
Diagnostic and Therapeutic Perspectives Associated to Cobalamin-Dependent Metabolism and Transcobalamins’ Synthesis in Solid Cancers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
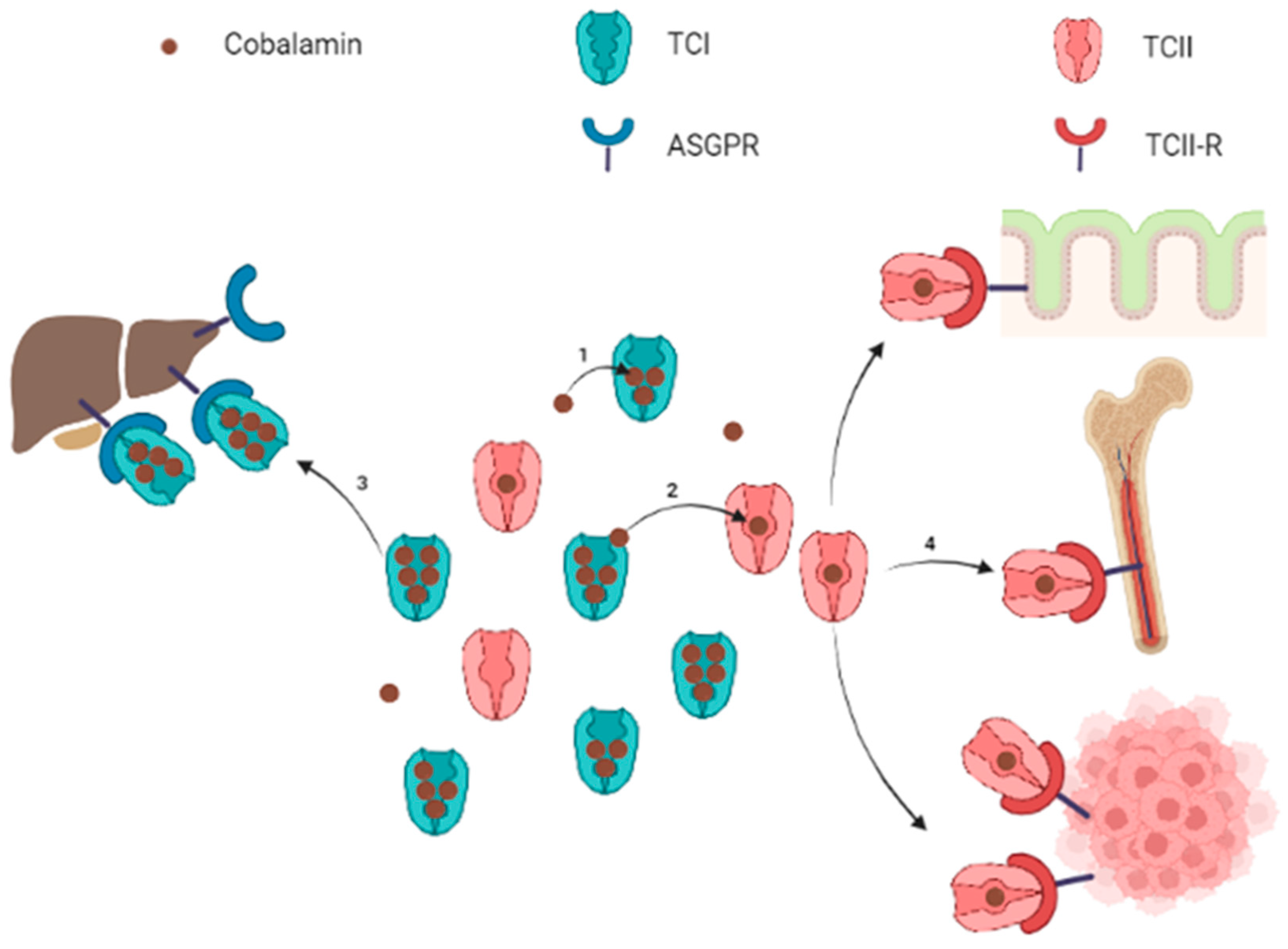
2. Cobalamin: From Absorption to Intracellular Metabolism
3. Relationship between Cobalamin, Transcobalamins and TCII-R in the Context of Solid Cancer
3.1. Measurements of Plasma Cobalamin and Transcobalamins in Clinical Practice
3.2. Association between tB12, TCI, TCII and the Diagnosis of Solid Cancers
3.3. Association between tB12, TCI, TCII and the Prognosis of Solid Cancers
3.4. Association between tB12, TCI, TCII Changes and the Course of Solid Cancers
3.5. Direction of the Causal Link between Plasma tB12, TCI or TCII Measurements and the Presence of Solid Cancers
3.6. Cobalamin Avidity and TCII-R Expression in Cancer Cells
3.7. TCN1 and TCN2 Gene Mutations and Expression in Cancer Cells
4. Implication of Cobalamin-Dependent Pathways in Tumor Initiation and Cell Proliferation
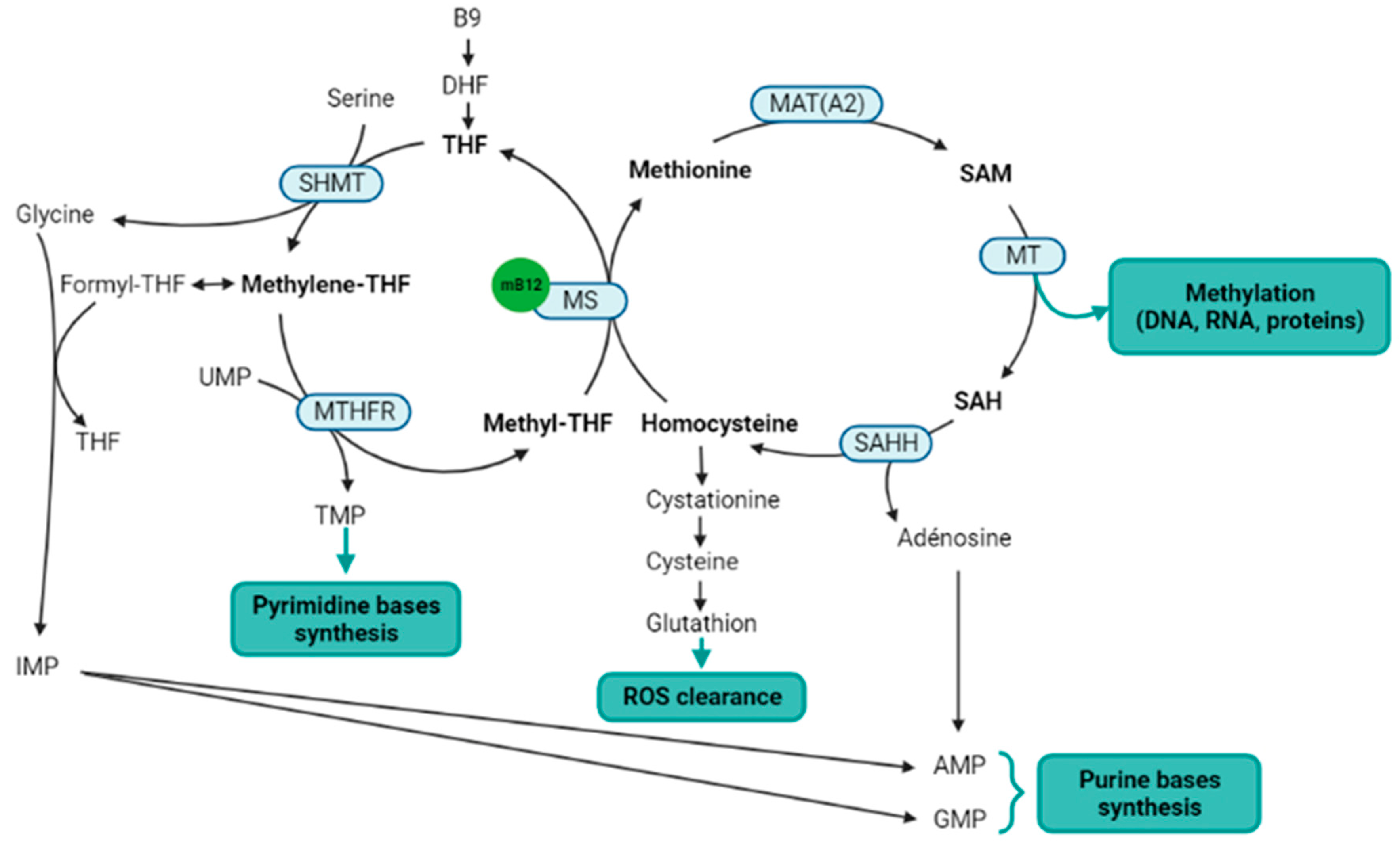
4.1. Methionine Synthesis and Methylation
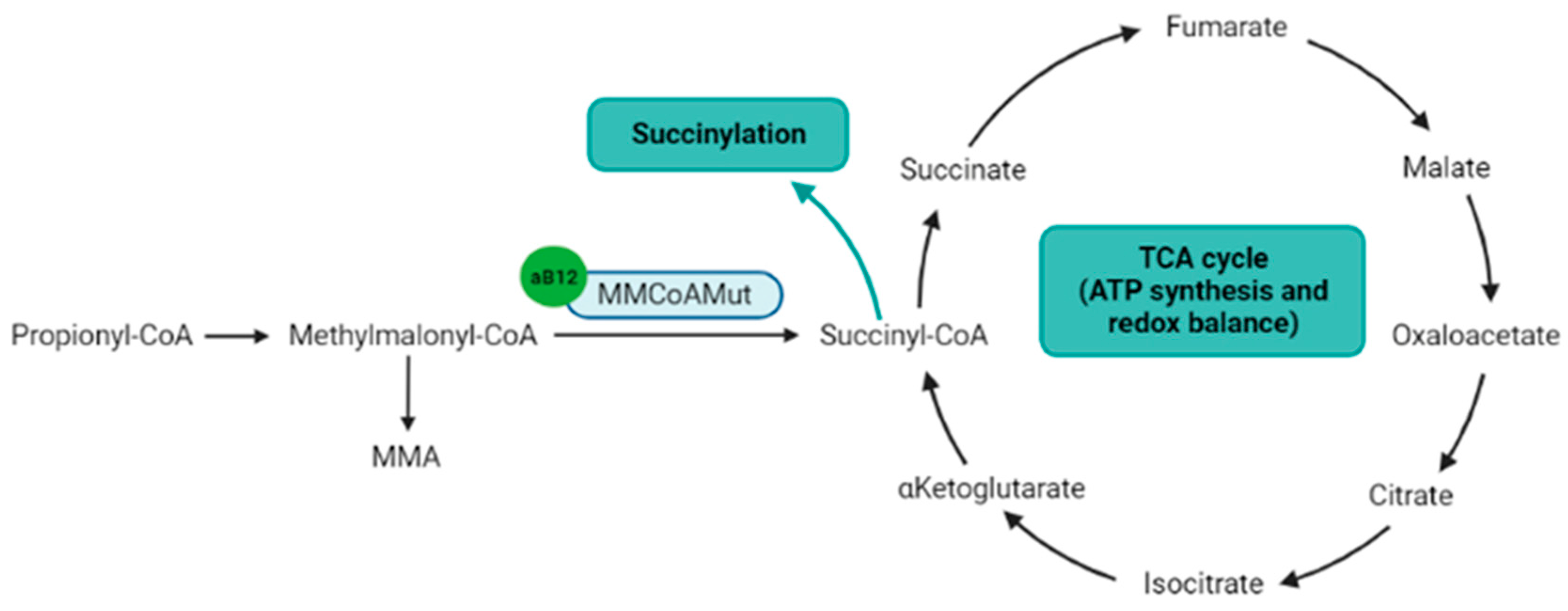
4.2. Succinylation
4.3. Other Functions Indirectly Related to the Cobalamin-Dependent Enzymes
5. Potential Therapeutic Uses Derived from Cobalamin Avidity and Methionine Needs of Cancer Cells
5.1. Using Cobalamin as a Vector for a Trojan Horse Effect
5.2. Inhibition of the One-Carbon Cycle
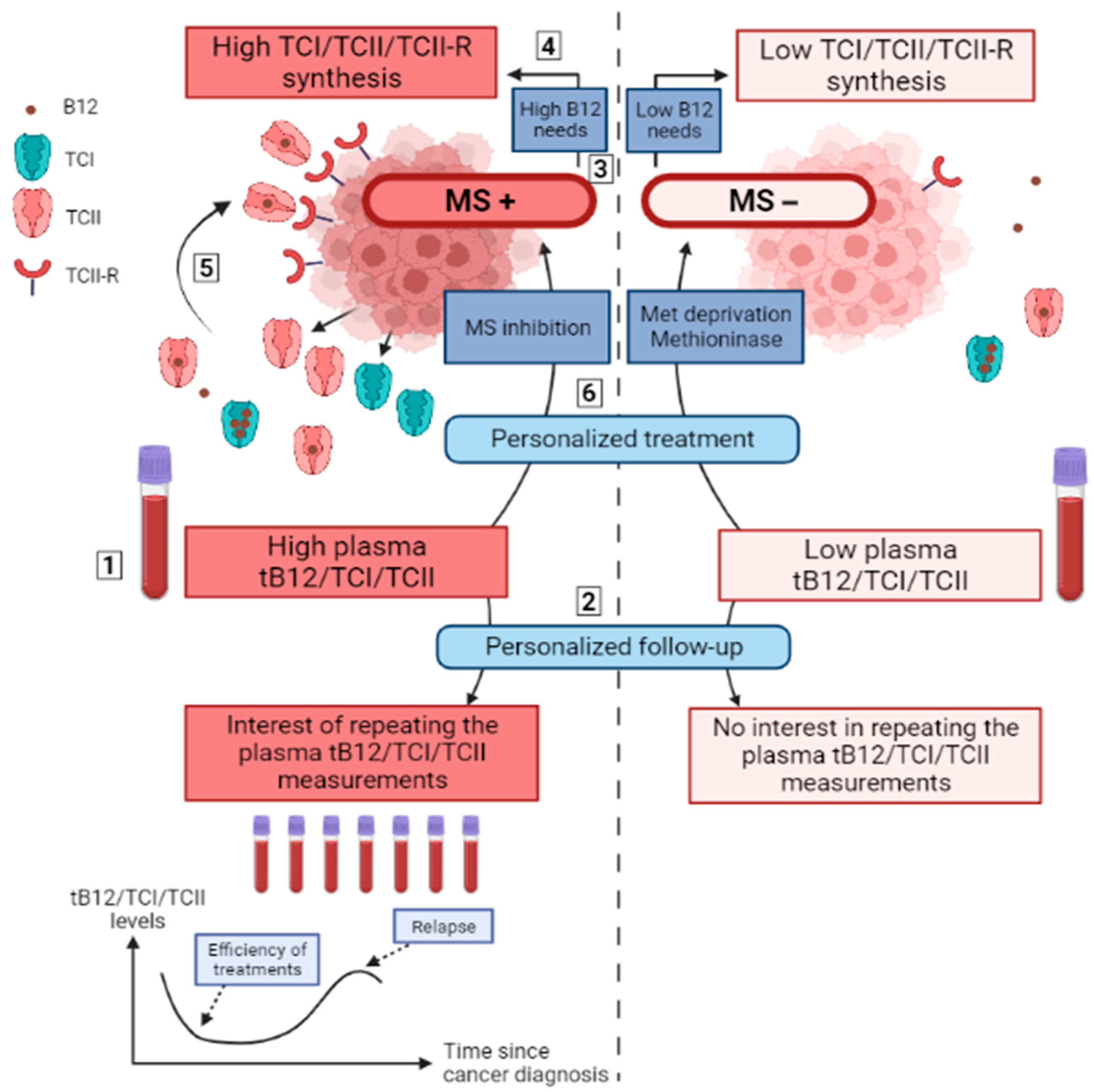
6. Remaining Questions, Hypotheses and Perspectives
6.1. Origin of Plasma TCI and TCII Elevation in the Context of Solid Cancer
6.2. Plasma tB12, TCI, and TCII as Biomarkers of Solid Cancers
6.3. Plasma tB12, TCI and TCII as Markers for Relapses
6.4. Plasma tB12/TCI/TCII Levels and Cancer Cell TCI/TCII/TCII-R Synthesis as Prognosis Markers
6.5. Plasma tB12/TCI/TCII and Cancer Cell TCI/TCII/TCII-R as Markers of a High Dependency on Cobalamin-Dependent Enzymes
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic Pathways Promoting Cancer Cell Survival and Growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Heiden, M.G.V.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, R.; Allen, L.H.; Bjørke-Monsen, A.-L.; Brito, A.; Guéant, J.-L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.-H.; et al. Vitamin B12 Deficiency. Nat. Rev. Dis. Primers 2017, 3, 17040. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.A.; Hogenkamp, H.P.C.; O’Connor, M.K.; Naylor, S.; Benson, L.M.; Hardyman, T.J.; Thorson, L.M. Biodistribution of Radiolabeled Adenosylcobalamin in Patients Diagnosed With Various Malignancies. Mayo Clin. Proc. 2000, 75, 568–580. [Google Scholar] [CrossRef]
- McLean, G.R.; Quadros, E.V.; Rothenberg, S.P.; Morgan, A.C.; Schrader, J.W.; Ziltener, H.J. Antibodies to Transcobalamin II Block In Vitro Proliferation of Leukemic Cells. Blood 1997, 89, 235–242. [Google Scholar] [CrossRef]
- Cooper, B.A.; Paranchych, W. Selective Uptake of Specifically Bound Cobalt-58 Vitamin B12 by Human and Mouse Tumour Cells. Nature 1961, 191, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Meyer, L.M.; Bertcher, R.W.; Cronkite, E.P.; Suarez, R.M.; Miller, I.F.; Mulzac, C.W.; Olivarreta, S.T. Co60 Vitamin B12 Binding Capacity of Serum in Persons with Hematologic Disorders, Various Medical Diseases and Neoplasms. Acta Med. Scand. 1961, 169, 557–575. [Google Scholar] [CrossRef]
- Hall, C.A.; Finkler, A.E. The dynamics of transcobalamin ii. a vitamin B12 binding substance in plasma. J. Lab. Clin. Med. 1965, 65, 459–468. [Google Scholar]
- Hall, C.A.; Finkler, A.E. A Second Vitamin B12-Binding Substance in Human Plasma. Biochim. Biophys. Acta 1963, 78, 234–236. [Google Scholar] [CrossRef]
- Carmel, R. Extreme Elevation of Serum Transcobalamin I in Patients with Metastatic Cancer. N. Engl. J. Med. 1975, 292, 282–284. [Google Scholar] [CrossRef]
- Kane, S.P.; Murray-Lyon, I.M.; Paradinas, F.J.; Johnson, P.J.; Williams, R.; Orr, A.H.; Kohn, J. Vitamin B12 Binding Protein as a Tumour Marker for Hepatocellular Carcinoma. Gut 1978, 19, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Gimsing, P.; Hippe, E. Increased Concentration of Transcobalamin I in a Patient with Metastatic Carcinoma of the Breast. Scand. J. Haematol. 1978, 21, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Arendt, J.F.B.; Pedersen, L.; Nexo, E.; Sørensen, H.T. Elevated Plasma Vitamin B12 Levels as a Marker for Cancer: A Population-Based Cohort Study. J. Natl. Cancer Inst. 2013, 105, 1799–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, J.F.H.; Sørensen, H.T.; Horsfall, L.J.; Petersen, I. Elevated Vitamin B12 Levels and Cancer Risk in UK Primary Care: A THIN Database Cohort Study. Cancer Epidemiol. Biomark. Prev. 2019, 28, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Urbanski, G.; Hamel, J.-F.; Prouveur, B.; Annweiler, C.; Ghali, A.; Cassereau, J.; Lozac’h, P.; Lavigne, C.; Lacombe, V. Strength of the Association of Elevated Vitamin B12 and Solid Cancers: An Adjusted Case-Control Study. J. Clin. Med. 2020, 9, 474. [Google Scholar] [CrossRef] [Green Version]
- Arendt, J.F.H.; Farkas, D.K.; Pedersen, L.; Nexo, E.; Sørensen, H.T. Elevated Plasma Vitamin B12 Levels and Cancer Prognosis: A Population-Based Cohort Study. Cancer Epidemiol. 2016, 40, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Arendt, J.F.B.; Nexo, E. Cobalamin Related Parameters and Disease Patterns in Patients with Increased Serum Cobalamin Levels. PLoS ONE 2012, 7, e45979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, J.F.B.; Nexo, E. Unexpected High Plasma Cobalamin/Proposal for a Diagnostic Strategy. Clin. Chem. Lab. Med. 2013, 51, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, V.; Chabrun, F.; Lacout, C.; Ghali, A.; Capitain, O.; Patsouris, A.; Lavigne, C.; Urbanski, G. Persistent Elevation of Plasma Vitamin B12 Is Strongly Associated with Solid Cancer. Sci. Rep. 2021, 11, 13361. [Google Scholar] [CrossRef]
- Huang, H.; Wang, J.; Zhang, J.; Cai, J.; Pi, J.; Xu, J.-F. Inspirations of Cobalt Oxide Nanoparticle Based Anticancer Therapeutics. Pharmaceutics 2021, 13, 1599. [Google Scholar] [CrossRef]
- Bauer, J.A. Effects of Interferon Beta on Transcobalamin II-Receptor Expression and Antitumor Activity of Nitrosylcobalamin. CancerSpectrum Knowl. Environ. 2002, 94, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Gupta, Y.; Kohli, D.V.; Jain, S.K. Vitamin B12-Mediated Transport: A Potential Tool for Tumor Targeting of Antineoplastic Drugs and Imaging Agents. Crit. Rev. Ther. Drug Carrier Syst. 2008, 25, 347–379. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M.; Tan, Y.; Li, S.; Han, Q.; Zavala, J.; Zavala, J. Pilot Phase I Clinical Trial of Methioninase on High-Stage Cancer Patients: Rapid Depletion of Circulating Methionine. In Methionine Dependence of Cancer and Aging; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1866, pp. 231–242. ISBN 978-1-4939-8795-5. [Google Scholar]
- Thivat, E.; Farges, M.-C.; Bacin, F.; D’Incan, M.; Mouret-Reynier, M.-A.; Cellarier, E.; Madelmont, J.-C.; Vasson, M.-P.; Chollet, P.; Durando, X. Phase II Trial of the Association of a Methionine-Free Diet with Cystemustine Therapy in Melanoma and Glioma. Anticancer Res. 2009, 29, 5235–5240. [Google Scholar] [PubMed]
- Durando, X.; Farges, M.-C.; Buc, E.; Abrial, C.; Petorin-Lesens, C.; Gillet, B.; Vasson, M.-P.; Pezet, D.; Chollet, P.; Thivat, E. Dietary Methionine Restriction with FOLFOX Regimen as First Line Therapy of Metastatic Colorectal Cancer: A Feasibility Study. Oncology 2010, 78, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Hoffman, R.M.; Bertino, J.R. Exploiting Methionine Restriction for Cancer Treatment. Biochem. Pharmacol. 2018, 154, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Kang, J.; Zhang, D. Microbial Production of Vitamin B12: A Review and Future Perspectives. Microb. Cell Fact. 2017, 16, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, F.; Bito, T. Vitamin B12 Sources and Microbial Interaction. Exp. Biol. Med. 2018, 243, 148–158. [Google Scholar] [CrossRef]
- Doscherholmen, A.; Hagen, P.S. A Dual Mechanism of Vitamin B12 Plasma Absorption1. J. Clin. Investig. 1957, 36, 1551–1557. [Google Scholar] [CrossRef]
- Lacombe, V.; Roquin, G.; Vinatier, E.; Lavigne, C.; Urbanski, G. Parietal Cell Antibodies: Evolution of Plasma Vitamin B12 during Oral Supplementation to Differentiate True and False Positives for Pernicious Anemia. Pol. Arch. Intern. Med. 2020, 130, 813–815. [Google Scholar] [CrossRef]
- Stabler, S.P. Vitamin B12 Deficiency. N. Engl. J. Med. 2013, 368, 149–160. [Google Scholar] [CrossRef]
- Johnston, J.; Bollekens, J.; Allen, R.H.; Berliner, N. Structure of the CDNA Encoding Transcobalamin I, a Neutrophil Granule Protein. J. Biol. Chem. 1989, 264, 15754–15757. [Google Scholar] [CrossRef]
- Quadros, E.V. Advances in the Understanding of Cobalamin Assimilation and Metabolism. Br. J. Haematol. 2010, 148, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, C.M.L.A.; Farquharson, J.; Bessent, R.G.; Adams, J.F. The Forms of Vitamin B12 on the Transcobalamins. Clin. Sci. 1977, 52, 215–218. [Google Scholar] [CrossRef]
- Nexø, E.; Andersen, J. Unsaturated and Cobalamin Saturated Transcobalamin I and II in Normal Human Plasma. Scand. J. Clin. Lab. Investig. 1977, 37, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Russell-Jones, G.J.; Alpers, D.H. Vitamin B12 Transporters. In Membrane Transporters as Drug Targets; Amidon, G.L., Sadée, W., Eds.; Pharmaceutical Biotechnology; Kluwer Academic Publishers: Boston, MA, USA, 2002; Volume 12, pp. 493–520. ISBN 978-0-306-46094-4. [Google Scholar]
- Pacifico, F.; Laviola, L.; Ulianich, L.; Porcellini, A.; Ventra, C.; Consiglio, E.; Avvedimento, V.E. Differential Expression of the Asialoglycoprotein Receptor in Discrete Brain Areas, in Kidney and Thyroid. Biochem. Biophys. Res. Commun. 1995, 210, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Zheng, J.; She, G.; Wei, X.; Zhang, X.; Shi, H.; Zhou, X. Expression Pattern of Asialoglycoprotein Receptor in Human Testis. Cell Tissue Res. 2013, 352, 761–768. [Google Scholar] [CrossRef]
- Mu, J.-Z.; Fallon, R.J.; Swanson, P.E.; Carroll, S.B.; Danaher, M.; Alpers, D.H. Expression of an Endogenous Asialoglycoprotein Receptor in a Human Intestinal Epithelial Cell Line, Caco-2. Biochim. Biophys. Acta—Mol. Cell Res. 1994, 1222, 483–491. [Google Scholar] [CrossRef]
- Witzigmann, D.; Quagliata, L.; Schenk, S.H.; Quintavalle, C.; Terracciano, L.M.; Huwyler, J. Variable Asialoglycoprotein Receptor 1 Expression in Liver Disease: Implications for Therapeutic Intervention: ASGR1 Expression in Liver Disease. Hepatol. Res. 2016, 46, 686–696. [Google Scholar] [CrossRef]
- Trerè, D.; Fiume, L.; De Giorgi, L.B.; Di Stefano, G.; Migaldi, M.; Derenzini, M. The Asialoglycoprotein Receptor in Human Hepatocellular Carcinomas: Its Expression on Proliferating Cells. Br. J. Cancer 1999, 81, 404–408. [Google Scholar] [CrossRef] [Green Version]
- Rothenberg, S.P.; Quadros, E.V. Transcobalamin II and the Membrane Receptor for the Transcobalamin II-Cobalamin Complex. Bailliere’s Clin. Haematol. 1995, 8, 499–514. [Google Scholar] [CrossRef]
- Li, N.; Seetharam, S.; Rosenblatt, D.S.; Seetharam, B. Expression of Transcobalamin II MRNA in Human Tissues and Cultured Fibroblasts from Normal and Transcobalamin II-Deficient Patients. Biochem. J. 1994, 301, 585–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fràter-Schröder, M.; Porck, H.J.; Erten, J.; Müller, M.R.; Steinmann, B.; Kierat, L.; Arwert, F. Synthesis and Secretion of the Human Vitamin B12-Binding Protein, Transcobalamin II, by Cultured Skin Fibroblasts and by Bone Marrow Cells. Biochim. Biophys. Acta 1985, 845, 421–427. [Google Scholar] [CrossRef]
- Begley, J.A.; Colligan, P.D.; Chu, R.C. Synthesis and Secretion of Transcobalamin II by Cultured Astrocytes Derived from Human Brain Tissue. J. Neurol. Sci. 1994, 122, 57–60. [Google Scholar] [CrossRef]
- Rabinowitz, R.; Rachmilewitz, B.; Rachmilewitz, M.; Schlesinger, M. Production of Transcobalamin II by Various Murine and Human Cells in Culture. Isr. J. Med. Sci. 1982, 18, 740–745. [Google Scholar] [PubMed]
- Lindemans, J.; Kroes, A.C.; van Geel, J.; van Kapel, J.; Schoester, M.; Abels, J. Uptake of Transcobalamin II-Bound Cobalamin by HL-60 Cells: Effects of Differentiation Induction. Exp. Cell Res. 1989, 184, 449–460. [Google Scholar] [CrossRef]
- Banerjee, R.V.; Matthews, R.G. Cobalamin-Dependent Methionine Synthase. FASEB J. 1990, 4, 1450–1459. [Google Scholar] [CrossRef]
- Newman, A.C.; Maddocks, O.D.K. One-Carbon Metabolism in Cancer. Br. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, S. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Machover, D. Recombinant Methioninase as a DNA Demethylation Agent. Methods Mol. Biol. 2019, 1866, 279–284. [Google Scholar] [CrossRef]
- Hasan, T.; Arora, R.; Bansal, A.K.; Bhattacharya, R.; Sharma, G.S.; Singh, L.R. Disturbed Homocysteine Metabolism Is Associated with Cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Tao, B.-B.; Cai, W.-J.; Zhu, Y.-C. H2S Is a Promoter of Angiogenesis: Identification of H2S “Receptors” and Its Molecular Switches in Vascular Endothelial Cells. Handb. Exp. Pharmacol. 2015, 230, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Warren, M.J.; Refsum, H. Vitamin B12. In Advances in Food and Nutrition Research; Elsevier: Amsterdam, The Netherlands, 2018; Volume 83, pp. 215–279. ISBN 978-0-12-811803-0. [Google Scholar]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The Emerging Role and Targetability of the TCA Cycle in Cancer Metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Alleyn, M.; Breitzig, M.; Lockey, R.; Kolliputi, N. The Dawn of Succinylation: A Posttranslational Modification. Am. J. Physiol.-Cell Physiol. 2018, 314, C228–C232. [Google Scholar] [CrossRef] [PubMed]
- Andres, E.; Serraj, K.; Zhu, J.; Vermorken, A.J.M. The Pathophysiology of Elevated Vitamin B12 in Clinical Practice. QJM 2013, 106, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Carmel, R.; Eisenberg, L. Serum Vitamin B12 and Transcobalamin Abnormalities in Patients with Cancer. Cancer 1977, 40, 1348–1353. [Google Scholar] [CrossRef]
- Clarke, R.; Sherliker, P.; Hin, H.; Nexo, E.; Hvas, A.M.; Schneede, J.; Birks, J.; Ueland, P.M.; Emmens, K.; Scott, J.M.; et al. Detection of Vitamin B12 Deficiency in Older People by Measuring Vitamin B12 or the Active Fraction of Vitamin B12, Holotranscobalamin. Clin. Chem. 2007, 53, 963–970. [Google Scholar] [CrossRef]
- Wickramasinghe, S.N.; Fida, S. Correlations between Holo-Transcobalamin II, Holo-Haptocorrin, and Total B12 in Serum Samples from Healthy Subjects and Patients. J. Clin. Pathol. 1993, 46, 537–539. [Google Scholar] [CrossRef]
- Carmel, R.; Hollander, D. Extreme Elevation of Transcobalamin II Levels in Multiple Myeloma and Other Disorders. Blood 1978, 51, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.-C.; Goldstein, B.Y.; Mu, L.; Cai, L.; You, N.-C.Y.; He, N.; Ding, B.-G.; Zhao, J.-K.; Yu, S.-Z.; Heber, D.; et al. Plasma Folate, Vitamin B12, and Homocysteine and Cancers of the Esophagus, Stomach, and Liver in a Chinese Population. Nutr. Cancer 2015, 67, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Collin, S.M. Folate and B12 in Prostate Cancer. Adv. Clin. Chem. 2013, 60, 1–63. [Google Scholar]
- Cui, L.-H.; Quan, Z.-Y.; Piao, J.-M.; Zhang, T.-T.; Jiang, M.-H.; Shin, M.-H.; Choi, J.-S. Plasma Folate and Vitamin B12 Levels in Patients with Hepatocellular Carcinoma. Int. J. Mol. Sci. 2016, 17, 1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsen, K.; Rode, A.; Nicoll, A.; Villadsen, G.; Espelund, U.; Lim, L.; Angus, P.; Arachchi, N.; Vilstrup, H.; Nexo, E.; et al. Vitamin B12 and Its Binding Proteins in Hepatocellular Carcinoma and Chronic Liver Diseases. Scand. J. Gastroenterol. 2014, 49, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Lo-Bisgaard, T.; Espelund, U.; Frystyk, J.; Rasmussen, T.R.; Nexo, E.; Arendt, J.F.H. Vitamin B12 and Its Binding Proteins in Patients with Non-Small Cell Lung Cancer Referred to Fast-Track Diagnostic Work-up for Lung Cancer. Scand. J. Clin. Lab. Investig. 2020, 80, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Geissbühler, P.; Mermillod, B.; Rapin, C.-H. Elevated Serum Vitamin B12 Levels Associated With CRP as a Predictive Factor of Mortality in Palliative Care Cancer Patients: A Prospective Study Over Five Years. J. Pain Symptom Manag. 2000, 20, 93–103. [Google Scholar] [CrossRef]
- Lacombe, V.; Patsouris, A.; Delattre, E.; Lacout, C.; Urbanski, G. Evolution of Plasma Vitamin B12 in Patients with Solid Cancers during Curative versus Supportive Care. Arch. Med. Sci. 2021, 17, 1811–1815. [Google Scholar] [CrossRef]
- Lildballe, D.L.; Nguyen, K.Q.T.; Poulsen, S.S.; Nielsen, H.O.; Nexo, E. Haptocorrin as Marker of Disease Progression in Fibrolamellar Hepatocellular Carcinoma. Eur. J. Surg. Oncol. 2011, 37, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Jensen, H.S.; Gimsing, P.; Pedersen, F.; Hippe, E. Transcobalamin II as an Indicator of Activity in Metastatic Renal Adenocarcinoma. Cancer 1983, 52, 1700–1704. [Google Scholar] [CrossRef]
- Rachmilewitz, B.; Sulkes, A.; Rachmilewitz, M.; Fuks, Z. Serum Transcobalamin II Levels in Breast Carcinoma Patients. Isr. J. Med. Sci. 1981, 17, 874–878. [Google Scholar]
- Wheeler, K.; Pritchard, J.; Luck, W.; Rossiter, M. Transcobalamin I as a “Marker” for Fibrolamellar Hepatoma. Med. Pediatric Oncol. 1986, 14, 227–229. [Google Scholar] [CrossRef]
- Frémont, S.; Champigneulle, B.; Gérard, P.; Felden, F.; Lambert, D.; Guéant, J.L.; Nicolas, J.P. Blood Transcobalamin Levels in Malignant Hepatoma. Tumour Biol. 1991, 12, 353–359. [Google Scholar] [CrossRef]
- Liu, G.; Wang, Y.; Yue, M.; Zhao, L.; Guo, Y.-D.; Liu, Y.; Yang, H.; Liu, F.; Zhang, X.; Zhi, L.; et al. High Expression of TCN1 Is a Negative Prognostic Biomarker and Can Predict Neoadjuvant Chemosensitivity of Colon Cancer. Sci. Rep. 2020, 10, 11951. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wei, Y.; Song, A.; Li, Y. Association Study between Genome-Wide Significant Variants of Vitamin B12 Metabolism and Gastric Cancer in a Han Chinese Population: Unexpected Role of Vitamin B12 Metabolism Genes. IUBMB Life 2016, 68, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanidi, A.; Carreras-Torres, R.; Larose, T.L.; Yuan, J.-M.; Stevens, V.L.; Weinstein, S.J.; Albanes, D.; Prentice, R.; Pettinger, M.; Cai, Q.; et al. Is High Vitamin B12 Status a Cause of Lung Cancer?: Is High Vitamin B12 Status a Cause of Lung Cancer? Int. J. Cancer 2019, 145, 1499–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakatsuki, Y.; Inada, M.; Kudo, H.; Oshio, G.; Masuda, T.; Miyake, T.; Kita, T. Immunological Characterization and Clinical Implication of Cobalamin Binding Protein in Human Gastric Cancer. Cancer Res. 1989, 49, 3122–3128. [Google Scholar]
- Sheppard, K.; Bradbury, D.A.; Davies, J.M.; Ryrie, D.R. Cobalamin and Folate Binding Proteins in Human Tumour Tissue. J. Clin. Pathol. 1984, 37, 1336–1338. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Seetharam, S.; Hammond, T.G.; Seetharam, B. Regulation of Expression of Transcobalamin II Receptor in the Rat. Biochem. J. 1995, 310, 923–929. [Google Scholar] [CrossRef] [Green Version]
- Oreshkin, A.E.; Gudkova, M.V.; Miasishcheva, N.V. The expression of plasmalemma transcobalamin-II receptors on human blood lymphocytes stimulated by mitogens. Biull. Eksp. Biol. Med. 1992, 114, 185–187. [Google Scholar] [CrossRef]
- Sysel, A.M.; Valli, V.E.; Nagle, R.B.; Bauer, J.A. Immunohistochemical Quantification of the Vitamin B12 Transport Protein (TCII), Cell Surface Receptor (TCII-R) and Ki-67 in Human Tumor Xenografts. Anticancer Res. 2013, 33, 4203–4212. [Google Scholar]
- Gick, G.G.; Arora, K.; Sequeira, J.M.; Nakayama, Y.; Lai, S.-C.; Quadros, E.V. Cellular Uptake of Vitamin B12: Role and Fate of TCblR/CD320, the Transcobalamin Receptor. Exp. Cell Res. 2020, 396, 112256. [Google Scholar] [CrossRef]
- Sah, B.-R.; Schibli, R.; Waibel, R.; von Boehmer, L.; Bläuenstein, P.; Nexo, E.; Johayem, A.; Fischer, E.; Müller, E.; Soyka, J.D.; et al. Tumor Imaging in Patients with Advanced Tumors Using a New 99mTc-Radiolabeled Vitamin B12 Derivative. J. Nucl. Med. 2014, 55, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Collins, D.A.; Hogenkamp, H.P.; Gebhard, M.W. Tumor Imaging via Indium 111-Labeled DTPA-Adenosylcobalamin. Mayo Clin. Proc. 1999, 74, 687–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuda-Wedagedara, A.N.W.; Workinger, J.L.; Nexo, E.; Doyle, R.P.; Viola-Villegas, N. 89Zr-Cobalamin PET Tracer: Synthesis, Cellular Uptake, and Use for Tumor Imaging. ACS Omega 2017, 2, 6314–6320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikotun, O.F.; Marquez, B.V.; Fazen, C.H.; Kahkoska, A.R.; Doyle, R.P.; Lapi, S.E. Investigation of a Vitamin B12 Conjugate as a PET Imaging Probe. ChemMedChem 2014, 9, 1244–1251. [Google Scholar] [CrossRef]
- Lai, S.-C.; Nakayama, Y.; Sequeira, J.M.; Quadros, E.V. Down-Regulation of Transcobalamin Receptor TCblR/CD320 by SiRNA Inhibits Cobalamin Uptake and Proliferation of Cells in Culture. Exp. Cell Res. 2011, 317, 1603–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.A.; Colligan, P.D.; Begley, J.A. Cyclic Activity of the Receptors of Cobalamin Bound to Transcobalamin II. J. Cell Physiol. 1987, 133, 187–191. [Google Scholar] [CrossRef]
- Amagasaki, T.; Green, R.; Jacobsen, D.W. Expression of Transcobalamin II Receptors by Human Leukemia K562 and HL-60 Cells. Blood 1990, 76, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ruberu, K.; Li, H.; Garner, B. Cell Type-Specific Modulation of Cobalamin Uptake by Bovine Serum. PLoS ONE 2016, 11, e0167044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskerstrand, T.; Riedel, B.; Ueland, P.M.; Seetharam, B.; Pezacka, E.H.; Gulati, S.; Bose, S.; Banerjee, R.; Berge, R.K.; Refsum, H. Disruption of a Regulatory System Involving Cobalamin Distribution and Function in a Methionine-Dependent Human Glioma Cell Line. J. Biol. Chem. 1998, 273, 20180–20184. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.-M.; Yao, C.-T.; Chang, Y.-T.; Chou, H.-L.; Chou, Y.-C.; Chen, K.-H.; Terng, H.-J.; Huang, C.-S.; Lee, C.-C.; Su, S.-L.; et al. Gene Expression Profiling of Colorectal Tumors and Normal Mucosa by Microarrays Meta-Analysis Using Prediction Analysis of Microarray, Artificial Neural Network, Classification, and Regression Trees. Dis. Markers 2014, 2014, 634123. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-Y.; Wei, Y.-C.; Tian, Y.-F.; Sun, D.-P.; Sheu, M.-J.; Yang, C.-C.; Lin, L.-C.; Lin, C.-Y.; Hsing, C.-H.; Li, W.-S.; et al. Overexpression of Transcobalamin 1 Is an Independent Negative Prognosticator in Rectal Cancers Receiving Concurrent Chemoradiotherapy. J. Cancer 2017, 8, 1330–1337. [Google Scholar] [CrossRef]
- Wang, Y.; Yue, C.; Fang, J.; Gong, L.; Lian, M.; Wang, R.; Feng, L.; Ma, H.; Ma, Z.; Liu, H. Transcobalamin I: A Novel Prognostic Biomarker of Neoadjuvant Chemotherapy in Locally Advanced Hypopharyngeal Squamous Cell Cancers. OncoTargets Ther. 2018, 11, 4253–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, M.; Scapoli, L.; Mattei, G.; Ugolini, G.; Montroni, I.; Zattoni, D.; Rosati, G.; Solmi, R. A Candidate Gene Study of One-Carbon Metabolism Pathway Genes and Colorectal Cancer Risk. Br. J. Nutr. 2013, 109, 984–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Xu, P.; Sowers, J.L.; Machuca, D.F.; Mirfattah, B.; Herring, J.; Tang, H.; Chen, Y.; Tian, B.; Brasier, A.R.; et al. Proteome Analysis of Hypoxic Glioblastoma Cells Reveals Sequential Metabolic Adaptation of One-Carbon Metabolic Pathways. Mol. Cell Proteom. 2017, 16, 1906–1921. [Google Scholar] [CrossRef] [Green Version]
- Aledo, J.C. Methionine in Proteins: The Cinderella of the Proteinogenic Amino Acids. Protein Sci. 2019, 28, 1785–1796. [Google Scholar] [CrossRef]
- Valley, C.C.; Cembran, A.; Perlmutter, J.D.; Lewis, A.K.; Labello, N.P.; Gao, J.; Sachs, J.N. The Methionine-Aromatic Motif Plays a Unique Role in Stabilizing Protein Structure. J. Biol. Chem. 2012, 287, 34979–34991. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Mosoni, L.; Berlett, B.S.; Stadtman, E.R. Methionine Residues as Endogenous Antioxidants in Proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 15036–15040. [Google Scholar] [CrossRef] [Green Version]
- Kanayama, A.; Inoue, J.-I.; Sugita-Konishi, Y.; Shimizu, M.; Miyamoto, Y. Oxidation of Ikappa Balpha at Methionine 45 Is One Cause of Taurine Chloramine-Induced Inhibition of NF-Kappa B Activation. J. Biol. Chem. 2002, 277, 24049–24056. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Keenan, M.M.; Wu, J.; Lin, C.-A.; Dubois, L.; Thompson, J.W.; Freedland, S.J.; Murphy, S.K.; Chi, J.-T. Comprehensive Profiling of Amino Acid Response Uncovers Unique Methionine-Deprived Response Dependent on Intact Creatine Biosynthesis. PLoS Genet. 2015, 11, e1005158. [Google Scholar] [CrossRef]
- Chen, H.; Rubin, E.; Zhang, H.; Chung, S.; Jie, C.C.; Garrett, E.; Biswal, S.; Sukumar, S. Identification of Transcriptional Targets of HOXA5. J. Biol. Chem. 2005, 280, 19373–19380. [Google Scholar] [CrossRef] [Green Version]
- Jeannotte, L.; Gotti, F.; Landry-Truchon, K. Hoxa5: A Key Player in Development and Disease. J. Dev. Biol. 2016, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Stasinopoulos, I.A.; Mironchik, Y.; Raman, A.; Wildes, F.; Winnard, P.; Raman, V. HOXA5-Twist Interaction Alters P53 Homeostasis in Breast Cancer Cells. J. Biol. Chem. 2005, 280, 2294–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordóñez-Morán, P.; Dafflon, C.; Imajo, M.; Nishida, E.; Huelsken, J. HOXA5 Counteracts Stem Cell Traits by Inhibiting Wnt Signaling in Colorectal Cancer. Cancer Cell 2015, 28, 815–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Nie, F.; Sun, M.; Xia, R.; Xie, M.; Lu, K.; Li, W. HOXA5 Indicates Poor Prognosis and Suppresses Cell Proliferation by Regulating P21 Expression in Non Small Cell Lung Cancer. Tumour. Biol. 2015, 36, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA Methylation and Cancer. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2010; Volume 70, pp. 27–56. ISBN 978-0-12-380866-0. [Google Scholar]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene Expression Regulation Mediated through Reversible M6A RNA Methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef]
- Bon, C.; Erdmann, D.; Halby, L.; Arimondo, P.B. Chemical targeting of DNA and histone methylation in cancer: Novelties, hopes and promises. Bull. Cancer 2019, 106, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Costello, J. DNA Methylation: An Epigenetic Mark of Cellular Memory. Exp. Mol. Med. 2017, 49, e322. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Kouzarides, T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A Decade of Exploring the Cancer Epigenome—Biological and Translational Implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Park, J.W.; Han, J.-W. Targeting Epigenetics for Cancer Therapy. Arch. Pharm. Res. 2019, 42, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Tiffen, J.; Gallagher, S.J.; Filipp, F.; Gunatilake, D.; Emran, A.A.; Cullinane, C.; Dutton-Register, K.; Aoude, L.; Hayward, N.; Chatterjee, A.; et al. EZH2 Cooperates with DNA Methylation to Downregulate Key Tumor Suppressors and IFN Gene Signatures in Melanoma. J. Investig. Dermatol. 2020, 140, 2442–2454.e5. [Google Scholar] [CrossRef]
- Martinez-Gamero, C.; Malla, S.; Aguilo, F. LSD1: Expanding Functions in Stem Cells and Differentiation. Cells 2021, 10, 3252. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Richards, J.S.; Shimada, M. Large-Scale DNA Demethylation Occurs in Proliferating Ovarian Granulosa Cells during Mouse Follicular Development. Commun. Biol. 2021, 4, 1334. [Google Scholar] [CrossRef] [PubMed]
- Methot, S.P.; Padeken, J.; Brancati, G.; Zeller, P.; Delaney, C.E.; Gaidatzis, D.; Kohler, H.; van Oudenaarden, A.; Großhans, H.; Gasser, S.M. H3K9me Selectively Blocks Transcription Factor Activity and Ensures Differentiated Tissue Integrity. Nat. Cell Biol. 2021, 23, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Borkiewicz, L. Histone 3 Lysine 27 Trimethylation Signature in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 12853. [Google Scholar] [CrossRef]
- Kim, H.G.; Kim, M.S.; Lee, Y.S.; Lee, E.H.; Kim, D.C.; Lee, S.-H.; Kim, Y.Z. Hypo-Trimethylation of Histone H3 Lysine 4 and Hyper-Tri/Dimethylation of Histone H3 Lysine 27 as Epigenetic Markers of Poor Prognosis in Patients with Primary Central Nervous System Lymphoma. Cancer Res. Treat. 2021. [Google Scholar] [CrossRef]
- Thivat, E.; Durando, X.; Demidem, A.; Farges, M.-C.; Rapp, M.; Cellarier, E.; Guenin, S.; D’Incan, M.; Vasson, M.-P.; Chollet, P. A Methionine-Free Diet Associated with Nitrosourea Treatment down-Regulates Methylguanine-DNA Methyl Transferase Activity in Patients with Metastatic Cancer. Anticancer Res. 2007, 27, 2779–2783. [Google Scholar]
- Mecham, J.O.; Rowitch, D.; Wallace, C.D.; Stern, P.H.; Hoffman, R.M. The Metabolic Defect of Methionine Dependence Occurs Frequently in Human Tumor Cell Lines. Biochem. Biophys. Res. Commun. 1983, 117, 429–434. [Google Scholar] [CrossRef]
- Judde, J.G.; Ellis, M.; Frost, P. Biochemical Analysis of the Role of Transmethylation in the Methionine Dependence of Tumor Cells. Cancer Res. 1989, 49, 4859–4865. [Google Scholar]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Kaiser, P. Methionine Dependence of Cancer. Biomolecules 2020, 10, 568. [Google Scholar] [CrossRef] [Green Version]
- Stern, P.H.; Wallace, C.D.; Hoffman, R.M. Altered Methionine Metabolism Occurs in All Members of a Set of Diverse Human Tumor Cell Lines. J. Cell Physiol. 1984, 119, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Epner, D.E. Molecular Mechanisms of Cell Cycle Block by Methionine Restriction in Human Prostate Cancer Cells. Nutr. Cancer 2000, 38, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M.; Jacobsen, S.J. Reversible Growth Arrest in Simian Virus 40-Transformed Human Fibroblasts. Proc. Natl. Acad. Sci. USA 1980, 77, 7306–7310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Yip, L.Y.; Lee, J.H.J.; Wu, Z.; Chew, H.Y.; Chong, P.K.W.; Teo, C.C.; Ang, H.Y.-K.; Peh, K.L.E.; Yuan, J.; et al. Methionine Is a Metabolic Dependency of Tumor-Initiating Cells. Nat. Med. 2019, 25, 825–837. [Google Scholar] [CrossRef]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.-W.; Pawel, B.R.; Zhang, J.; Finley, L.W.S.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine Catabolism Regulates Mitochondrial Redox Control during Hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [Green Version]
- Miyo, M.; Konno, M.; Colvin, H.; Nishida, N.; Koseki, J.; Kawamoto, K.; Tsunekuni, K.; Nishimura, J.; Hata, T.; Takemasa, I.; et al. The Importance of Mitochondrial Folate Enzymes in Human Colorectal Cancer. Oncol. Rep. 2017, 37, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of Lysine Succinylation as a New Post-Translational Modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Gao, P. Metabolic Reprogramming and Epigenetic Modifications on the Path to Cancer. Protein Cell 2021, 1–43. [Google Scholar] [CrossRef]
- Smestad, J.; Erber, L.; Chen, Y.; Maher, L.J. Chromatin Succinylation Correlates with Active Gene Expression and Is Perturbed by Defective TCA Cycle Metabolism. iScience 2018, 2, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Guo, Y.R.; Liu, K.; Yin, Z.; Liu, R.; Xia, Y.; Tan, L.; Yang, P.; Lee, J.-H.; Li, X.-J.; et al. KAT2A Coupled with the α-KGDH Complex Acts as a Histone H3 Succinyltransferase. Nature 2017, 552, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Yuan, Y.; Yuan, H.; Wang, J.; Yun, H.; Geng, Y.; Zhao, M.; Li, L.; Weng, Y.; Liu, Z.; et al. Histone Acetyltransferase 1 Is a Succinyltransferase for Histones and Non-histones and Promotes Tumorigenesis. EMBO Rep. 2021, 22, e50967. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, C.; Li, X.; Shen, J.; Xu, Y.; Shi, H.; Mu, X.; Pan, J.; Zhao, T.; Li, M.; et al. CPT1A-Mediated Succinylation of S100A10 Increases Human Gastric Cancer Invasion. J. Cell Mol. Med. 2019, 23, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Ning, X.; Yu, C.; Ji, X.; Jin, Y.; McNutt, M.A.; Yin, Y. Succinylation-Dependent Mitochondrial Translocation of PKM2 Promotes Cell Survival in Response to Nutritional Stress. Cell Death Dis. 2019, 10, 170. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA Damage Response in Cancer. Redox Biol. 2018, 25, 101084. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of Reactive Oxygen Species Levels and Radioresistance in Cancer Stem Cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Patwardhan, R.S.; Sharma, D.; Checker, R.; Thoh, M.; Sandur, S.K. Spatio-Temporal Changes in Glutathione and Thioredoxin Redox Couples during Ionizing Radiation-Induced Oxidative Stress Regulate Tumor Radio-Resistance. Free Radic. Res. 2015, 49, 1218–1232. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Akram, M. Citric Acid Cycle and Role of Its Intermediates in Metabolism. Cell Biochem. Biophys. 2014, 68, 475–478. [Google Scholar] [CrossRef]
- Scagliola, A.; Mainini, F.; Cardaci, S. The Tricarboxylic Acid Cycle at the Crossroad Between Cancer and Immunity. Antioxid. Redox Signal. 2020, 32, 834–852. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.-X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.-P.; Kunst, H.; Devilee, P.; Cremers, C.W.R.J.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a Gene Required for Flavination of Succinate Dehydrogenase, Is Mutated in Paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Multiple Leiomyoma Consortium Germline Mutations in FH Predispose to Dominantly Inherited Uterine Fibroids, Skin Leiomyomata and Papillary Renal Cell Cancer. Nat. Genet. 2002, 30, 406–410. [CrossRef] [PubMed]
- Green, A.; Beer, P. Somatic Mutations of IDH1 and IDH2 in the Leukemic Transformation of Myeloproliferative Neoplasms. N. Engl. J. Med. 2010, 362, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional Triggers of Oncogenic Signalling Cascades. Free. Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Deng, L.; Chen, Z.; Chen, Z.; Yu, J.; Liu, H.; Li, T.; Lin, T.; Chen, H.; Zhao, M.; et al. Vitamin B12-Conjugated Sericin Micelles for Targeting CD320-Overexpressed Gastric Cancer and Reversing Drug Resistance. Nanomedicine 2019, 14, 353–370. [Google Scholar] [CrossRef]
- Chen, Z.; Liang, Y.; Feng, X.; Liang, Y.; Shen, G.; Huang, H.; Chen, Z.; Yu, J.; Liu, H.; Lin, T.; et al. Vitamin-B12-Conjugated PLGA-PEG Nanoparticles Incorporating MiR-532-3p Induce Mitochondrial Damage by Targeting Apoptosis Repressor with Caspase Recruitment Domain (ARC) on CD320-Overexpressed Gastric Cancer. Mater. Sci. Eng. C 2021, 120, 111722. [Google Scholar] [CrossRef]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Gómez Padilla, P.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Zavala, J.; Xu, M.; Zavala, J.; Hoffman, R.M. Serum Methionine Depletion without Side Effects by Methioninase in Metastatic Breast Cancer Patients. Anticancer Res. 1996, 16, 3937–3942. [Google Scholar]
- Sugimura, T.; Birnbaum, S.M.; Winitz, M.; Greenstein, J.P. Quantitative Nutritional Studies with Water-Soluble, Chemically Defined Diets. VIII. The Forced Feeding of Diets Each Lacking in One Essential Amino Acid. Arch. Biochem. Biophys. 1959, 81, 448–455. [Google Scholar] [CrossRef]
- Epner, D.E.; Morrow, S.; Wilcox, M.; Houghton, J.L. Nutrient Intake and Nutritional Indexes in Adults with Metastatic Cancer on a Phase I Clinical Trial of Dietary Methionine Restriction. Nutr. Cancer 2002, 42, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, T.; Goseki, N.; Endo, M. Doxorubicin and Vincristine with Methionine Depletion Contributed to Survival in the Yoshida Sarcoma Bearing Rats. Anticancer Res. 1998, 18, 25–31. [Google Scholar] [PubMed]
- Taguchi, T.; Kosaki, G.; Onodera, T.; Endo, M.; Nakagawara, G.; Sano, K.; Kaibara, N.; Kakegawa, T.; Nakano, S.; Kurihara, M. A controlled study of AO-90, a methionine-free intravenous amino acid solution, in combination with 5-fluorouracil and mitomycin C in advanced gastric cancer patients (surgery group evaluation). Gan Kagaku Ryoho 1995, 22, 753–764. [Google Scholar]
- Hoffman, R.M. Clinical Studies of Methionine-Restricted Diets for Cancer Patients. In Methionine Dependence of Cancer and Aging; Hoffman, R.M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1866, pp. 95–105. ISBN 978-1-4939-8795-5. [Google Scholar]
- Goseki, N.; Maruyama, M.; Nagai, K.; Kando, F.; Endo, M.; Shimoju, K.; Wada, Y. Clinical evaluation of anticancer effect of methionine-depleting total parenteral nutrition with 5-fluorouracil and/or mitomycin C. Gan Kagaku Ryoho 1995, 22, 1028–1035. [Google Scholar]
- Kitamura, S.; Ohtani, T.; Kurihara, M.; Kosaki, G.; Akazawa, S.; Sasaki, T.; Takahashi, H.; Nakano, S.; Tokunaga, K. A controlled study of AO-90, a methionine-free intravenous amino acid solution, in combination with 5-fluorouracil and mitomycin C in advanced gastric cancer patients (internal medicine group evaluation). Gan Kagaku Ryoho 1995, 22, 765–775. [Google Scholar]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [Green Version]
- Kalev, P.; Hyer, M.L.; Gross, S.; Konteatis, Z.; Chen, C.-C.; Fletcher, M.; Lein, M.; Aguado-Fraile, E.; Frank, V.; Barnett, A.; et al. MAT2A Inhibition Blocks the Growth of MTAP-Deleted Cancer Cells by Reducing PRMT5-Dependent MRNA Splicing and Inducing DNA Damage. Cancer Cell 2021, 39, 209–224.e11. [Google Scholar] [CrossRef]
- Konteatis, Z.; Travins, J.; Gross, S.; Marjon, K.; Barnett, A.; Mandley, E.; Nicolay, B.; Nagaraja, R.; Chen, Y.; Sun, Y.; et al. Discovery of AG-270, a First-in-Class Oral MAT2A Inhibitor for the Treatment of Tumors with Homozygous MTAP Deletion. J. Med. Chem. 2021, 64, 4430–4449. [Google Scholar] [CrossRef]
- Fehr, J.; De Vecchi, P. Transcobalamin II: A Marker for Macrophage/Histiocyte Proliferation. Am. J. Clin. Pathol. 1985, 84, 291–296. [Google Scholar] [CrossRef]
- Serraj, K.; Mecili, M.; Housni, I.; Andrès, E. Hypervitaminémie B12: Physiopathologie et intérêt en pratique clinique. La Presse Méd. 2011, 40, 1120–1127. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacombe, V.; Lenaers, G.; Urbanski, G. Diagnostic and Therapeutic Perspectives Associated to Cobalamin-Dependent Metabolism and Transcobalamins’ Synthesis in Solid Cancers. Nutrients 2022, 14, 2058. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14102058
Lacombe V, Lenaers G, Urbanski G. Diagnostic and Therapeutic Perspectives Associated to Cobalamin-Dependent Metabolism and Transcobalamins’ Synthesis in Solid Cancers. Nutrients. 2022; 14(10):2058. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14102058
Chicago/Turabian StyleLacombe, Valentin, Guy Lenaers, and Geoffrey Urbanski. 2022. "Diagnostic and Therapeutic Perspectives Associated to Cobalamin-Dependent Metabolism and Transcobalamins’ Synthesis in Solid Cancers" Nutrients 14, no. 10: 2058. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14102058