and Denaturing Gradient Gel Electrophoresis. Appl. Environ. Microbiol. 2001, 67, 2578–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masco, L.; Ventura, M.; Zink, R.; Huys, G.; Swings, J. Polyphasic taxonomic analysis of Bifidobacterium animalis and Bifidobacterium lactis reveals relatedness at the subspecies level: Reclassification of Bifidobacterium animalis as Bifidobacterium animalis subsp. animalis subsp. nov. and Bifidobacterium lactis as Bifidobacterium animalis subsp. lactis subsp. nov. Int. J. Syst. Evol. Microbiol. 2004, 54, 1137–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Helmstetter, C.E. Relationship between ftsZ gene expression and chromosome replication in Escherichia coli. J. Bacteriol. 1994, 176, 6100–6106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, E.; Amir, I.; Zafran, M.; Gophna, U.; Samra, Z.; Pitlik, S.; Bishara, J. The correlation between Clostridium-difficile infection and human gut concentrations of Bacteroidetes phylum and clostridial species. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 33, 377–383. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline | Endpoint | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phylum | FOS | Eco0% | Eco4% | |||||||||
| Firmicutes | 54.005 | ± | 0.635 b | 41.997 | ± | 1.111 a | 63.470 | ± | 0.651 c | 41.509 | ± | 0.596 a |
| Bacteroidetes | 33.997 | ± | 0.741 a | 23.957 | ± | 0.870 b | 17.418 | ± | 0.422 c | 33.264 | ± | 0.529 a |
| Actinobacteria | 7.537 | ± | 0.613 a | 27.832 | ± | 1.232 b | 6.338 | ± | 0.738 a | 16.696 | ± | 1.058 c |
| Proteobacteria | 1.762 | ± | 0.193 a | 3.577 | ± | 0.544 b | 11.628 | ± | 1.344 c | 5.571 | ± | 0.798 b |
| Verrucomicrobia | 1.775 | ± | 0.218 a | 1.175 | ± | 0.128 b | 0.207 | ± | 0.065 c | 1.910 | ± | 0.478 a |
| Euryarchaeota | 0.145 | ± | 0.023 a | 0.010 | ± | 0.002 c | 0.076 | ± | 0.006 b | 0.030 | ± | 0.004 c |
| Fusobacteria | 0.009 | ± | 0.001 a | 0.001 | ± | 0.000 a | 0.001 | ± | 0.000 a | 0.084 | ± | 0.014 b |
| Synergistetes | 0.011 | ± | 0.002 a | 0.001 | ± | 0.000 b | 0.007 | ± | 0.002 a | 0.001 | ± | 0.000 b |
| Tenericutes | 0.009 | ± | 0.001 a | >0.001 | ± | 0.000 b | 0.001 | ± | 0.000 a | >0.001 | ± | 0.000 b |
| Crenarchaeota | 0.001 | ± | 0.000 a | >0.001 | ± | 0.000 b | >0.001 | ± | 0.000 b | 0.001 | ± | 0.000 a |
| Bacteria; Other | 0.637 | ± | 0.098 a | 0.038 | ± | 0.009 c | 0.071 | ± | 0.012 b | 0.107 | ± | 0.021 b |
| Archaea; Other | 0.004 | ± | 0.001 a | 0.001 | ± | 0.000 a | 0.002 | ± | 0.000 a | 0.001 | ± | 0.000 a |
| Unclassified | 0.030 | ± | 0.005 a | 0.005 | ± | 0.001 b | 0.017 | ± | 0.005 a | 0.018 | ± | 0.006 a |
| F/B 1 | 1.589 | ± | 0.053 * | 1.753 | ± | 0.017 * | 3.644 | ± | 0.051 § | 1.247 | ± | 0.012 * |
| Time (h) | Eubacteria | Bifidobacteriaceae | Lactobacillales | Enterobacteriaceae | Escherichia coli | Clostridiaceae | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FOS | 0 | 9.16 | ± | 0.16 a | 6.77 | ± | 0.11 a | 7.36 | ± | 0.09 a | 8.60 | ± | 0.07 b | 4.08 | ± | 0.03 ab | 7.15 | ± | 0.11 a |
| 5 | 9.32 | ± | 0.10 a | 6.99 | ± | 0.12 ab | 7.76 | ± | 0.10 ab | 8.62 | ± | 0.13 b | 4.40 | ± | 0.10 b | 7.22 | ± | 0.04 a | |
| 10 | 9.77 | ± | 0.09 ab | 7.48 | ± | 0.09 b | 8.31 | ± | 0.09 b | 8.51 | ± | 0.02 ab | 4.62 | ± | 0.07 b | 7.67 | ± | 0.03 ab | |
| 24 | 10.09 | ± | 0.28 b | 8.81 | ± | 0.23 c | 8.79 | ± | 0.11 b | 8.05 | ± | 0.06 a | 3.62 | ± | 0.07 a | 7.34 | ± | 0.30 a | |
| Eco 0% | 0 | 9.12 | ± | 0.25 a | 6.47 | ± | 0.08 a | 7.11 | ± | 0.09 a | 8.71 | ± | 0.08 b | 4.00 | ± | 0.07 ab | 7.11 | ± | 0.11 a |
| 5 | 9.00 | ± | 0.11 a | 6.71 | ± | 0.09 a | 7.65 | ± | 0.11 ab | 8.91 | ± | 0.12 bc | 4.40 | ± | 0.08 b | 7.35 | ± | 0.11 a | |
| 10 | 9.41 | ± | 0.26 a | 6.68 | ± | 0.09 a | 7.90 | ± | 0.14 ab | 9.14 | ± | 0.11 bc | 4.92 | ± | 0.11 bc | 7.95 | ± | 0.21 b | |
| 24 | 9.57 | ± | 0.07 ab | 6.27 | ± | 0.08 a | 7.71 | ± | 0.11 ab | 9.44 | ± | 0.23 c | 5.13 | ± | 0.21 c | 8.10 | ± | 0.10 b | |
| Eco 4% | 0 | 9.02 | ± | 0.12 a | 6.77 | ± | 0.10 a | 7.24 | ± | 0.10 a | 8.40 | ± | 0.06 ab | 4.31 | ± | 0.07 b | 7.01 | ± | 0.10 a |
| 5 | 9.22 | ± | 0.08 a | 7.10 | ± | 0.10 ab | 7.36 | ± | 0.11 a | 8.62 | ± | 0.11 b | 4.17 | ± | 0.17 ab | 7.23 | ± | 0.20 a | |
| 10 | 9.70 | ± | 0.09 ab | 7.74 | ± | 0.09 b | 7.98 | ± | 0.21 b | 8.70 | ± | 0.08 b | 4.22 | ± | 0.16 ab | 7.47 | ± | 0.10 ab | |
| 24 | 10.03 | ± | 0.20 b | 8.55 | ± | 0.15 c | 8.80 | ± | 0.14 b | 9.16 | ± | 0.19 bc | 3.92 | ± | 0.11 a | 8.01 | ± | 0.19 b | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nissen, L.; Casciano, F.; Chiarello, E.; Di Nunzio, M.; Bordoni, A.; Gianotti, A. Colonic In Vitro Model Assessment of the Prebiotic Potential of Bread Fortified with Polyphenols Rich Olive Fiber. Nutrients 2021, 13, 787. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13030787
Nissen L, Casciano F, Chiarello E, Di Nunzio M, Bordoni A, Gianotti A. Colonic In Vitro Model Assessment of the Prebiotic Potential of Bread Fortified with Polyphenols Rich Olive Fiber. Nutrients. 2021; 13(3):787. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13030787
Chicago/Turabian StyleNissen, Lorenzo, Flavia Casciano, Elena Chiarello, Mattia Di Nunzio, Alessandra Bordoni, and Andrea Gianotti. 2021. "Colonic In Vitro Model Assessment of the Prebiotic Potential of Bread Fortified with Polyphenols Rich Olive Fiber" Nutrients 13, no. 3: 787. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13030787