
Glucose Stimulates Gut Motility in Fasted and Fed Conditions: Potential Involvement of a Nitric Oxide Pathway

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Isotonic Contraction
2.3. Gene Expression
2.4. Longitudinal Muscle and Adherent Myenteric Plexus (LMMP) Dissection and Myenteric Neuronal Cells Culture
2.5. NO Real Time Measurement in Neuronal Cells
2.6. Statistics
3. Results
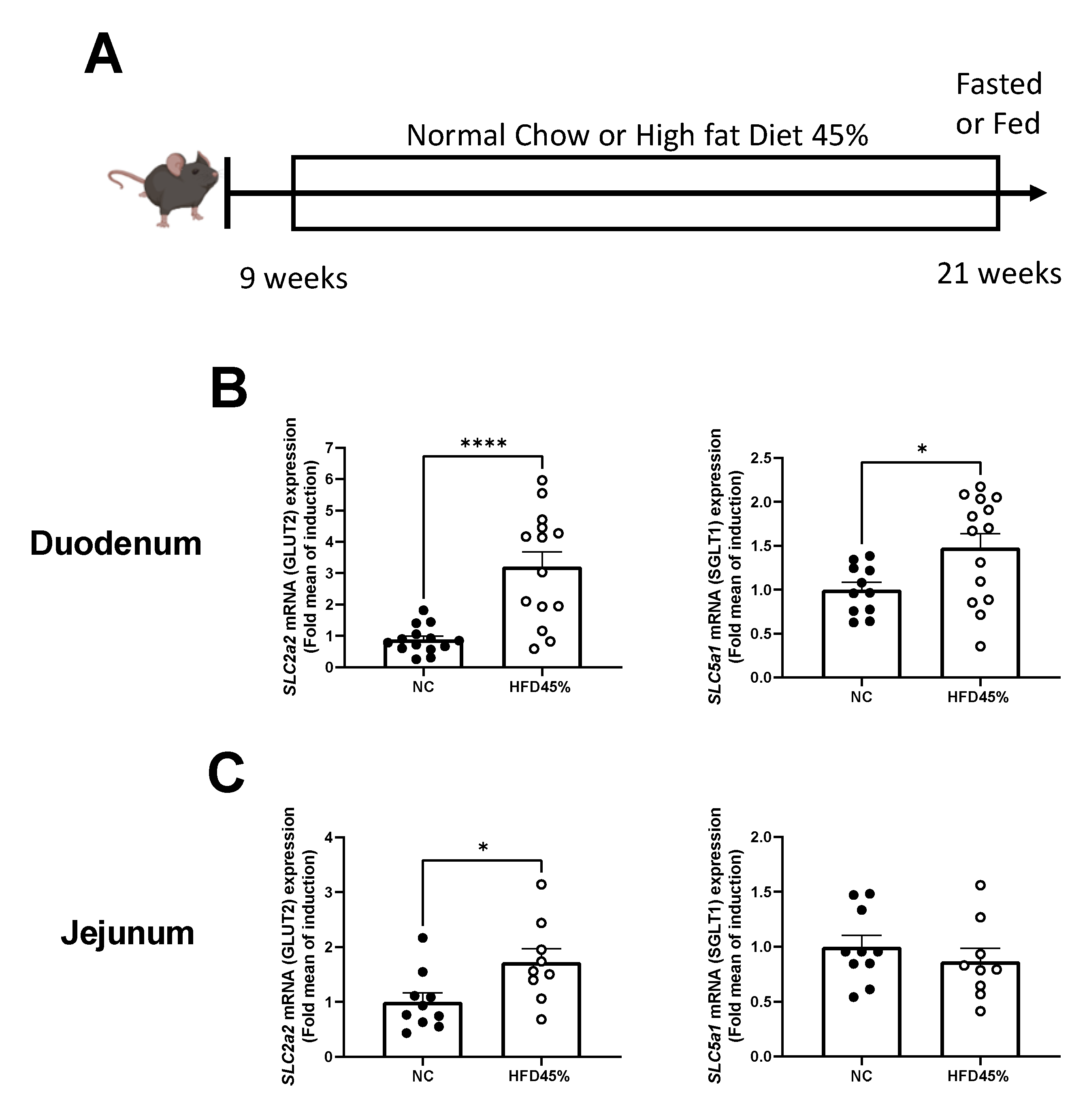
3.1. HFD Increases the Expression of Glucose Transporters in the Intestine
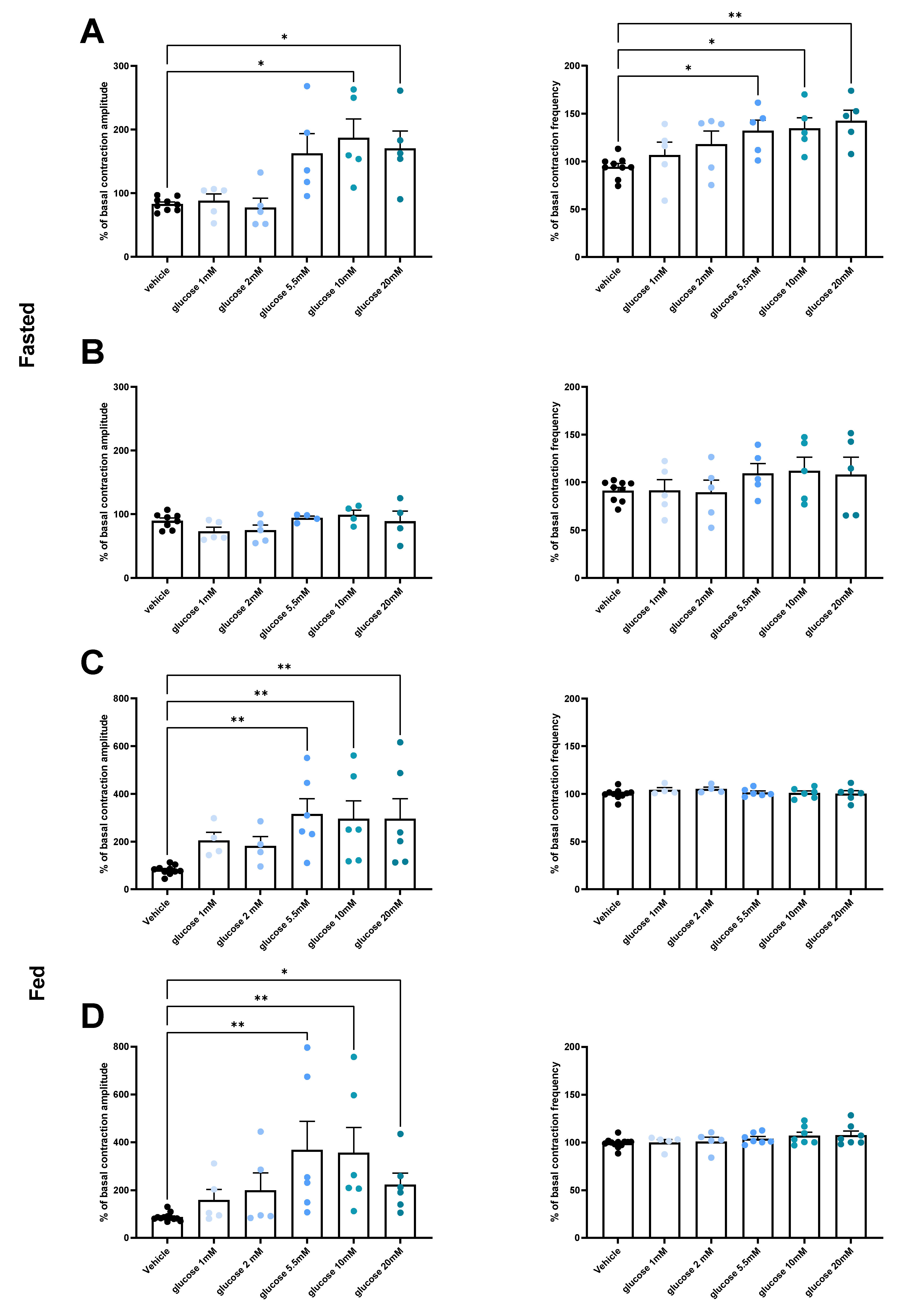
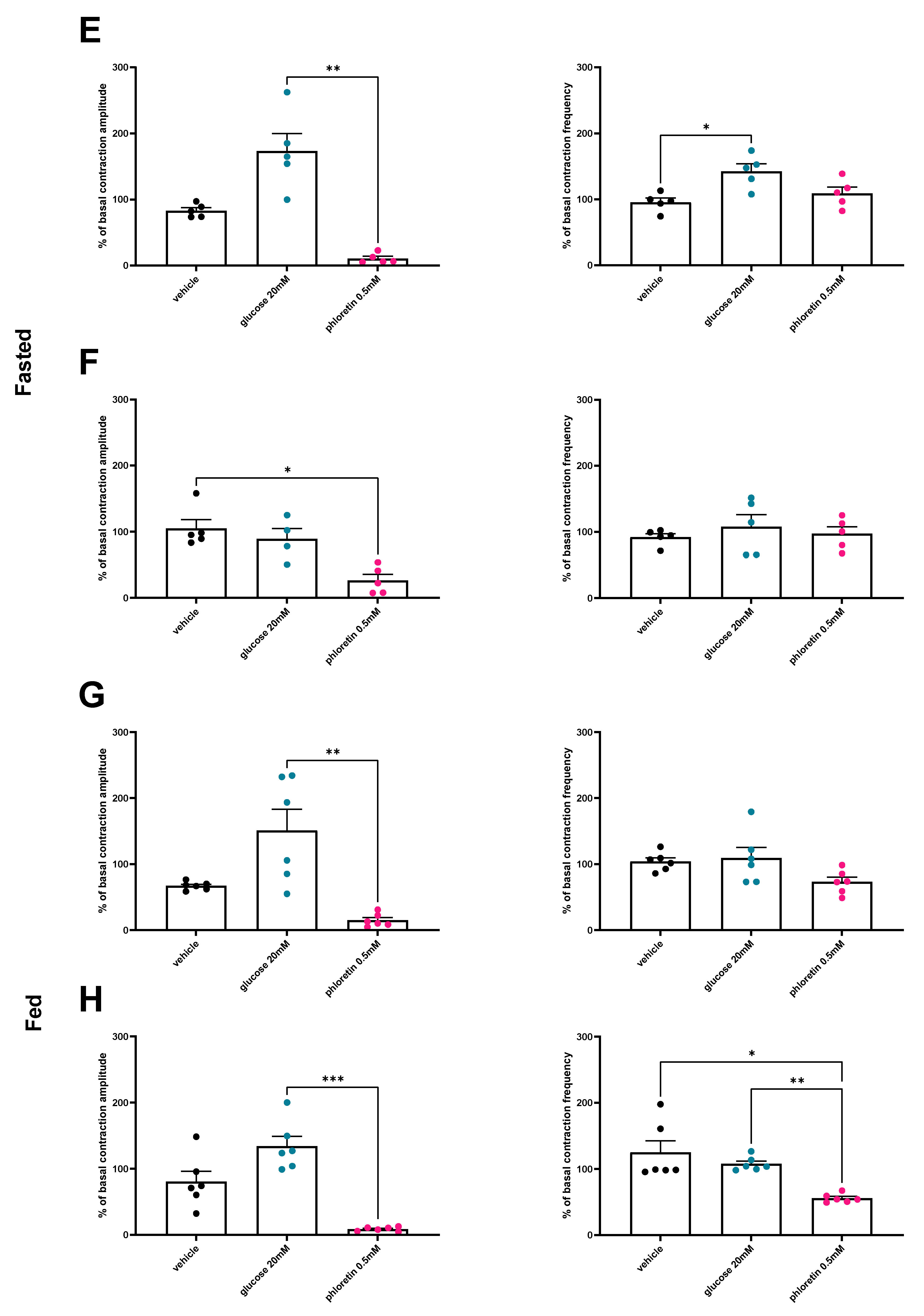
3.2. Under Normal Diet Conditions, Glucose Induces Changes in Duodenal and Jejunal Contractility via a GLUT2-Dependent Mechanism
3.3. Glucose Decreases NO Release from Neuronal Cells of the LMMP In Vitro
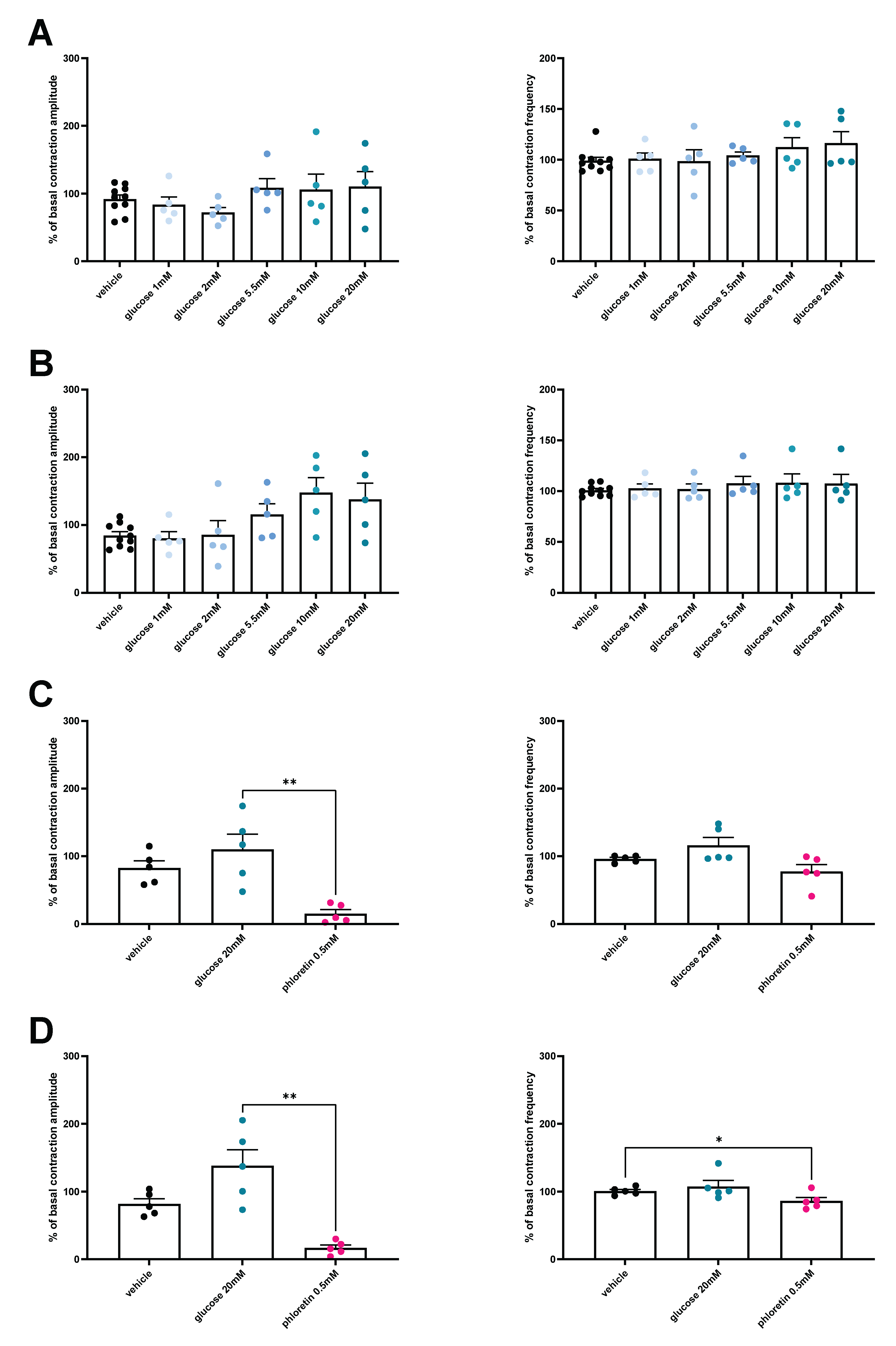
3.4. HFD Is Associated with a Loss of Glucose-Induced Contractility
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richards, P.; Thornberry, N.A.; Pinto, S. The Gut–Brain Axis: Identifying New Therapeutic Approaches for Type 2 Diabetes, Obesity, and Related Disorders. Mol. Metab. 2021, 46, 101175. [Google Scholar] [CrossRef] [PubMed]
- Duparc, T.; Naslain, D.; Colom, A.; Muccioli, G.G.; Massaly, N.; Delzenne, N.M.; Valet, P.; Cani, P.D.; Knauf, C. Jejunum Inflammation in Obese and Diabetic Mice Impairs Enteric Glucose Detection and Modifies Nitric Oxide Release in the Hypothalamus. Antioxid. Redox Signal. 2011, 14, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knauf, C.; Cani, P.D.; Perrin, C.; Iglesias, M.A.; Maury, J.F.; Bernard, E.; Benhamed, F.; Grémeaux, T.; Drucker, D.J.; Kahn, C.R.; et al. Brain Glucagon-like Peptide-1 Increases Insulin Secretion and Muscle Insulin Resistance to Favor Hepatic Glycogen Storage. J. Clin. Investig. 2005, 115, 3554–3563. [Google Scholar] [CrossRef] [PubMed]
- Knauf, C.; Cani, P.D.; Kim, D.H.; Iglesias, M.A.; Chabo, C.; Waget, A.; Colom, A.; Rastrelli, S.; Delzenne, N.M.; Drucker, D.J.; et al. Role of Central Nervous System Glucagon-like Peptide-1 Receptors in Enteric Glucose Sensing. Diabetes 2008, 57, 2603–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, M.B.; Beverly, J.L. Nitric Oxide’s Role in Glucose Homeostasis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R590–R591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittmann, T. Neuropathy in the Gut: Gut Motility Disorders in Patients with Diabetes Mellitus. Orv. Hetil. 2012, 153, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Azpiroz, F.; Malagelada, C. Diabetic Neuropathy in the Gut: Pathogenesis and Diagnosis. Diabetologia 2016, 59, 404–408. [Google Scholar] [CrossRef]
- Abot, A.; Wemelle, E.; Laurens, C.; Paquot, A.; Pomie, N.; Carper, D.; Bessac, A.; Mas Orea, X.; Fremez, C.; Fontanie, M.; et al. Identification of New Enterosynes Using Prebiotics: Roles of Bioactive Lipids and Mu-Opioid Receptor Signalling in Humans and Mice. Gut 2021, 70, 1078–1087. [Google Scholar] [CrossRef]
- Knauf, C.; Abot, A.; Wemelle, E.; Cani, P.D. Targeting the Enteric Nervous System to Treat Metabolic Disorders? “Enterosynes” as Therapeutic Gut Factors. Neuroendocrinology 2020, 110, 139–146. [Google Scholar] [CrossRef]
- Fournel, A.; Drougard, A.; Duparc, T.; Marlin, A.; Brierley, S.M.; Castro, J.; Le-Gonidec, S.; Masri, B.; Colom, A.; Lucas, A.; et al. Apelin Targets Gut Contraction to Control Glucose Metabolism via the Brain. Gut 2017, 66, 258–269. [Google Scholar] [CrossRef] [Green Version]
- de Vos, W.M.; Tilg, H.; van Hul, M.; Cani, P.D. Gut Microbiome and Health: Mechanistic Insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Weber, C. Neurogastroenterology: Improving Glucose Tolerance via the Gut-Brain Axis. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Sababi, M.; Bengtsson, U.H. Enhanced Intestinal Motility Influences Absorption in Anaesthetized Rat. Acta Physiol. Scand. 2001, 172, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Cefalo, C.M.A.; Cinti, F.; Moffa, S.; Impronta, F.; Sorice, G.P.; Mezza, T.; Pontecorvi, A.; Giaccari, A. Sotagliflozin, the First Dual SGLT Inhibitor: Current Outlook and Perspectives. Cardiovasc. Diabetol. 2019, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abot, A.; Lucas, A.; Bautzova, T.; Bessac, A.; Fournel, A.; Le-Gonidec, S.; Valet, P.; Moro, C.; Cani, P.D.; Knauf, C. Galanin Enhances Systemic Glucose Metabolism through Enteric Nitric Oxide Synthase-Expressed Neurons. Mol. Metab. 2018, 10, 100–108. [Google Scholar] [CrossRef]
- Wahba, G.; Hebert, A.E.; Grynspan, D.; Staines, W.; Schock, S. A Rapid and Efficient Method for Dissociated Cultures of Mouse Myenteric Neurons. J. Neurosci. Methods 2016, 261, 110–116. [Google Scholar] [CrossRef]
- Fried, S.; Wemelle, E.; Cani, P.D.; Knauf, C. Interactions between the Microbiota and Enteric Nervous System during Gut-Brain Disorders. Neuropharmacology 2021, 197, 108721. [Google Scholar] [CrossRef]
- Dimidi, E.; Christodoulides, S.; Scott, S.M.; Whelan, K. Mechanisms of Action of Probiotics and the Gastrointestinal Microbiota on Gut Motility and Constipation. Adv. Nutr. 2017, 8, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Pan, J.; Liu, Y.; Wang, H.; Zhou, W.; Wang, X. Intestinal Crosstalk between Microbiota and Serotonin and Its Impact on Gut Motility. Curr. Pharm. Biotechnol. 2018, 19, 190–195. [Google Scholar] [CrossRef]
- Hindson, J. Enteric Neuron Regulation of Gut Motility by the Microbiota. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 194–195. [Google Scholar] [CrossRef]
- Čamborová, P.; Hubka, P.; Šulková, I.; Hulín, I. The Pacemaker Activity of Interstitial Cells of Cajal and Gastric Electrical Activity. Physiol. Res. 2003, 52, 275–284. [Google Scholar] [PubMed]
- Wei, R.; Parsons, S.P.; Huizinga, J.D. Network Properties of Interstitial Cells of Cajal Affect Intestinal Pacemaker Activity and Motor Patterns, According to a Mathematical Model of Weakly Coupled Oscillators. Exp. Physiol. 2017, 102, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Foong, D.; Zhou, J.; Zarrouk, A.; Ho, V.; O’Connor, M.D. Understanding the Biology of Human Interstitial Cells of Cajal in Gastrointestinal Motility. Int. J. Mol. Sci. 2020, 21, 4540. [Google Scholar] [CrossRef] [PubMed]
- Sayegh, A.I.; Covasa, M.; Ritter, R.C. Intestinal Infusions of Oleate and Glucose Activate Distinct Enteric Neurons in the Rat. Auton. Neurosci. Basic Clin. 2004, 115, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.M. Spontaneous Electrical Activity and Rhythmicity in Gastrointestinal Smooth Muscles. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1124, pp. 3–46. [Google Scholar]
- Sharp, P.A.; Boyer, S.; Srai, S.K.S.; Baldwin, S.A.; Debnam, E.S. Early Diabetes-Induced Changes in Rat Jejunal Glucose Transport and the Response to Insulin. J. Endocrinol. 1997, 154, 19–25. [Google Scholar] [CrossRef]
- Omar, A.A.; le Gall, M.; Poitou, C.; Monteiro-Sepulved, M.; Houllier, A.; Château, D.; Veyrie, N.; Hugol, D.; Tordjman, J.; Serradas, P.; et al. Impaired GLUT2 Trafficking in Enterocytes of Human Obese Subjects and Fat-Fed Mice. Genes Nutr. 2010, 5, S64. [Google Scholar]
- Ait-Omar, A.; Monteiro-Sepulveda, M.; Poitou, C.; le Gall, M.; Cotillard, A.; Gilet, J.; Garbin, K.; Houllier, A.; Château, D.; Lacombe, A.; et al. GLUT2 Accumulation in Enterocyte Apical and Intracellular Membranes: A Study in Morbidly Obese Human Subjects and Ob/Ob and High Fat-Fed Mice. Diabetes 2011, 60, 2598–2607. [Google Scholar] [CrossRef] [Green Version]
- Dyer, J.; Wood, I.S.; Palejwala, A.; Ellis, A.; Shirazi-Beechey, S.P. Expression of Monosaccharide Transporters in Intestine of Diabetic Humans. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G241–G248. [Google Scholar] [CrossRef] [Green Version]
- Gough, N.R. Targeting the Duodenum to Control Diabetes. Sci. Signal. 2015, 8, ec134. [Google Scholar] [CrossRef]
- Fournel, A.; Marlin, A.; Abot, A.; Pasquio, C.; Cirillo, C.; Cani, P.D.; Knauf, C. Glucosensing in the Gastrointestinal Tract: Impact on Glucose Metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G645–G658. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Hprt1 | gccagactttgttggatttgaa | gcttgcgaccttgaccatct |
| Slc2a2 (GLUT2) | tggaaggatcaaagcaatgttg | catcaagagggctccagtcaa |
| Slc5a1(SGLT1) | ggcttctccacctgcctcata | tagggcatccaggagatggtgt |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wemelle, E.; Carneiro, L.; Abot, A.; Lesage, J.; Cani, P.D.; Knauf, C. Glucose Stimulates Gut Motility in Fasted and Fed Conditions: Potential Involvement of a Nitric Oxide Pathway. Nutrients 2022, 14, 2176. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14102176
Wemelle E, Carneiro L, Abot A, Lesage J, Cani PD, Knauf C. Glucose Stimulates Gut Motility in Fasted and Fed Conditions: Potential Involvement of a Nitric Oxide Pathway. Nutrients. 2022; 14(10):2176. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14102176
Chicago/Turabian StyleWemelle, Eve, Lionel Carneiro, Anne Abot, Jean Lesage, Patrice D. Cani, and Claude Knauf. 2022. "Glucose Stimulates Gut Motility in Fasted and Fed Conditions: Potential Involvement of a Nitric Oxide Pathway" Nutrients 14, no. 10: 2176. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14102176