
Inhibition of Antiestrogen-Promoted Pro-Survival Autophagy and Tamoxifen Resistance in Breast Cancer through Vitamin D Receptor

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. High VDR Expression Is Associated with Longer Recurrence-Free Survival in TAM-Treated Breast Cancer Patients
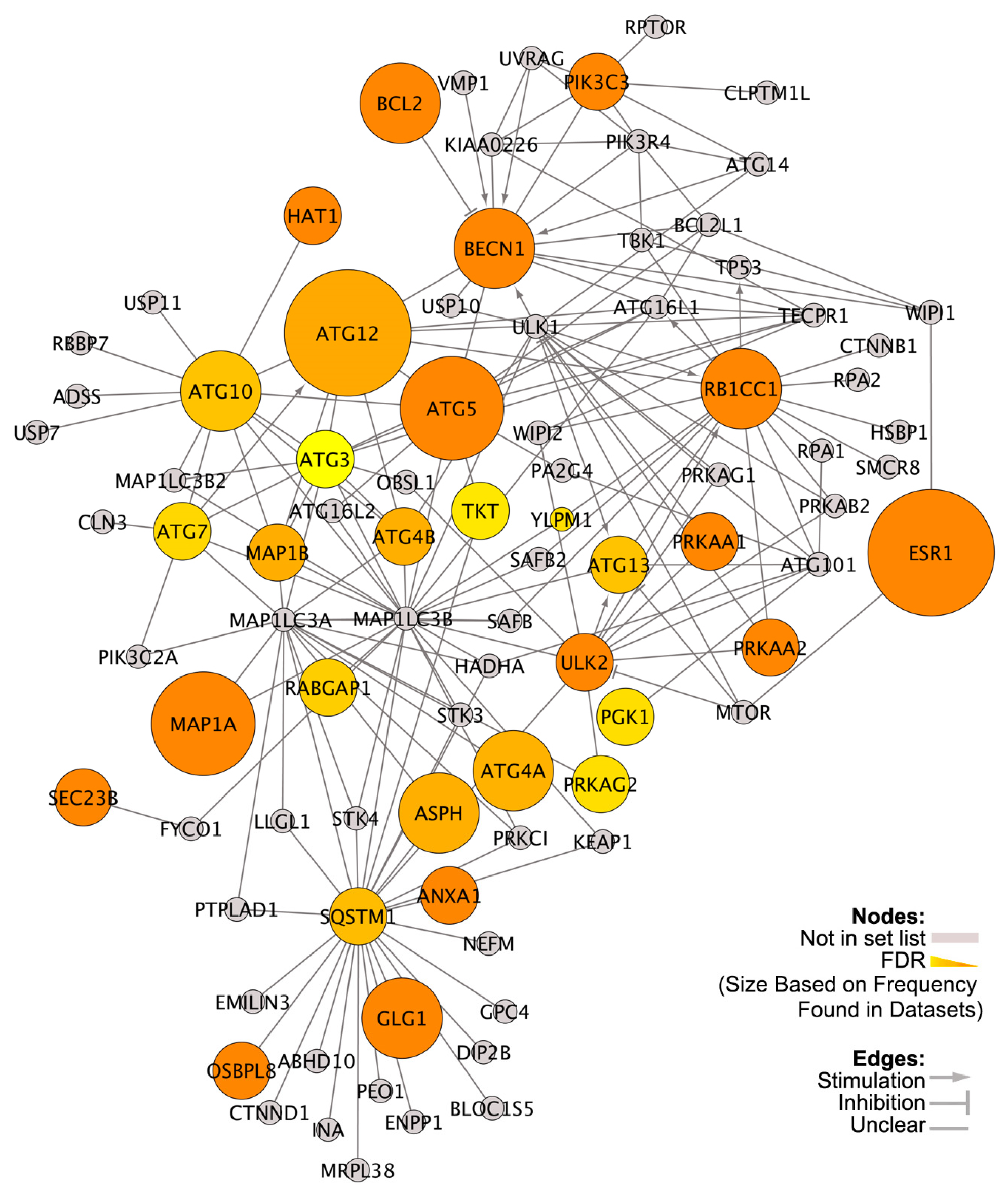
3.2. Differentially Expressed Genes (DEGs) in High versus Low VDR-Expressing ER+ Breast Cancers
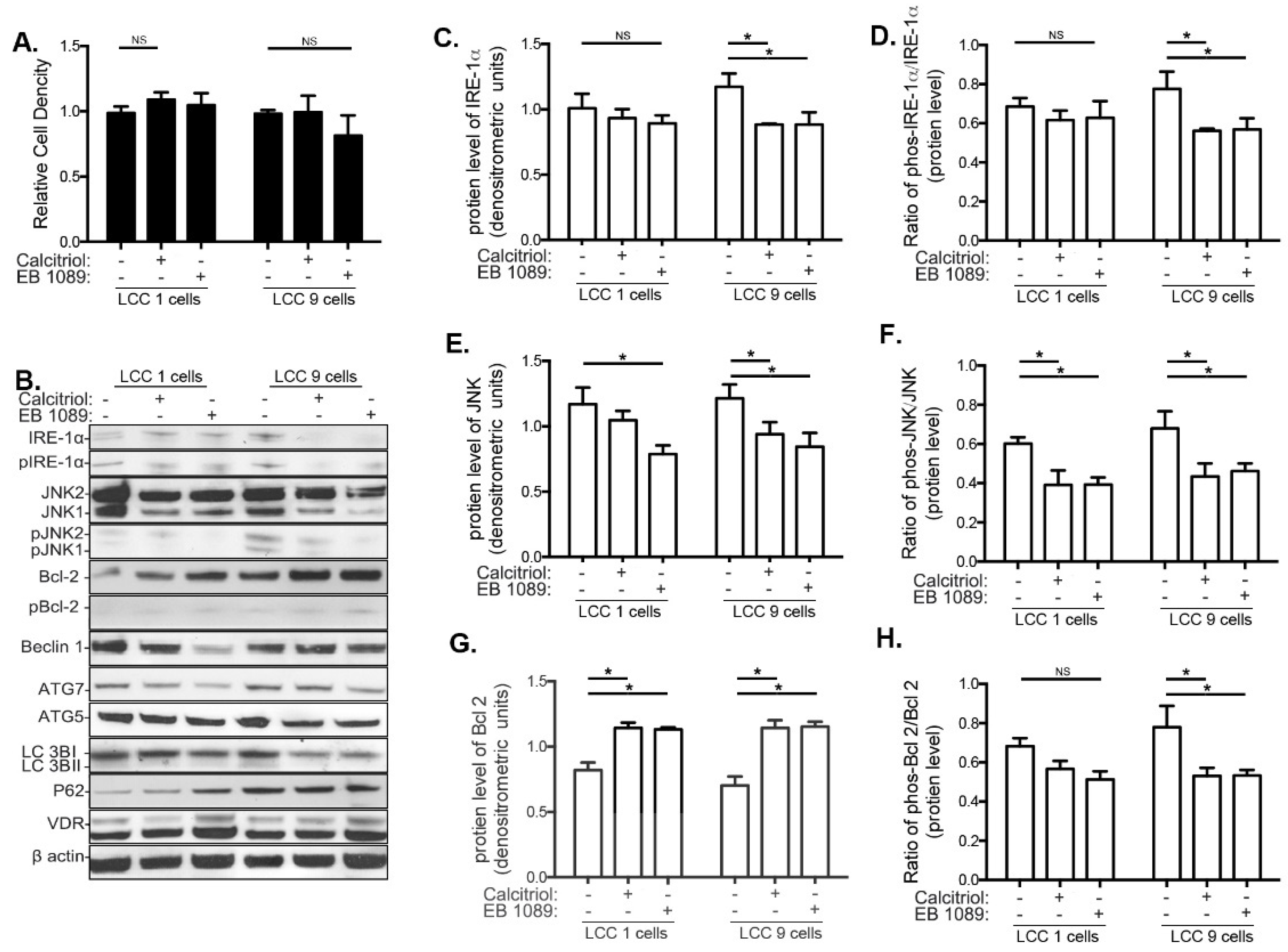
3.3. Effects of Calcitriol and EB1089 on Cancer Cell Proliferation, UPR and Autophagy in LCC1 and LCC9 Human Breast Cancer Cell Lines
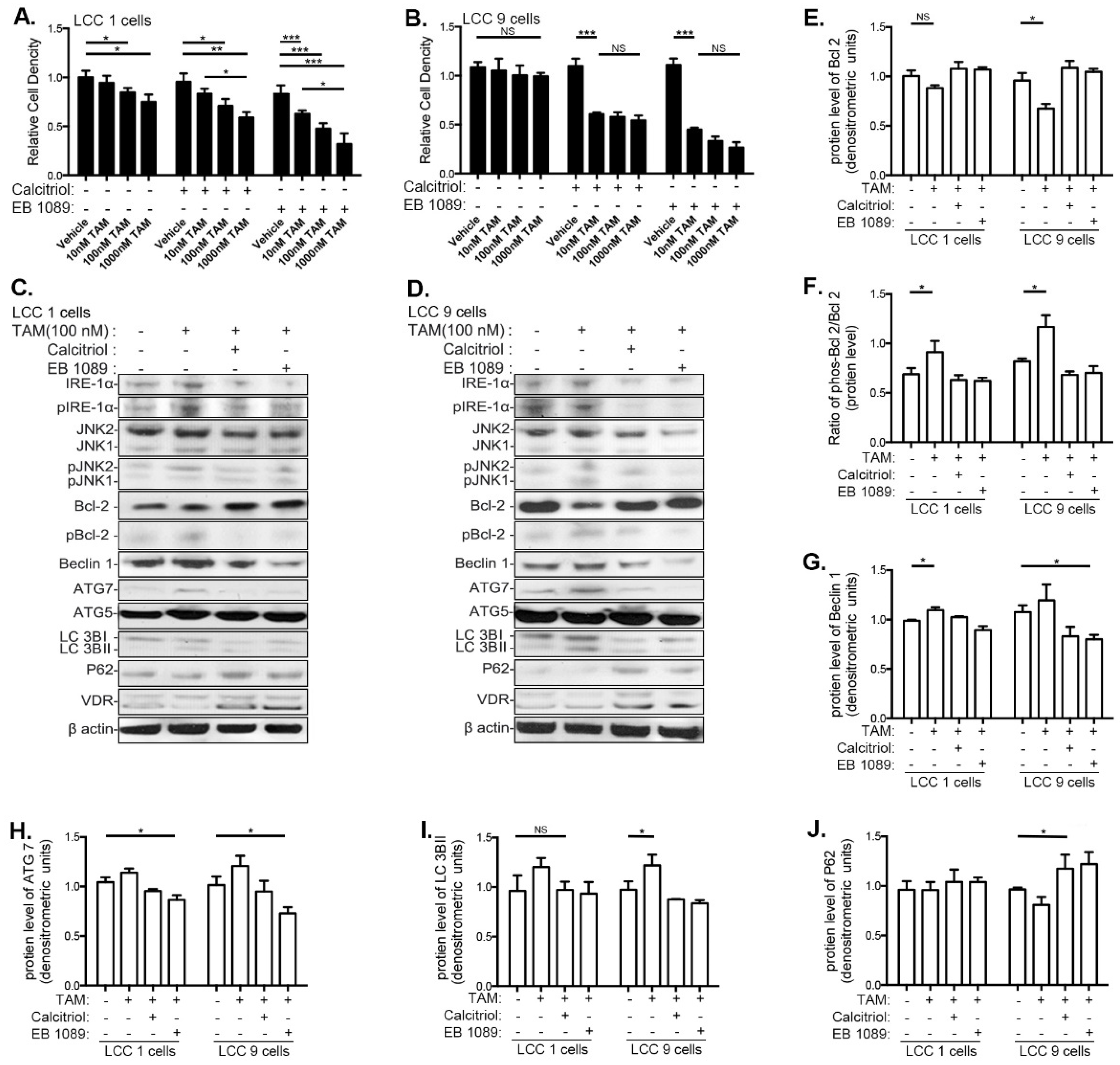
3.4. VitD Restores TAM Sensitivity and Inhibits the UPR and Autophagy in Human Breast Cancer Cell Lines
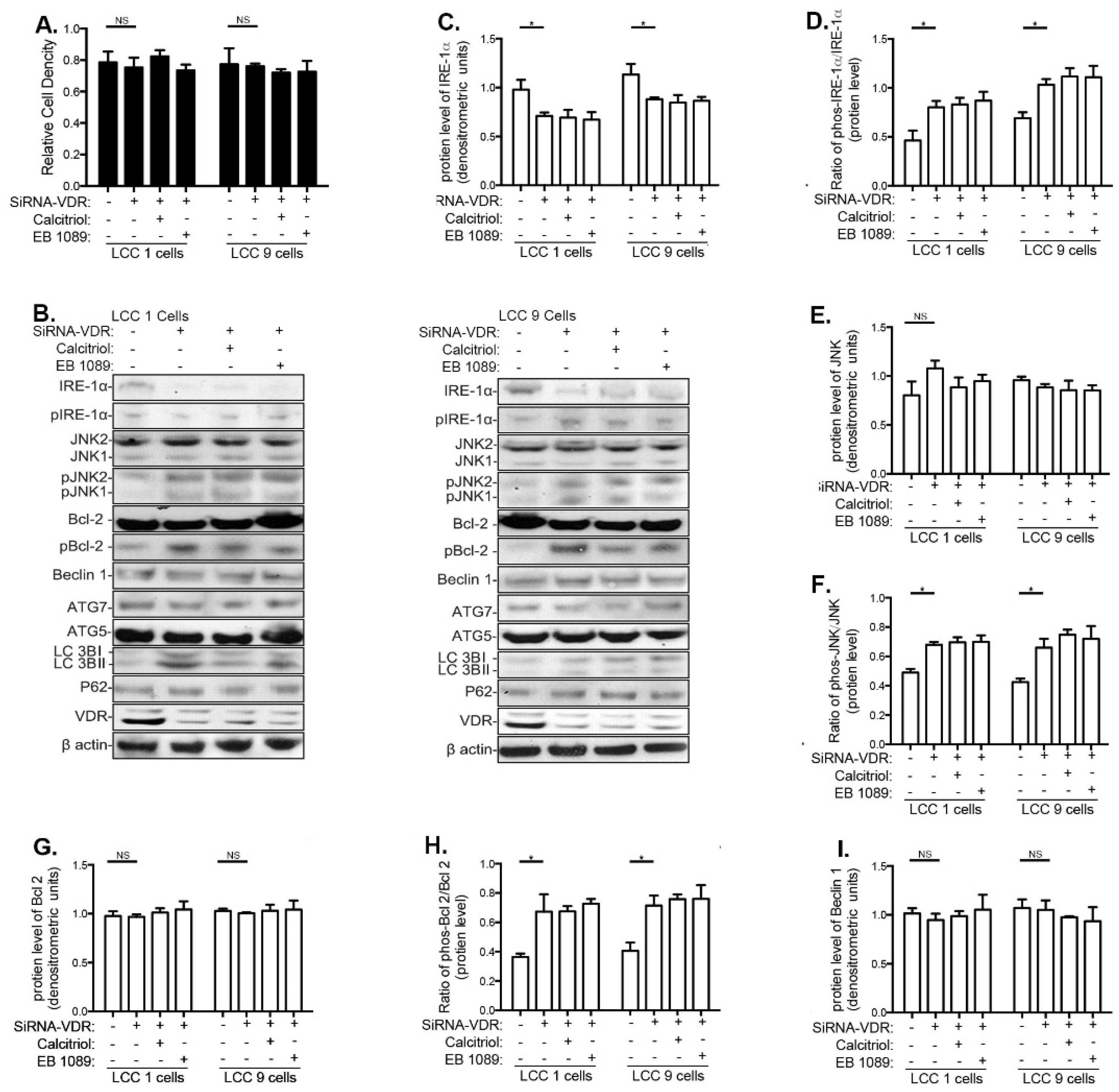
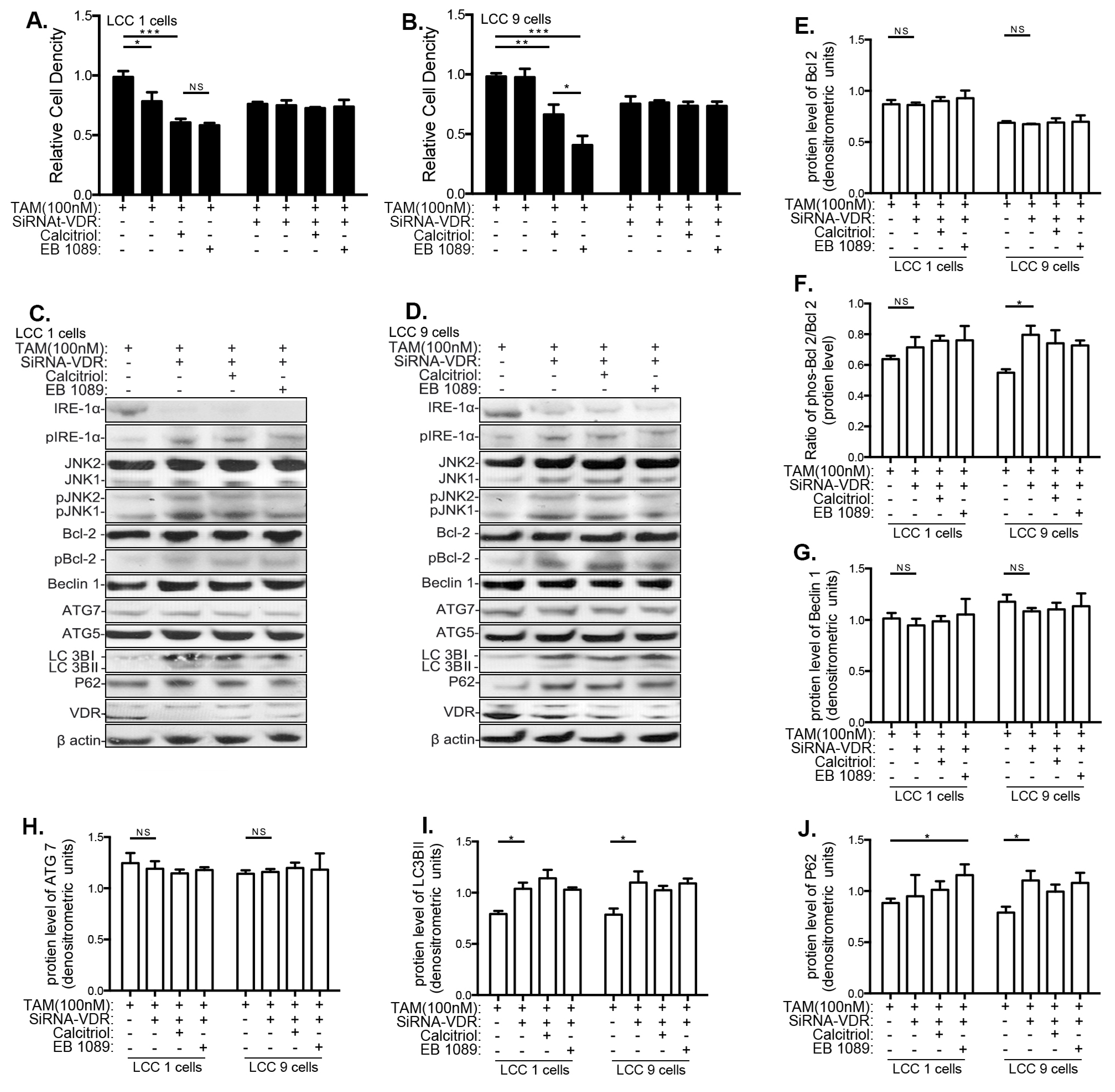
3.5. Effects of VitD Are Mediated by VDR Expression
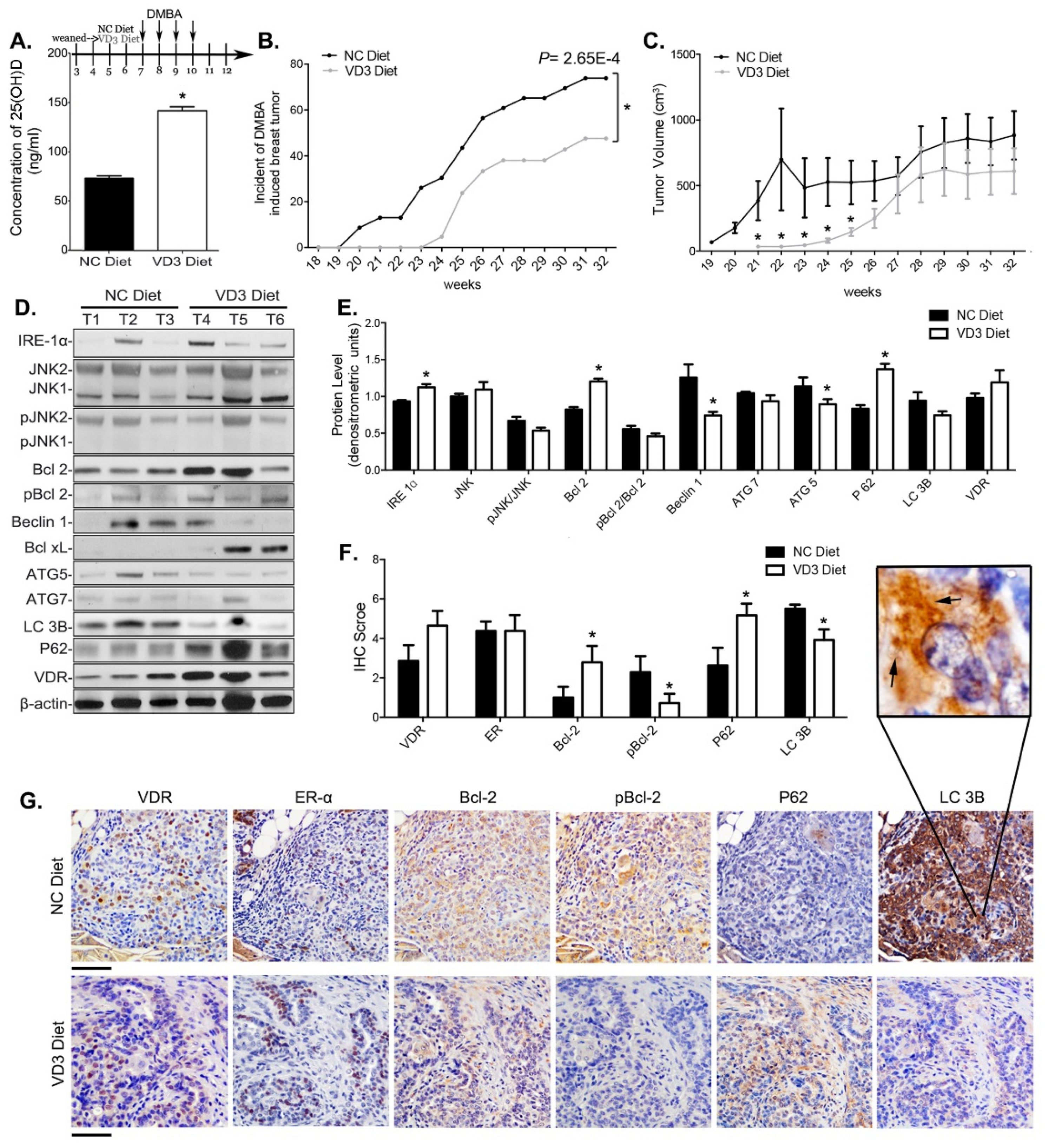
3.6. VD3 Exposure Reduced Mammary Cancer Risk in Mice and Inhibited Survival Autophagy in Their Mammary Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef]
- Lim, E.; Metzger-Filho, O.; Winer, E.P. The natural history of hormone receptor-positive breast cancer. Oncology 2012, 26, 688–694, 696. [Google Scholar]
- DeSantis, C.E.; Ma, J.; Sauer, A.G.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA A Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Alencar, V.H.M.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013, 381, 805–816. [Google Scholar] [CrossRef]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer–An overview and update. Mol. Cell. Endocrinol. 2015, 418, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Cook, K.L.; Hu, R.; Facey, C.O.; Tavassoly, I.; Schwartz, J.L.; Baumann, W.T.; Tyson, J.J.; Xuan, J.; Wang, Y.; et al. Endoplasmic Reticulum Stress, the Unfolded Protein Response, Autophagy, and the Integrated Regulation of Breast Cancer Cell Fate. Cancer Res. 2012, 72, 1321–1331. [Google Scholar] [CrossRef]
- Hoyerhansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Samaddar, J.S.; Gaddy, V.T.; Duplantier, J.; Thandavan, S.P.; Shah, M.; Smith, M.J.; Browning, D.; Rawson, J.; Smith, S.B.; Barrett, J.T.; et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol. Cancer Ther. 2008, 7, 2977–2987. [Google Scholar] [CrossRef]
- Cook, K.L.; Clarke, P.A.G.; Parmar, J.; Hu, R.; Schwartz-Roberts, J.L.; Abu-Asab, M.; Wärri, A.; Baumann, W.T.; Clarke, R. Knockdown of estrogen receptor-α induces autophagy and inhibits antiestrogen-mediated unfolded protein response activation, promoting ROS-induced breast cancer cell death. FASEB J. 2014, 28, 3891–3905. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Crawford, A.C.; Riggins, R.B.; Shajahan, A.N.; Zwart, A.; Clarke, R. Co-Inhibition of BCL-W and BCL2 Restores Antiestrogen Sensitivity through BECN1 and Promotes an Autophagy-Associated Necrosis. PLoS ONE 2010, 5, e8604. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pedro, J.M.B.-S.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Bristol, M.L.; Di, X.; Beckman, M.J.; Wilson, E.N.; Henderson, S.C.; Maiti, A.; Fan, Z.; Gewirtz, D.A. Dual functions of autophagy in the response of breast tumor cells to radiation: Cytoprotective autophagy with radiation alone and cytotoxic autophagy in radiosensitization by vitamin D3. Autophagy 2012, 8, 739–753. [Google Scholar] [CrossRef]
- Wilson, E.N.; Bristol, M.L.; Di, X.; Maltese, W.A.; Koterba, K.; Beckman, M.J.; Gewirtz, D.A. A Switch between Cytoprotective and Cytotoxic Autophagy in the Radiosensitization of Breast Tumor Cells by Chloroquine and Vitamin D. Horm. Cancer 2011, 2, 272–285. [Google Scholar] [CrossRef]
- DeMasters, G.; Di, X.; Newsham, I.; Shiu, R.; Gewirtz, D.A. Potentiation of radiation sensitivity in breast tumor cells by the vitamin D3 analogue, EB 1089, through promotion of autophagy and interference with proliferative recovery. Mol. Cancer Ther. 2006, 5, 2786–2797. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Hamberg, K.; Binderup, E. Seocalcitol (EB 1089) A Vitamin D Analogue of Anti-cancer Potential. Background, Design, Synthesis, Pre-clinical and Clinical Evaluation. Curr. Pharm. Des. 2000, 6, 803–828. [Google Scholar] [CrossRef] [PubMed]
- Gocek, E.; Studzinski, G.P. Vitamin D and differentiation in cancer. Crit. Rev. Clin. Lab. Sci. 2009, 46, 190–209. [Google Scholar] [CrossRef] [PubMed]
- Andruska, N.; Zheng, X.; Yang, X.; Helferich, W.G.; Shapiro, D.J. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor α-positive breast cancer. Oncogene 2014, 34, 3760–3769. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Eads, D.; Naczki, C.; Northrup, S.; Chen, T.; Koumenis, C. 19-nor-1α,25-Dihydroxyvitamin D2(Paricalcitol) inhibits the proliferation of human pancreatic cancer cells in vitro and in vivo. Cancer Biol. Ther. 2008, 7, 430–436. [Google Scholar] [CrossRef]
- Tavera-Mendoza, L.E.; Westerling, T.; Libby, E.; Marusyk, A.; Cato, L.; Cassani, R.; Cameron, L.A.; Ficarro, S.B.; Marto, J.A.; Klawitter, J.; et al. Vitamin D receptor regulates autophagy in the normal mammary gland and in luminal breast cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2186–E2194. [Google Scholar] [CrossRef] [PubMed]
- Høyer-Hansen, M.; Bastholm, L.; Mathiasen, I.S.; Elling, F.; Jaattela, M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 2005, 12, 1297–1309. [Google Scholar] [CrossRef]
- Zella, L.A.; Meyer, M.B.; Nerenz, R.D.; Lee, S.M.; Martowicz, M.L.; Pike, J.W. Multifunctional Enhancers Regulate Mouse and Human Vitamin D Receptor Gene Transcription. Mol. Endocrinol. 2010, 24, 128–147. [Google Scholar] [CrossRef]
- Wiese, R.; Uhland-Smith, A.; Ross, T.; Prahl, J.; DeLuca, H. Up-regulation of the vitamin D receptor in response to 1,25-dihydroxyvitamin D3 results from ligand-induced stabilization. J. Biol. Chem. 1992, 267, 20082–20086. [Google Scholar] [CrossRef]
- Davoodi, F.; Brenner, R.V.; Evans, S.R.; Schumaker, L.M.; Shabahang, M.; Nauta, R.J.; Buras, R.R. Modulation of vitamin D receptor and estrogen receptor by 1,25(OH)2-vitamin D3 in T-47D human breast cancer cells. J. Steroid Biochem. Mol. Biol. 1995, 54, 147–153. [Google Scholar] [CrossRef]
- Symmans, W.F.; Hatzis, C.; Sotiriou, C.; Andre, F.; Peintinger, F.; Regitnig, P.; Daxenbichler, G.; Desmedt, C.; Domont, J.; Marth, C.; et al. Genomic Index of Sensitivity to Endocrine Therapy for Breast Cancer. J. Clin. Oncol. 2010, 28, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sieuwerts, A.M.; McGreevy, M.; Casey, G.; Cufer, T.; Paradiso, A.; Harbeck, N.; Span, P.N.; Hicks, D.G.; Crowe, J.; et al. The 76-gene signature defines high-risk patients that benefit from adjuvant tamoxifen therapy. Breast Cancer Res. Treat 2009, 116, 303–309. [Google Scholar] [CrossRef]
- Loi, S.; Haibe-Kains, B.; Desmedt, C.; Lallemand, F.; Tutt, A.M.; Gillet, C.; Ellis, P.; Harris, A.; Bergh, J.; Foekens, J.A.; et al. Definition of Clinically Distinct Molecular Subtypes in Estrogen Receptor–Positive Breast Carcinomas Through Genomic Grade. J. Clin. Oncol. 2007, 25, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-J.; Wang, Z.; Ryan, P.D.; Isakoff, S.J.; Barmettler, A.; Fuller, A.; Muir, B.; Mohapatra, G.; Salunga, R.; Tuggle, J.; et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell 2004, 5, 607–616. [Google Scholar] [CrossRef]
- Türei, D.; Földvári-Nagy, L.; Fazekas, D.; Módos, D.; Kubisch, J.; Kadlecsik, T.; Demeter, A.; Lenti, K.F.-N.L.; Csermely, P.; Vellai, T.; et al. Autophagy Regulatory Network—A systems-level bioinformatics resource for studying the mechanism and regulation of autophagy. Autophagy 2014, 11, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Brünner, N.; Boulay, V.; Fojo, A.; Freter, C.E.; Lippman, M.E.; Clarke, R. Acquisition of hormone-independent growth in MCF-7 cells is accompanied by increased expression of estrogen-regulated genes but without detectable DNA amplifications. Cancer Res. 1993, 53, 283–290. [Google Scholar]
- Brünner, N.; Boysen, B.; Jirus, S.; Skaar, T.C.; Holst-Hansen, C.; Lippman, J.; Frandsen, T.; Spang-Thomsen, M.; Fuqua, S.A.; Clarke, R. MCF7/LCC9: An antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res. 1997, 57, 3486–3493. [Google Scholar]
- Pawitan, Y.; Bjöhle, J.; Amler, L.; Borg, A.-L.; Egyhazi, S.; Hall, P.; Han, X.; Holmberg, L.; Huang, F.; Klaar, S.; et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: Derived and validated in two population-based cohorts. Breast Cancer Res. 2005, 7, R953–R964. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.; O’Prey, J.; Fricker, M.; Ryan, K.M. Hypoxia-selective macroautophagy and cell survival signaled by autocrine PDGFR activity. Genes Dev. 2009, 23, 1283–1288. [Google Scholar] [CrossRef]
- Sui, X.; Jin, L.; Huang, X.; Geng, S.; He, C.; Hu, X. p53 signaling and autophagy in cancer: A revolutionary strategy could be developed for cancer treatment. Autophagy 2011, 7, 565–571. [Google Scholar] [CrossRef]
- Liu, J.; Debnath, J. The Evolving, Multifaceted Roles of Autophagy in Cancer. Adv. Cancer Res. 2016, 130, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Hanley, D.A.; Davison, K.S. Vitamin D insufficiency in North America. J. Nutr. 2005, 135, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Pierrot-Deseilligny, C.; Souberbielle, J.-C. Vitamin D and multiple sclerosis: An update. Mult. Scler. Relat. Disord. 2017, 14, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J. Vitamin D and Cardiovascular Disease. Annu. Rev. Med. 2016, 67, 261–272. [Google Scholar] [CrossRef]
- Berridge, M.J. Vitamin D and Depression: Cellular and Regulatory Mechanisms. Pharmacol. Rev. 2017, 69, 80–92. [Google Scholar] [CrossRef]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef]
- Cui, Y.; Rohan, T.E. Vitamin D, calcium, and breast cancer risk: A review. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1427–1437. [Google Scholar] [CrossRef]
- Yin, L.; Grandi, N.; Raum, E.; Haug, U.; Arndt, V.; Brenner, H. Meta-analysis: Serum vitamin D and breast cancer risk. Eur. J. Cancer 2010, 46, 2196–2205. [Google Scholar] [CrossRef]
- Peppone, L.J.; Rickles, A.S.; Janelsins, M.C.; Insalaco, M.R.; Skinner, K.A. The Association Between Breast Cancer Prognostic Indicators and Serum 25-OH Vitamin D Levels. Ann. Surg. Oncol. 2012, 19, 2590–2599. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, Y.M.; Ko, B.S.; Lee, J.W.; Yu, J.H.; Son, B.H.; Gong, G.-Y.; Kim, S.B.; Ahn, S.H. Vitamin D Deficiency is Correlated with Poor Outcomes in Patients with Luminal-type Breast Cancer. Ann. Surg. Oncol. 2010, 18, 1830–1836. [Google Scholar] [CrossRef]
- Yao, S.; Sucheston, L.E.; Millen, A.E.; Johnson, C.S.; Trump, D.L.; Nesline, M.K.; Davis, W.; Hong, C.-C.; McCann, S.E.; Hwang, H.; et al. Pretreatment Serum Concentrations of 25-Hydroxyvitamin D and Breast Cancer Prognostic Characteristics: A Case-Control and a Case-Series Study. PLoS ONE 2011, 6, e17251. [Google Scholar] [CrossRef]
- Yao, S.; Kwan, M.L.; Ergas, I.J.; Roh, J.M.; Cheng, T.D.; Hong, C.C.; McCann, S.E.; Tang, L.; Davis, W.; Liu, S.; et al. Association of Serum Level of Vitamin D at Diagnosis With Breast Cancer Survival: A Case-Cohort Analysis in the Pathways Study. JAMA Oncol. 2017, 3, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, M.; Redaniel, M.T.; Martin, R.M. The effect of pre-diagnostic vitamin D supplementation on cancer survival in women: A cohort study within the UK Clinical Practice Research Datalink. BMC Cancer 2015, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Madden, J.M.; Murphy, L.; Zgaga, L.; Bennett, K. De novo vitamin D supplement use post-diagnosis is associated with breast cancer survival. Breast Cancer Res. Treat. 2018, 172, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Cook, N.R.; Lee, I.-M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, S.L.; Baggerly, C.A.; French, C.B.; Baggerly, L.L.; Garland, C.F.; Gorham, E.D.; Hollis, B.W.; Trump, D.L.; Lappe, J.M. Breast cancer risk markedly lower with serum 25-hydroxyvitamin D concentrations > 60 vs. <20 ng/ml (150 vs. 50 nmol/L): Pooled analysis of two randomized trials and a prospective cohort. PLoS ONE 2018, 13, e0199265. [Google Scholar] [CrossRef]
- Lundqvist, J.; Yde, C.W.; Lykkesfeldt, A.E. 1alpha,25-dihydroxyvitamin D3 inhibits cell growth and NFkappaB signaling in tamoxifen-resistant breast cancer cells. Steroids 2014, 85, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.S.; Heiberg, I.; Lykkesfeldt, A.E. Anti-oestrogen resistant human breast cancer cell lines are more sensitive towards treatment with the vitamin D analogue EB1089 than parent MCF-7 cells. Br. J. Cancer 2001, 84, 686–690. [Google Scholar] [CrossRef]
- Christensen, G.L.; Jepsen, J.; Fog, C.; Christensen, I.; Lykkesfeldt, A. Sequential Versus Combined Treatment of Human Breast Cancer Cells with Antiestrogens and the Vitamin D Analogue EB1089 and Evaluation of Predictive Markers for Vitamin D Treatment. Breast Cancer Res. Treat. 2004, 85, 53–63. [Google Scholar] [CrossRef]
- Wijngaarden, T.V.-V.; Pols, H.A.P.; Buurman, C.J.; Birkenhäger, J.C.; Van Leeuwen, J.P.T.M. Combined effects of 1,25-dihydroxyvitamin D3 and tamoxifen on the growth of MCF-7 and ZR-75-1 human breast cancer cells. Breast Cancer Res. Treat. 1994, 29, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Wijngaarden, T.V.-V.; Pols, H.A.; Buurman, C.J.; Bemd, G.J.V.D.; Dorssers, L.C.; Birkenhäger, J.C.; Van Leeuwen, J.P. Inhibition of breast cancer cell growth by combined treatment with vitamin D3 analogues and tamoxifen. Cancer Res. 1994, 54, 5711–5717. [Google Scholar]
- Welsh, J. Induction of apoptosis in breast cancer cells in response to vitamin D and antiestrogens. Biochem. Cell Biol. 1994, 72, 537–545. [Google Scholar] [CrossRef] [PubMed]
- James, S.Y.; Mackay, A.G.; Binderup, L.; Colston, K.W. Effects of a new synthetic vitamin D analogue, EB1089, on the oestrogen-responsive growth of human breast cancer cells. J. Endocrinol. 1994, 141, 555–563. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Cook, K.L.; Yu, W.; Jin, L.; Bouker, K.B.; Clarke, R.; Hilakivi-Clarke, L. Inhibition of Antiestrogen-Promoted Pro-Survival Autophagy and Tamoxifen Resistance in Breast Cancer through Vitamin D Receptor. Nutrients 2021, 13, 1715. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13051715
Li Y, Cook KL, Yu W, Jin L, Bouker KB, Clarke R, Hilakivi-Clarke L. Inhibition of Antiestrogen-Promoted Pro-Survival Autophagy and Tamoxifen Resistance in Breast Cancer through Vitamin D Receptor. Nutrients. 2021; 13(5):1715. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13051715
Chicago/Turabian StyleLi, Ye, Katherine L Cook, Wei Yu, Lu Jin, Kerrie B Bouker, Robert Clarke, and Leena Hilakivi-Clarke. 2021. "Inhibition of Antiestrogen-Promoted Pro-Survival Autophagy and Tamoxifen Resistance in Breast Cancer through Vitamin D Receptor" Nutrients 13, no. 5: 1715. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13051715