
Exploratory Data Analysis of Cell and Mitochondrial High-Fat, High-Sugar Toxicity on Human HepG2 Cells
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Chemicals
2.2. Fatty Acid Preparation
2.3. Cell culture and FA Treatments
2.4. Nile Red Staining
2.5. Phospholipid Analysis
2.6. Intracellular Oxidative Stress
2.7. Mitochondrial Morphology Imaging
2.8. Mitochondrial DNA Copy Number Measurements
2.9. Western Blotting Analysis
2.10. BN-PAGE in-Gel Activity of Complex I
2.11. Cellular Oxygen Consumption Rate Measurements
2.12. Measurement of Caspase 8 and 9-Like Activities
2.13. Measurement of Caspase 3/7-Like Activity
2.14. Cell Metabolic Activity
2.15. Cell Mass
2.16. Intracellular ATP Levels
2.17. Computational Data Analysis
2.18. Statistics
3. Results
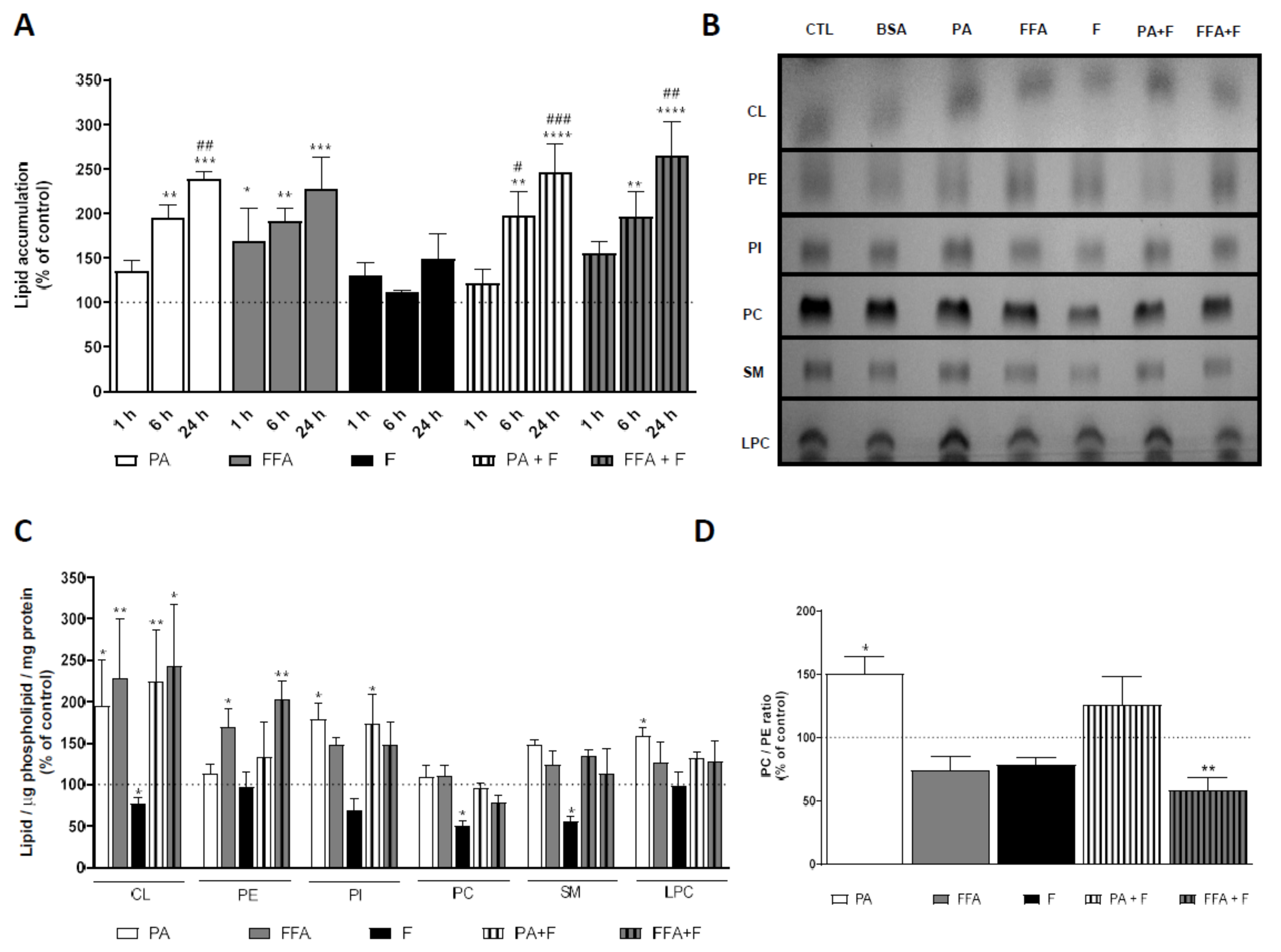
3.1. Supra-Physiological Concentrations of FA Increase the Accumulation of Lipid Droplets
3.2. Fructose Treatment Increased HepG2 Fructokinase Protein Levels
3.3. Supra-Physiological Concentrations of fa Altered Mitochondrial Phospholipid Content
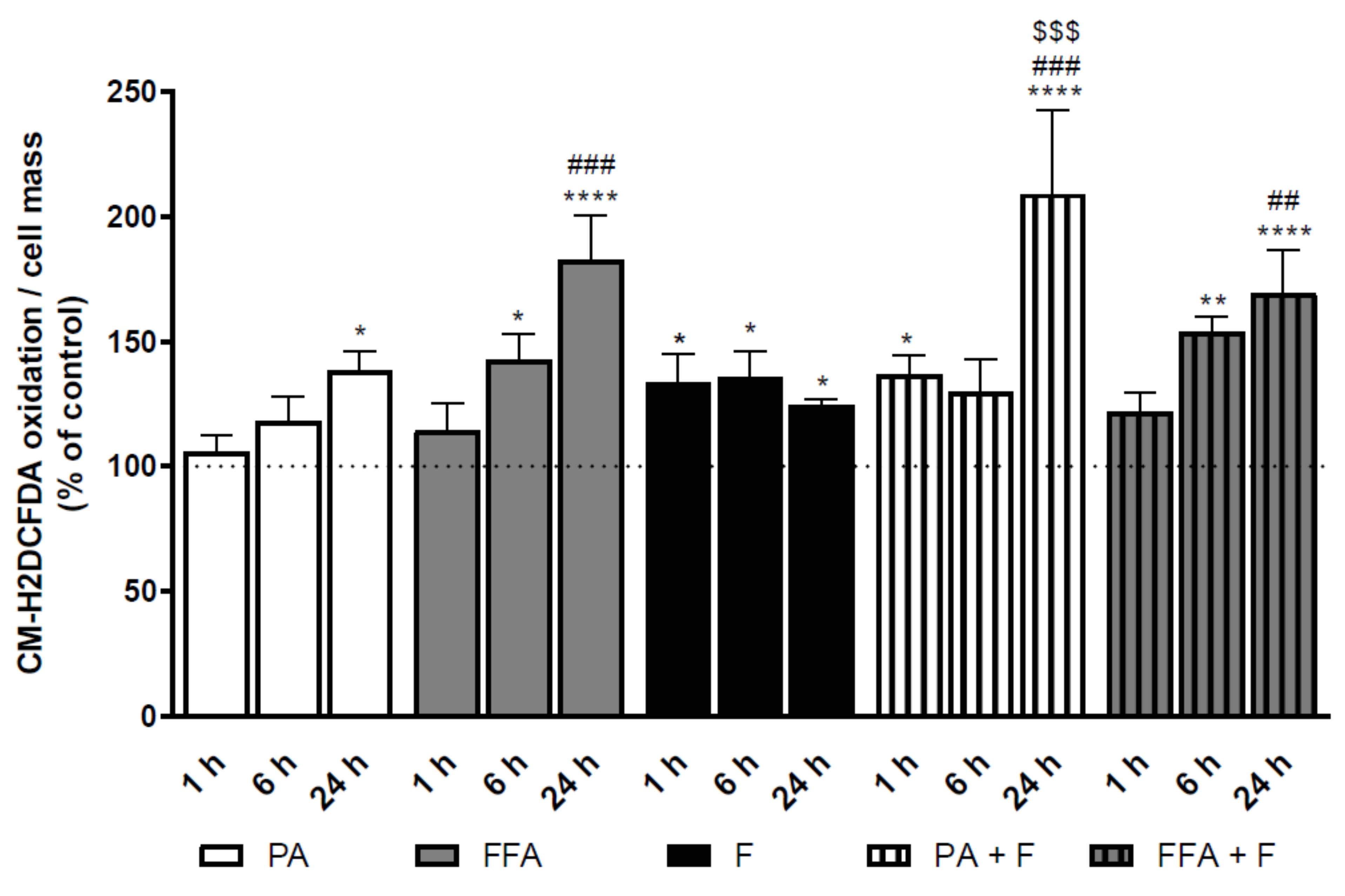
3.4. Supra-Physiological Concentrations of fa Time-Dependently Increase Cm-H2dcfda Oxidation
3.5. Supra-Physiological Concentrations of FA Altered Mitochondrial Membrane Potential (ΔΨm) and Induced Changes in Mitochondrial Morphology
3.6. Supra-Physiological Concentrations of Unsaturated FA Significantly Increase mtDNA Copy Number
3.7. Supra-Physiological Concentrations of FA Altered Level of OXPHOS Subunits
3.8. Supra-Physiological Concentrations of FA Altered Native Mitochondrial Electron Transport Complex I Activity
3.9. Supra-Physiological Concentrations of FA Time-Dependently Decrease Oxygen Consumption Rates (OCR) and Increased Extracellular Acidification Rates (ECAR)
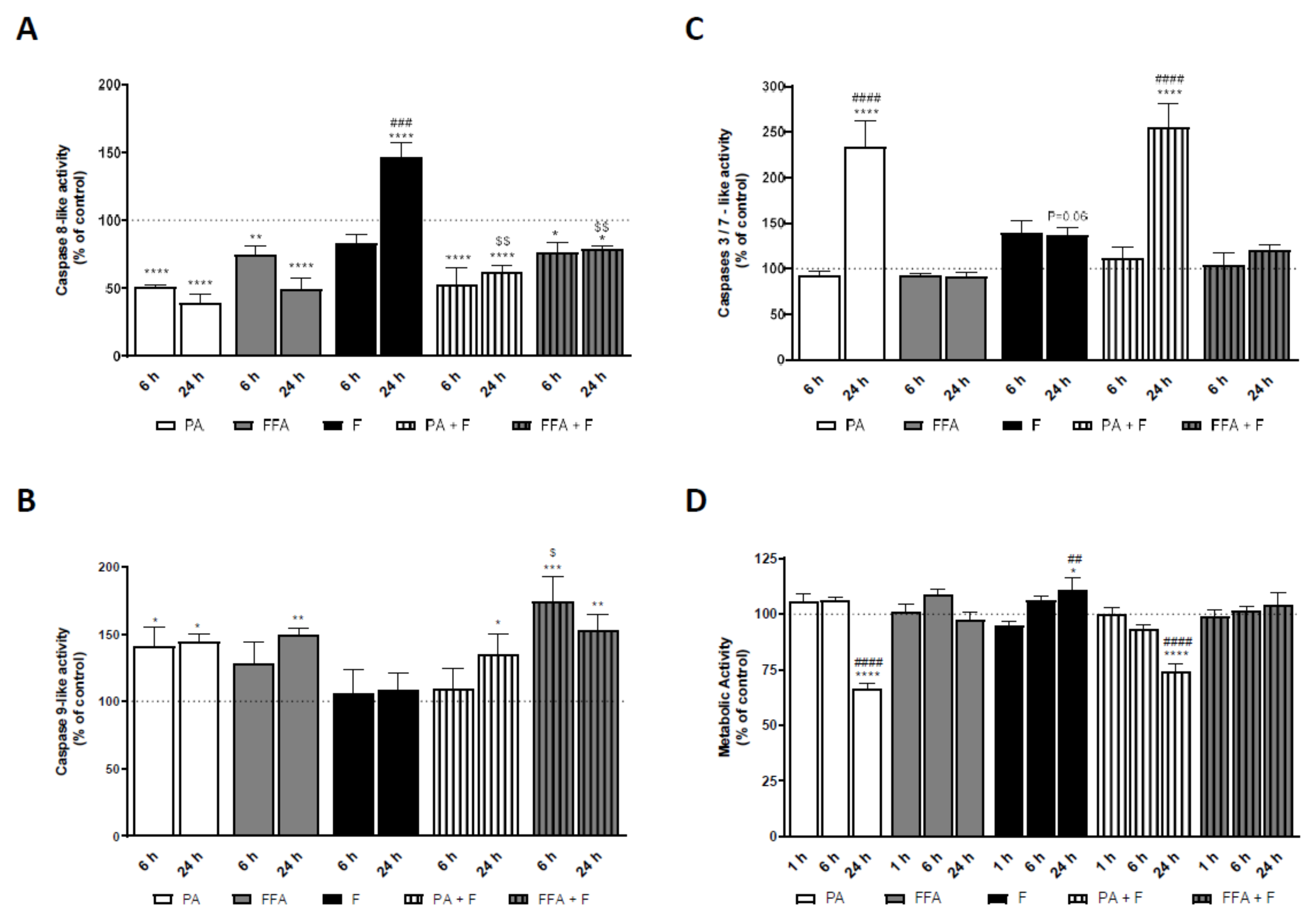
3.10. Supra-Physiological Concentrations of FA Induced Caspases Activation in HepG2 Cells Which Follows ROS and Mitochondrial Dysfunction
3.11. Supra-Physiological Concentrations of Saturated FA Time-Dependently Decrease Intracellular ATP Levels
3.12. Supra-Physiological Concentrations of FA Time-Dependently Decrease Cell Metabolic Activity and Mass of Human Hepatocarcinoma Cells
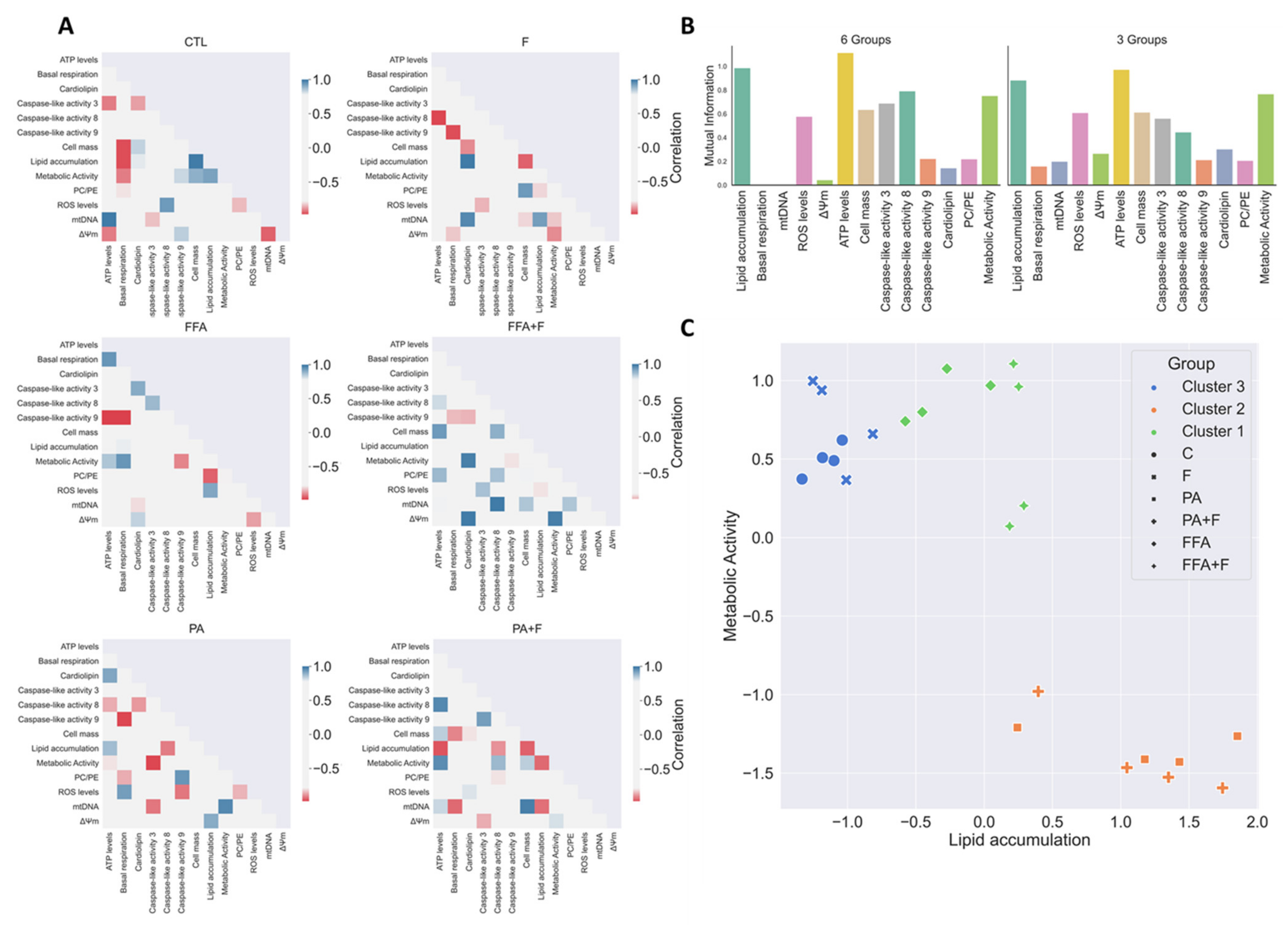
3.13. Exploratory Data Analysis Separated PA and FFA Regimens By Identifying a Subset Of Critical Mitochondrial Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation 2019, 103, 22–27. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Park, E.-J.; Lee, A.Y.; Park, S.; Kim, J.-H.; Cho, M.-H. Multiple pathways are involved in palmitic acid-induced toxicity. Food Chem. Toxicol. 2014, 67, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Metab. 2006, 291, E275–E281. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wei, Y.; Pagliassotti, M.J. Saturated Fatty Acids Promote Endoplasmic Reticulum Stress and Liver Injury in Rats with Hepatic Steatosis. Endocrinology 2006, 147, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Šrámek, J.; Němcová-Fürstová, V.; Kovář, J. Kinase Signaling in Apoptosis Induced by Saturated Fatty Acids in Pancreatic β-Cells. Int. J. Mol. Sci. 2016, 17, 1400. [Google Scholar] [CrossRef] [Green Version]
- Egnatchik, R.A.; Leamy, A.K.; Jacobson, D.A.; Shiota, M.; Young, J.D. ER calcium release promotes mitochondrial dysfunction and hepatic cell lipotoxicity in response to palmitate overload. Mol. Metab. 2014, 3, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Berk, M.; McIntyre, T.M.; Gores, G.J.; Feldstein, A.E. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 2007, 47, 1495–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazanave, S.C.; Elmi, N.A.; Akazawa, Y.; Bronk, S.F.; Mott, J.L.; Gores, G.J. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am. J. Physiol. Liver Physiol. 2010, 299, G236–G243. [Google Scholar] [CrossRef] [Green Version]
- Egnatchik, R.A.; Leamy, A.K.; Noguchi, Y.; Shiota, M.; Young, J.D. Palmitate-induced Activation of Mitochondrial Metabolism Promotes Oxidative Stress and Apoptosis in H4IIEC3 Rat Hepatocytes. Metabolism 2014, 63, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Pardo, V.; González-Rodríguez, Á.; Guijas, C.; Balsinde, J.; Valverde, Á.M. Opposite Cross-Talk by Oleate and Palmitate on Insulin Signaling in Hepatocytes through Macrophage Activation. J. Biol. Chem. 2015, 290, 11663–11677. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-T.; Chen, W.; Cheng, K.-C.; Ku, P.-M.; Yeh, C.-H.; Cheng, J.-T. Oleic acid activates peroxisome proliferator-activated receptor δ to compensate insulin resistance in steatotic cells. J. Nutr. Biochem. 2012, 23, 1264–1270. [Google Scholar] [CrossRef]
- Yang, J.; Fernández-Galilea, M.; Martínez-Fernández, L.; González-Muniesa, P.; Pérez-Chávez, A.; Martínez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Dong, L.; Yang, X.; Shi, H.; Zhang, L. α-Linolenic acid prevents endoplasmic reticulum stress-mediated apoptosis of stearic acid lipotoxicity on primary rat hepatocytes. Lipids Health Dis. 2011, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Chang, Y.; Shi, Z.; Han, X.; Han, Y.; Yao, Q.; Hu, Z.; Cui, H.; Zheng, L.; Han, T.; et al. ω-3 PUFAs ameliorate liver fibrosis and inhibit hepatic stellate cells proliferation and acti-vation by promoting YAP/TAZ degradation. Sci. Rep. 2016, 6, 1–14. [Google Scholar]
- Moreira, A.R.S.; Rüschenbaum, S.; Schefczyk, S.; Hendgen-Cotta, U.; Rassaf, T.; Broering, R.; Hardtke-Wolenski, M.; Buitrago-Molina, L.E. 9-PAHSA Prevents Mitochondrial Dysfunction and Increases the Viability of Steatotic Hepatocytes. Int. J. Mol. Sci. 2020, 21, 8279. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Carbohydrate intake and nonalcoholic fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 446–452. [Google Scholar] [CrossRef]
- Agoun, H.; Semiane, N.; Mallek, A.; Bellahreche, Z.; Hammadi, S.; Madjerab, M.; Abdlalli, M.; Khalkhal, A.; Dahmani, Y. High-carbohydrate diet-induced metabolic disorders in Gerbillus tarabuli (a new model of non-alcoholic fatty-liver disease). Protective effects of 20-hydroxyecdysone. Arch. Physiol. Biochem. 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kakisaka, K.; Cazanave, S.C.; Fingas, C.D.; Guicciardi, M.E.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Gores, G.J. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am. J. Physiol. Liver Physiol. 2012, 302, G77–G84. [Google Scholar] [CrossRef] [Green Version]
- Kurek, K.; Piotrowska, D.M.; Wiesiołek-Kurek, P.; Łukaszuk, B.; Chabowski, A.; Górski, J.; Żendzian-Piotrowska, M. Inhibition of ceramide de novo synthesis reduces liver lipid accumula-tion in rats with nonalcoholic fatty liver disease. Liver Int. 2014, 34, 1074–1083. [Google Scholar] [CrossRef]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab. 2012, 23, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Arguello, G.; Balboa, E.; Arrese, M.; Zanlungo, S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1765–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, D.M.; Afonso, M.B.; Rodrigues, P.M.; Simão, A.L.; Pereira, D.M.; Borralho, P.M.; Rodrigues, C.M.; Castro, R.E. c-Jun N-Terminal Kinase 1/c-Jun Activation of the p53/MicroRNA 34a/Sirtuin 1 Pathway Contributes to Apoptosis Induced by Deoxycholic Acid in Rat Liver. Mol. Cell. Biol. 2014, 34, 1100–1120. [Google Scholar] [CrossRef] [Green Version]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef] [Green Version]
- Sobrecases, H.; Lê, K.-A.; Bortolotti, M.; Schneiter, P.; Ith, M.; Kreis, R.; Boesch, C.; Tappy, L. Effects of short-term overfeeding with fructose, fat and fructose plus fat on plasma and hepatic lipids in healthy men. Diabetes Metab. 2010, 36, 244–246. [Google Scholar] [CrossRef]
- Yao, H.-R.; Liu, J.; Plumeri, D.; Cao, Y.-B.; He, T.; Lin, L.; Li, Y.; Jiang, Y.-Y.; Li, J.; Shang, J. Lipotoxicity in HepG2 cells triggered by free fatty acids. Am. J. Transl. Res. 2011, 3, 284–291. [Google Scholar] [PubMed]
- Vecchione, G.; Grasselli, E.; Cioffi, F.; Baldini, F.; Oliveira, P.J.; Sardão, V.A.; Cortese, K.; Lanni, A.; Voci, A.; Portincasa, P.; et al. The Nutraceutic Silybin Counteracts Excess Lipid Accumulation and Ongoing Oxidative Stress in an In Vitro Model of Non-Alcoholic Fatty Liver Disease Progression. Front. Nutr. 2017, 4, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einer, C.; Hohenester, S.; Wimmer, R.; Wottke, L.; Artmann, R.; Schulz, S.; Gosmann, C.; Simmons, A.; Leitzinger, C.; Eberhagen, C.; et al. Data on chow, liver tissue and mitochondrial fatty acid compositions as well as mitochondrial proteome changes after feeding mice a western diet for 6–24 weeks. Data Br. 2017, 15, 163–169. [Google Scholar] [CrossRef]
- McMillian, M.K.; Grant, E.R.; Zhong, Z.; Parker, J.B.; Li, L.; Zivin, R.A.; Burczynski, M.E.; Johnson, M.D. Nile red binding to HepG2 cells: An improved assay for in vitro studies of hepa-tosteatosis. Vitr. Mol. Toxicol. J. Basic Appl. Res. 2001, 14, 177–190. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Rouser, G.; Fleischer, S.; Yamamoto, A. Two dimensional thin layer chromatographic separation of polar lipids and determi-nation of phospholipids by phosphorus analysis of spots. Lipids 1970, 5, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Cherra, S.J., III; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijtmans, L.G.J.; Henderson, N.S.; Holt, I.J. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods 2002, 26, 327–334. [Google Scholar] [CrossRef]
- Silva, F.S.; Starostina, I.G.; Ivanova, V.V.; Rizvanov, A.A.; Oliveira, P.J.; Pereira, S.P. Determination of metabolic viability and cell mass using a tandem resaz-urin/sulforhodamine B assay. Curr. Protoc. Toxicol. 2016, 68, 2–24. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.C. Mutual Information between Discrete and Continuous Data Sets. PLoS ONE 2014, 9, e87357. [Google Scholar] [CrossRef]
- MacQueen, J. Some methods for classification and analysis of multivariate observations. In Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability, Berkeley, CA, USA, 21 June–18 July 1965; Volume 1, pp. 281–297. [Google Scholar]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 51–56. [Google Scholar]
- Van Der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy array: A structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.; Oliphant, T.; Peterson, P. SciPy: Open Source Scientific Tools for Python. 2001. Available online: http://www.scipy.org/ (accessed on 12 April 2021).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Schultz, A.; Neil, D.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Hepatic Adverse Effects of Fructose Consumption Independent of Overweight/Obesity. Int. J. Mol. Sci. 2013, 14, 21873–21886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Zhong, W.; Zhang, W.; Zhou, Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: Role of zinc deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G205–G214. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonal-coholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Martínez-Romero, A.; Jiménez, N.; Castell, J.V.; O’Connor, J.-E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Zhao, L.; Guo, X.; Wang, O.; Zhang, H.; Wang, Y.; Zhou, F.; Liu, J.; Ji, B. Fructose and glucose combined with free fatty acids induce metabolic disorders in HepG2 cell: A new model to study the impacts of high-fructose/sucrose and high-fat diets in vitro. Mol. Nutr. Food Res. 2016, 60, 909–921. [Google Scholar] [CrossRef]
- Sasi, U.S.; Sindhu, G.; Raghu, K.G. Fructose-palmitate based high calorie induce steatosis in HepG2 cells via mitochondrial dysfunction: An in vitro approach. Toxicol In Vitro 2020, 68, 104952. [Google Scholar] [CrossRef]
- Busch, S.J.; Barnhart, R.L.; Martin, G.A.; Flanagan, M.A.; Jackson, R.L. Differential Regulation of Hepatic Triglyceride Lipase and Reductase Gene Expression in a Human Hepatoma Cell Line, HepG2. J. Biol. Chem. 1990, 265, 22474–22479. [Google Scholar] [CrossRef]
- Shi, X.; Li, J.; Zou, X.; Greggain, J.; Rødkær, S.V.; Færgeman, N.J.; Liang, B.; Watts, J.L. Regulation of lipid droplet size and phospholipid composition by stearoyl-CoA desaturase. J. Lipid Res. 2013, 54, 2504–2514. [Google Scholar] [CrossRef] [Green Version]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Mesmin, B. Mitochondrial lipid transport and biosynthesis: A complex balance. J. Cell Biol. 2016, 214, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Benga, G.; Hodârnâu, A.; Böhm, B.; Borza, V.; Tilinca, R.; Dancea, S.; Petrescu, I.; Ferdinand, W. Human liver mitochondria: Relation of a particular lipid composition to the mobility of spin-labelled lipids. Eur. J. Biochem. 1978, 84, 625–633. [Google Scholar] [CrossRef]
- Osman, C.; Voelker, D.R.; Langer, T. Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 2011, 192, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Crimi, M.; Esposti, M.D. Apoptosis-induced changes in mitochondrial lipids. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 551–557. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Van der Veen, J.N.; Lingrell, S.; da Silva, R.P.; Jacobs, R.L.; Vance, D.E. The Concentration of Phosphatidylethanolamine in Mitochondria Can Modulate ATP Production and Glucose Metabolism in Mice. Diabetes 2014, 63, 2620–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, B.M.; Ma, D.W.; Simons, B.; Noureldin, S.A.; Therapondos, G.; Guindi, M.; Sherman, M.; Allard, J.P. Nonalcoholic fatty liver disease is associated with lower hepatic and erythrocyte ratios of phosphatidylcholine to phosphatidylethanolamine. Appl. Physiol. Nutr. Metab. 2013, 38, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Van Meer, G.; de Kroon, A.I. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasseva, G.; Bai, H.D.; Davidescu, M.; Haromy, A.; Michelakis, E.; Vance, J.E. Phosphatidylethanolamine Deficiency in Mammalian Mitochondria Impairs Oxida-tive Phosphorylation and Alters Mitochondrial Morphology. J. Biol. Chem. 2013, 288, 4158–4173. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, G.; Shamni, O.; Avrahami, Y.; Cohen, O.; Broner, E.C.; Filippov-Levy, N.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Beta cell response to nutrient overload involves phospholipid remodelling and lipid peroxidation. Diabetologia 2015, 58, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, L.; Gurley, E.; Studer, E.; Shang, J.; Wang, T.; Wang, C.; Yan, M.; Jiang, Z.; Hylemon, P.B.; et al. Prevention of free fatty acid-induced hepatic lipotoxicity by 18β-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology 2008, 47, 1905–1915. [Google Scholar] [CrossRef]
- Bereiter-Hahn, J.; Vöth, M. Dynamics of mitochondria in living cells. Microsc. Res. Tech. 1994, 219, 198–219. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Galloway, C.A.; Yoon, Y. Mitochondrial Morphology in Metabolic Diseases. Antioxid. Redox Signal. 2013, 19, 415–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ruiz, I.; Solís-Muñoz, P.; Fernández-Moreira, D.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in nonalcoholic steatohepatitis. DMM Dis. Model Mech. 2015, 8, 183–191. [Google Scholar]
- Suárez-Rivero, J.M.; Villanueva-Paz, M.; De La Cruz-Ojeda, P.; De La Mata, M.; Cotán, D.; Oropesa-Ávila, M.; De Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, R.A.; Garcia-Smith, R.; Bisoffi, M.; Conn, C.A.; Trujillo, K.A. Conjugated linoleic acid or omega 3 fatty acids increase mitochondrial bio-synthesis and metabolism in skeletal muscle cells. Lipids Health Dis. 2012, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-S.; Shin, Y.; Moon, S.; Kim, S.; Kim, Y. Effects of Eicosapentaenoic Acid and Docosahexaenoic Acid on Mitochondrial DNA Replication and PGC-1α Gene Expression in C2C12Muscle Cells. Prev. Nutr. Food Sci. 2016, 21, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Patková, J.; Anděl, M.; Trnka, J. Palmitate-Induced Cell Death and Mitochondrial Respiratory Dysfunction in Myoblasts are Not Prevented by Mitochondria-Targeted Antioxidants. Cell. Physiol. Biochem. 2014, 33, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, J.; Burgueño, A.L.; Rosselli, M.S.; Gianotti, T.F.; Lago, N.R.; Pirola, C.J.; Sookoian, S. High fat diet-induced liver steatosis promotes an increase in liver mitochondrial biogenesis in response to hypoxia. J. Cell. Mol. Med. 2010, 15, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.N.; Czajka, A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013, 13, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Aoun, M.; Fouret, G.; Michel, F.; Bonafos, B.; Ramos, J.; Cristol, J.-P.; Carbonneau, M.-A.; Coudray, C.; Feillet-Coudray, C. Dietary fatty acids modulate liver mitochondrial cardiolipin content and its fatty acid composition in rats with non alcoholic fatty liver disease. J. Bioenerg. Biomembr. 2012, 44, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Fouret, G.; Gaillet, S.; LeComte, J.; Bonafos, B.; Djohan, F.; Barea, B.; Badia, E.; Coudray, C.; Feillet-Coudray, C. 20-Week follow-up of hepatic steatosis installation and liver mitochondrial structure and activity and their interrelation in rats fed a high-fat–high-fructose diet. Br. J. Nutr. 2018, 119, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001, 509, 435–438. [Google Scholar] [CrossRef] [Green Version]
- Meissen, J.K.; Hirahatake, K.M.; Adams, S.H.; Fiehn, O. Temporal metabolomic responses of cultured HepG2 liver cells to high fructose and high glucose exposures. Metabolomics 2015, 11, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.D.; Xu, S.; Choi, S.-K.; Ha, C.-M.; Thoudam, T.; Cha, S.-K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.-K.; Park, K.-S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [PubMed]
- Spitz, D.R.; Kinter, M.T.; Kehrer, J.P.; Roberts, R.J. The Effect of Monosaturated and Polyunsaturated Fatty Acids on Oxygen Toxicity in Cultured Cells. Pediatr. Res. 1992, 32, 366–372. [Google Scholar] [CrossRef] [Green Version]
- Joseph, L.C.; Barca, E.; Subramanyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NAPDH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PLoS ONE 2016, 11, e0145750. [Google Scholar] [CrossRef]
- Cha, S.H.; Wolfgang, M.; Tokutake, Y.; Chohnan, S.; Lane, M.D. Differential effects of central fructose and glucose on hypothalamic malonyl—CoA and food intake. Proc. Natl. Acad. Sci. USA 2008, 105, 16871–16875. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Luedde, T. Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Kominami, K.; Nakabayashi, J.; Nagai, T.; Tsujimura, Y.; Chiba, K.; Kimura, H.; Miyawaki, A.; Sawasaki, T.; Yokota, H.; Manabe, N.; et al. Biochimica et Biophysica Acta The molecular mechanism of apoptosis upon caspase-8 activation: Quantitative experimental validation of a mathematical model. BBA Mol. Cell Res. 2012, 1823, 1825–1840. [Google Scholar]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef] [Green Version]
- Sorice, M.; Circella, A.; Cristea, I.M.; Garofalo, T.; Di Renzo, L.; Alessandri, C.; Valesini, G.; Degli Esposti, M. Cardiolipin and its metabolites move from mitochondria to other cellular membranes during death receptor-mediated apoptosis. Cell Death Differ. 2004, 11, 1133–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goonesinghe, A.; Mundy, E.S.; Smith, M.; Khosravi-Far, R.; Martinou, J.-C.; Degli Esposti, M. Pro-apoptotic Bid induces membrane perturbation by inserting selected lysolipids into the bilayer. Biochem. J. 2005, 387, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, J.R.; Andresen, T.L.; Feldborg, L.N.; Duelund, L.; Ipsen, J.H. Understanding detergent effects on lipid membranes: A model study of lysolipids. Biophys. J. 2010, 98, 2199–2205. [Google Scholar] [CrossRef] [Green Version]
- Lauber, K.; Bohn, E.; Kröber, S.M.; Xiao, Y.-J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic Cells Induce Migration of Phagocytes via Caspase-3-Mediated Release of a Lipid Attraction Signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, R.; Franken, H.; Dammeier, S.; Rosenbaum, L.; Kantartzis, K.; Peter, A.; Zell, A.; Adam, P.; Li, J.; Xu, G.; et al. Circulating Lysophosphatidylcholines Are Markers of a Metabolically Benign Nonalcoholic Fatty Liver. Diabetes Care 2013, 36, 2331–2338. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xiao, Y.; Tang, L.; Zhong, F.; Huang, G.; Xu, J.-M.; Xu, A.-M.; Dai, R.-P.; Zhou, Z.-G. Adipocyte Fatty Acid-Binding Protein Promotes Palmitate-Induced Mitochondrial Dysfunction and Apoptosis in Macrophages. Front. Immunol. 2018, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase Functions in Cell Death and Disease: Figure 1. Cold Spring Harb. Perspect. Biol. 2015, 7, a026716. [Google Scholar] [CrossRef] [Green Version]
- Kučera, O.; Lotkova, H.; Sobotka, O.; Červinková, Z. The Effect of D-Galactosamine on Lean and Steatotic Rat Hepatocytes in Primary Culture. Physiol. Res. 2015, 64, S637–S646. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, L.; Liu, X.; Luo, R.; Liao, G.; Li, L.; Liu, J.; Cheng, J.; Lu, Y.; Chen, Y. Oleic acid protects saturated fatty acid mediated lipotoxicity in hepatocytes and rat of non-alcoholic steatohepatitis. Life Sci. 2018, 203, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, M.; Sgroi, A.; Veyrat-Durebex, C.; Rubbia-Brandt, L.; Buhler, L.H.; Foti, M. Unsaturated fatty acids inhibit the expression of tumor suppressor phosphatase and tensin homolog (PTEN) via microRNA-21 up-regulation in hepatocytes. Hepatology 2008, 49, 1176–1184. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amorim, R.; Simões, I.C.M.; Veloso, C.; Carvalho, A.; Simões, R.F.; Pereira, F.B.; Thiel, T.; Normann, A.; Morais, C.; Jurado, A.S.; et al. Exploratory Data Analysis of Cell and Mitochondrial High-Fat, High-Sugar Toxicity on Human HepG2 Cells. Nutrients 2021, 13, 1723. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13051723
Amorim R, Simões ICM, Veloso C, Carvalho A, Simões RF, Pereira FB, Thiel T, Normann A, Morais C, Jurado AS, et al. Exploratory Data Analysis of Cell and Mitochondrial High-Fat, High-Sugar Toxicity on Human HepG2 Cells. Nutrients. 2021; 13(5):1723. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13051723
Chicago/Turabian StyleAmorim, Ricardo, Inês C. M. Simões, Caroline Veloso, Adriana Carvalho, Rui F. Simões, Francisco B. Pereira, Theresa Thiel, Andrea Normann, Catarina Morais, Amália S. Jurado, and et al. 2021. "Exploratory Data Analysis of Cell and Mitochondrial High-Fat, High-Sugar Toxicity on Human HepG2 Cells" Nutrients 13, no. 5: 1723. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13051723