Regressive Effect of Myricetin on Hepatic Steatosis in Mice Fed a High-Fat Diet


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Indirect Calorimetric Analysis
2.3. Plasma Biochemical Analysis
2.4. Hepatic Oxidative Stress Biomarker Determination
2.5. Liver Histology
2.6. Hepatic Lipid Content Determination
2.7. Nimblegen Gene Chip Microarray
2.8. Real-Time Quantitative RT-PCR Analyses
2.9. Western Blotting
2.10. Statistical Analysis
3. Results
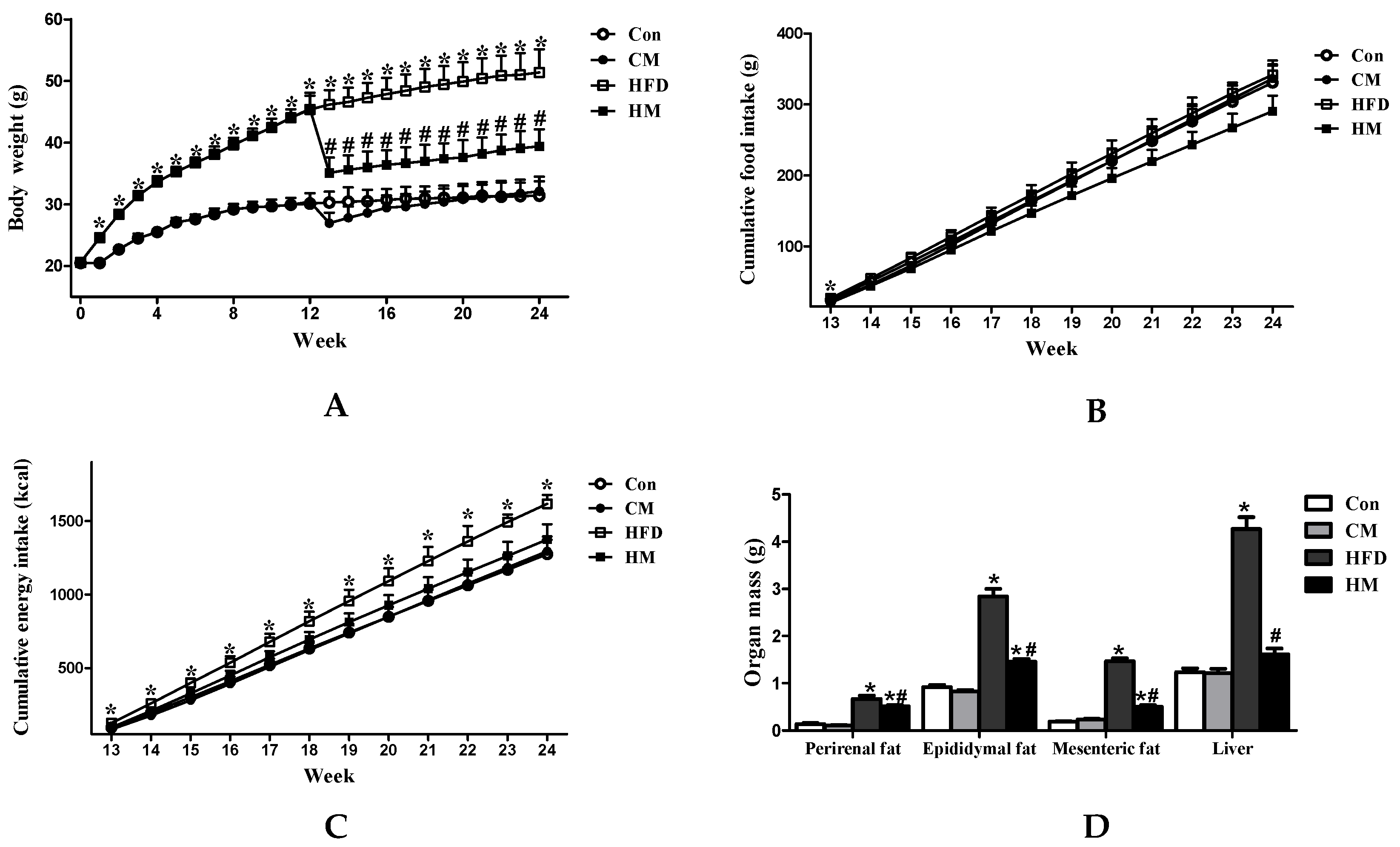
3.1. Effects of Myricetin on Body Weight, Food Intake, and Tissue Weight in HFD-Fed Mice
3.2. Effects of Myricetin on FBG and Plasma Parameters in HFD-Fed Mice
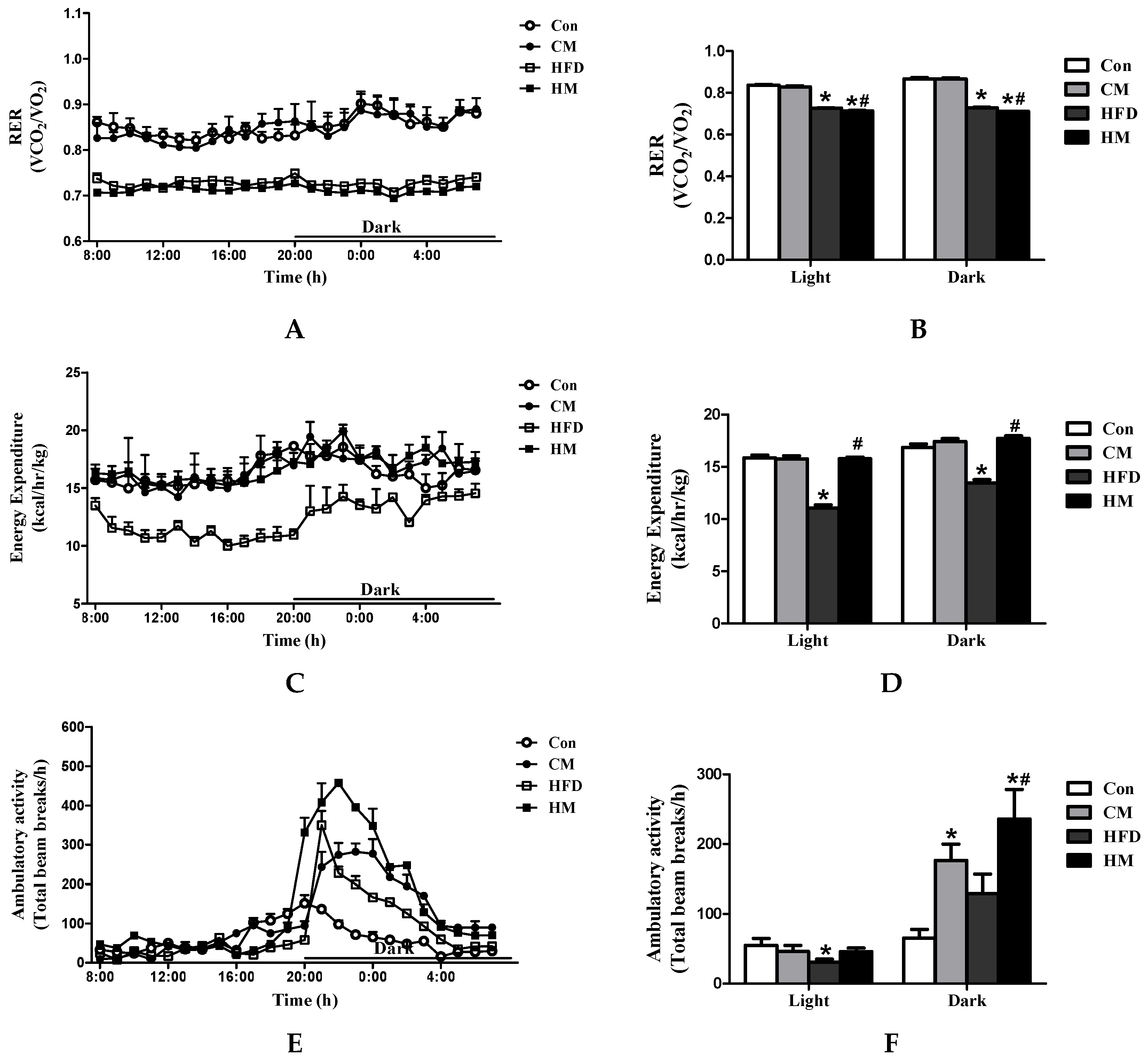
3.3. Effects of Myricetin on RER, Energy Expenditure and Ambulatory Activities in HFD-Fed Mice
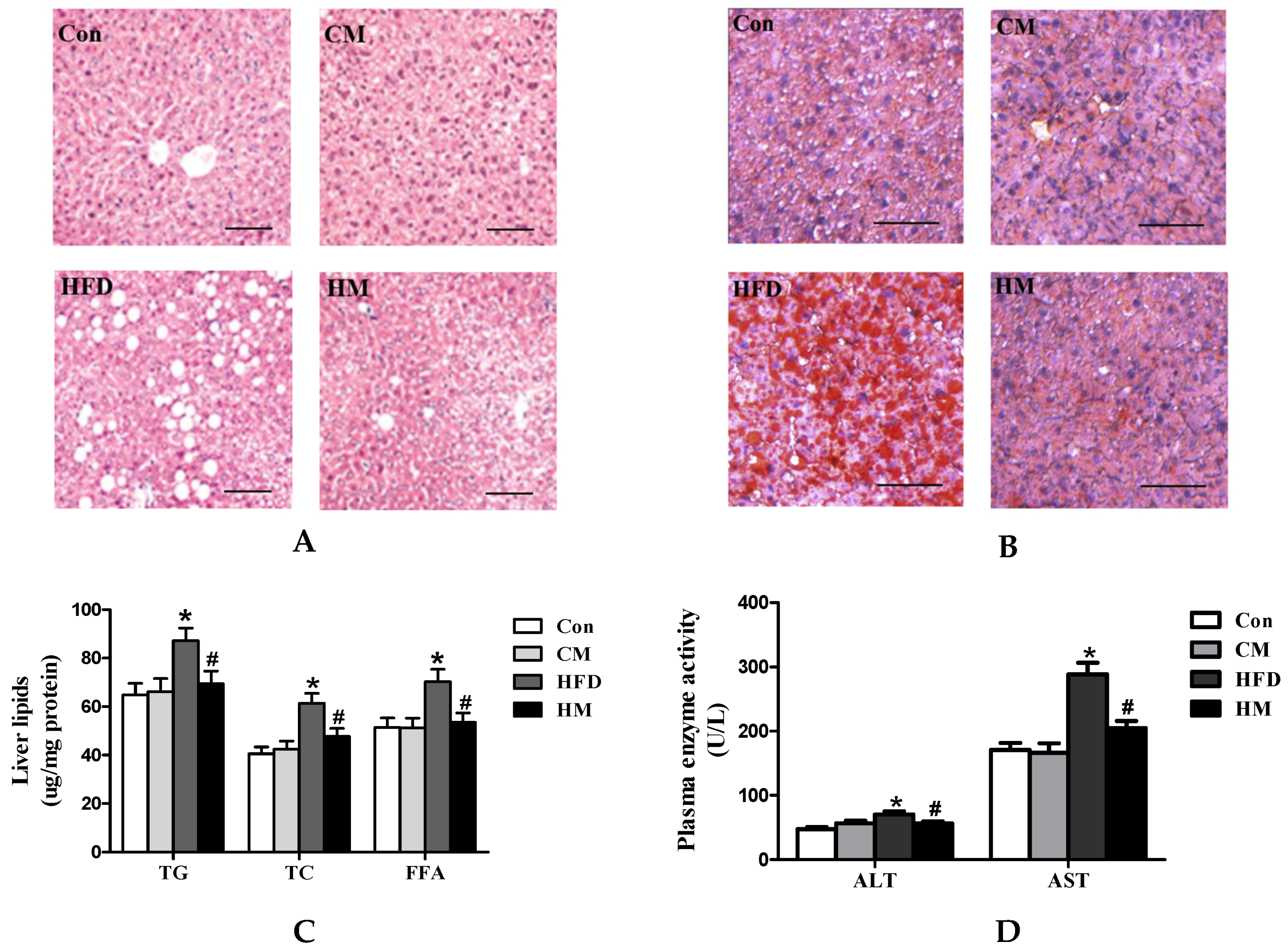
3.4. Effects of Myricetin on Hepatic Steatosis and Liver Function in HFD-Fed Mice
3.5. Effects of Myricetin on Hepatic Biological Pathways in HFD-Fed Mice
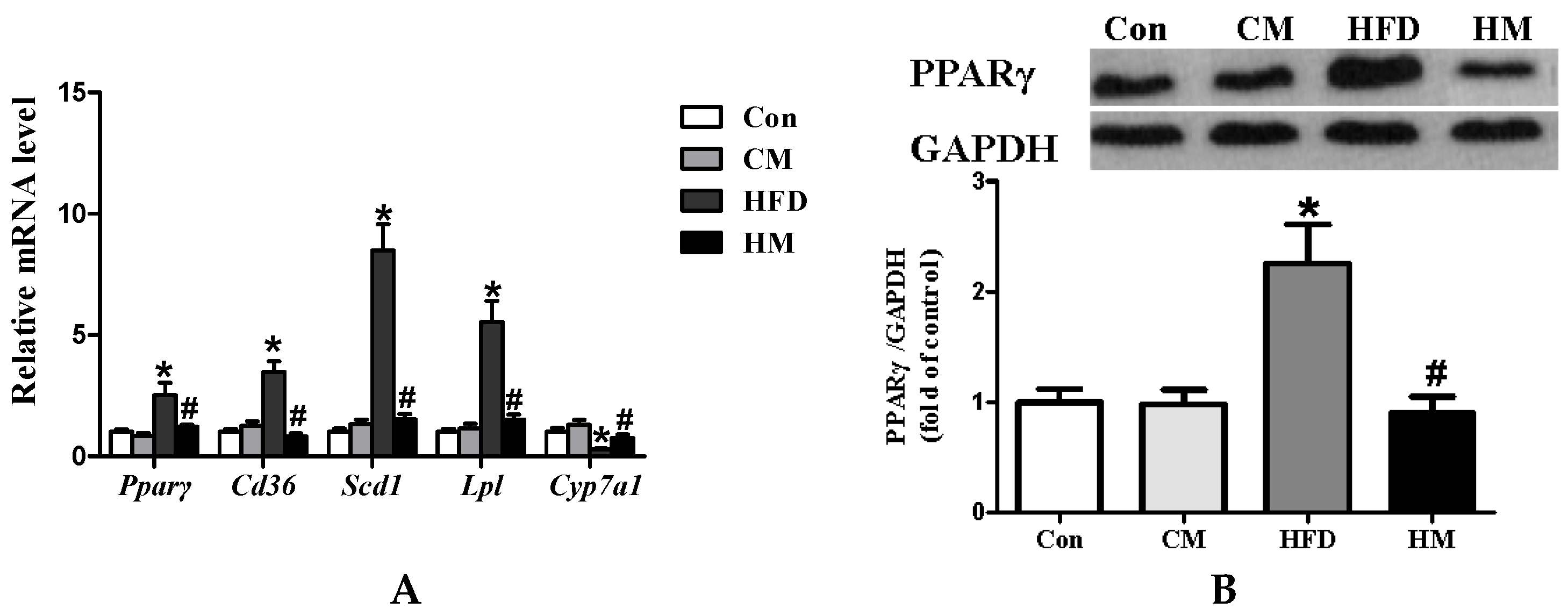
3.6. Effects of Myricetin on Expressions of PPAR Signaling Pathway-Related Genes and PPARγ Protein Expression
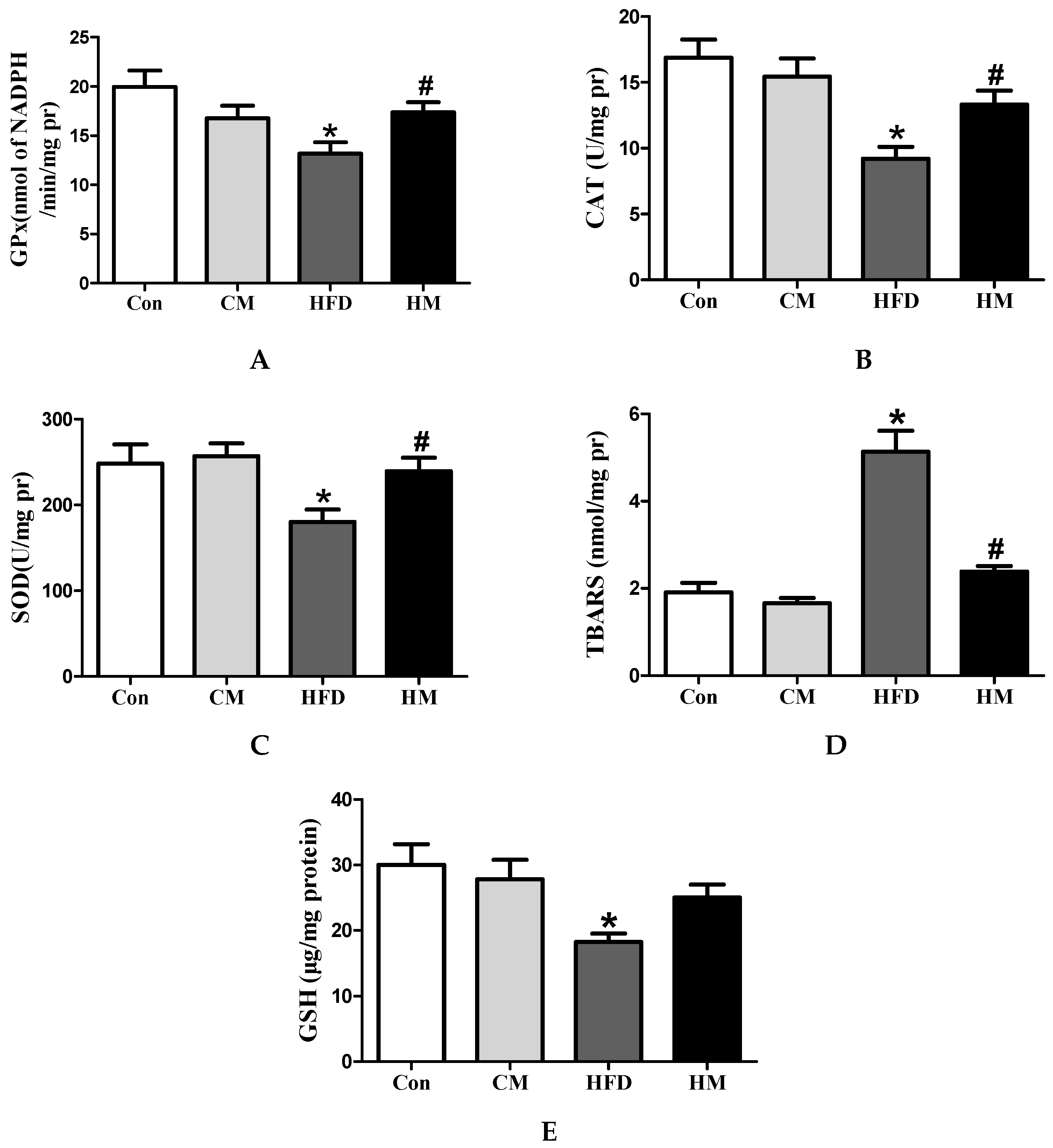
3.7. Effects of Myricetin on Hepatic Redox Status in HFD-Fed Mice
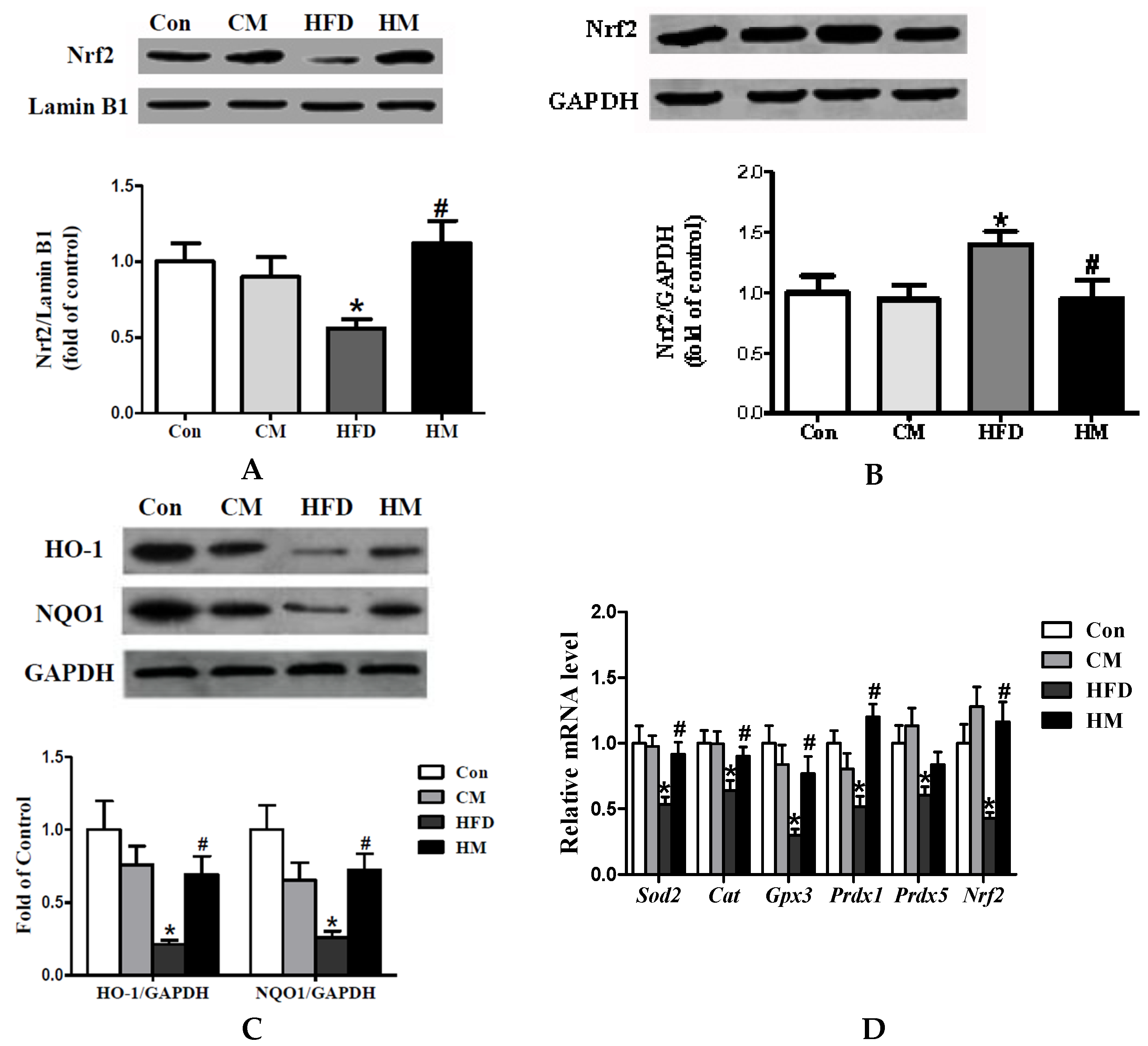
3.8. Effects of Myricetin on Expressions of Oxidative Stress-Related Genes and Nrf2 Pathway
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Friedman, J. Diabetes: Fat in all the wrong places. Nature 2002, 415, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Seppala-Lindroos, A.; Vehkavaara, S.; Hakkinen, A.-M.; Goto, T.; Westerbacka, J.; Sovijarvi, A.; Halavaara, J.; Yki-Jarvinen, H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J. Clin. Endocr. Metab. 2002, 87, 3023–3028. [Google Scholar] [CrossRef] [PubMed]
- Westerbacka, J.; Lammi, K.; Hakkinen, A.-M.; Rissanen, A.; Salminen, I.; Aro, A.; Yki-Jarvinen, H. Dietary fat content modifies liver fat in overweight nondiabetic subjects. J. Clin. Endocr. Metab. 2005, 90, 2804–2809. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Mehta, K.; van Thiel, D.H.; Shah, N.; Mobarhan, S. Nonalcoholic fatty liver disease: Pathogenesis and the role of antioxidants. Nutr. Rev. 2002, 60, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M.; Lee, Y.S.; Shin, D.M.; Lee, S.; Yoo, K.S.; Lee, M.K.; Lee, J.H.; Kim, S.Y.; Lee, Y.M.; Hong, J.T.; et al. Green tomato extract attenuates high-fat-diet-induced obesity through activation of the AMPK pathway in C57BL/6 mice. J. Nutr. Biochem. 2013, 24, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Ganji, S.H.; Kukes, G.D.; Lambrecht, N.; Kashyap, M.L.; Kamanna, V.S. Therapeutic role of niacin in the prevention and regression of hepatic steatosis in rat model of nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G320–G327. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A dietary molecule with diverse biological activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Tzeng, T.-F.; Liou, S.-S.; Chang, Y.-S.; Liu, I.-M. Myricetin increases hepatic peroxisome proliferator-activated receptor α protein expression and decreases plasma lipids and adiposity in rats. Evid.-Based Complement. Altern. Med. 2012, 2012, 787152. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-N.; Kang, M.-J.; Lee, S.-J.; Kim, J.-I. Ameliorative effect of myricetin on insulin resistance in mice fed a high-fat, high-sucrose diet. Nutr. Res. Pract. 2014, 8, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Meng, Y.; Zhao, Y.; Hu, Y.; Ren, D.; Yang, X. Myricetin derived from Hovenia dulcis Thunb. Ameliorates vascular endothelial dysfunction and liver injury in high choline-fed mice. Food Funct. 2015, 6, 1620–1634. [Google Scholar] [CrossRef] [PubMed]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Khatri, P.; Sellamuthu, S.; Malhotra, P.; Amin, K.; Done, A.; Draghici, S. Recent additions and improvements to the Onto-Tools. Nucleic Acids Res. 2005, 33, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Khatri, P.; Voichita, C.; Kattan, K.; Ansari, N.; Khatri, A.; Georgescu, C.; Tarca, A.L.; Draghici, S. Onto-Tools: New additions and improvements in 2006. Nucleic Acids Res. 2007, 35, W206–W211. [Google Scholar] [CrossRef] [PubMed]
- Lemberger, T.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: A nuclear receptor signaling pathway in lipid physiology. Annu. Rev. Cell. Dev. Biol. 1996, 12, 335–363. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.J.; Li, Y.-Y.; Nie, Y.Q.; Ma, J.-X.; Lu, L.-G.; Shi, S.-L.; Chen, M.-H.; Hu, P.-J. Prevalence of fatty liver disease and its risk factors in the population of south China. World. J. Gastroenterol. 2007, 13, 6419–6424. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.G.; Gordon, F.D.; Chopra, S. Nonalcoholic steatohepatitis. Ann. Intern. Med. 1997, 126, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Deeg, M.A.; Crabb, D.W. Systemic levels of lipid peroxidation and its metabolic and dietary correlates in patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2004, 99, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Tortosa, M.C.; Ramirez-Tortosa, C.L.; Mesa, M.D.; Granados, S.; Gil, A.; Quiles, J.L. Curcumin ameliorates rabbits’s steatohepatitis via respiratory chain, oxidative stress, and TNF-α. Free Radic. Biol. Med. 2009, 47, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K.; Takayama, F.; Mankura, M.; Hidaka, Y.; Egashira, T.; Ogino, T.; Kawasaki, H.; Mori, A. Beneficial effects of fermented green tea extract in a rat model of non-alcoholic steatohepatitis. J. Clin. Biochem. Nutr. 2009, 44, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Chen, J.; Tanigawa, S.; Hou, D.X. Microarray and pathway analysis highlight Nrf2/ARE-mediated expression profiling by polyphenolic myricetin. Mol. Nutr. Food Res. 2013, 57, 435–446. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, R.A.; Markiewski, M.M.; Taub, R.; Lambris, J.D. A high-fat diet impairs liver regeneration in C57BL/6 mice through overexpression of the NF-κB inhibitor, IKBα. Hepatology 2005, 42, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Ohtake, T.; Motomura, W.; Takahashi, N.; Hosoki, Y.; Miyoshi, S.; Suzuki, Y.; Saito, H.; Kohgo, Y.; Okumura, T. Increased expression of PPARγ in high fat diet-induced liver steatosis in mice. Biochem. Biophys. Res. Commun. 2005, 336, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.-H.; Gavrilova, O.; Ward, J.M.; Brewer, B., Jr.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of PPARγ in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Investig. 2003, 111, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor γ contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [PubMed]
- Pardina, E.; Baena-Fustegueras, J.A.; Llamas, R.; Catalan, R.; Galard, R.; Lecube, A.; Fort, J.M.; Llobera, M.; Allende, H.; Vargas, V. Lipoprotein lipase expression in livers of morbidly obese patients could be responsible for liver steatosis. Obes. Surg. 2009, 19, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.K.; Qian, K.; Ogimoto, K.; Morton, G.J.; Wisse, B.E.; Agrawal, N.; McDonald, T.O.; Schwartz, M.W.; Dichek, H.L. Mice lacking hepatic lipase are lean and protected against diet-induced obesity and hepatic steatosis. Endocrinology 2010, 151, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARγ in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. BBA-Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M. Protective mechanisms against reactive oxygen species. In The Liver: Biology and Pathobiology, 4th ed.; Arias, I.M., Boyer, J.L., Chisari, F.V., Fausto, N., Schachter, D., Shafritz, D.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1994; pp. 443–459. [Google Scholar]
- Tang, W.; Jiang, Y.F.; Ponnusamy, M.; Diallo, M. Role of Nrf2 in chronic liver disease. World. J. Gastroenterol. 2014, 20, 13079–13087. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, W.; Liu, Y.; Sun, Y.; Li, Y.; Yao, Q.; Li, J.; Zhang, Q.; Gao, Y.; Gao, L. Alpha-lipoic acid improves high-fat diet-induced hepatic steatosis by modulating the transcription factors SREBP-1, FoxO1 and Nrf2 via the SIRT1/LKB1/AMPK pathway. J. Nutr. Biochem. 2014, 25, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Ziros, P.G.; Psyrogiannis, A.I.; Papavassiliou, A.G.; Kyriazopoulou, V.E.; Sykiotis, G.P.; Habeos, I.G. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes 2011, 60, 2465–2473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, Z.; Wu, S.; Li, X.; Sang, Y.; Li, J.; Niu, Y.; Ding, H. Myricetin alleviates cuprizone-induced behavioral dysfunction and demyelination in mice by Nrf2 pathway. Food Funct. 2016, 7, 4332–4342. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Hanselmann, C.; Gassmann, M.G.; Keller, U.; Born-Berclaz, C.; Chan, K.; Kan, Y.W.; Werner, S. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol. Cell. Biol. 2002, 22, 5492–5505. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Polvani, S.; Tarocchi, M.; Galli, A. PPARγ and Oxidative Stress: Con (β) Catenating NRF2 and FOXO. PPAR Res. 2012, 2012, 641087. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Leung, L.; Xue, P.; Wang, W.; Hou, Y.; Liu, D.; Yehuda-Shnaidman, E.; Lee, C.; Lau, J.; Kurtz, T.W. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J. Biol. Chem. 2010, 285, 9292–9300. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Con | CM | HFD | HM | |
|---|---|---|---|---|
| Fasting blood glucose (mg/dL) | 113.96 ± 11.51 | 117.24 ± 12.23 | 166.19 ± 15.79 * | 123.35 ± 12.55 # |
| Plasma insulin (μIU/mL) | 14.47 ± 0.75 | 15.28 ± 1.93 | 20.63 ± 2.06 * | 14.94 ± 1.46 # |
| HOMA-IR | 4.05 ± 0.45 | 4.47 ± 0.87 | 8.67 ± 1.39 * | 4.61 ± 0.60 # |
| Plasma TG (mmol/L) | 2.53 ± 0.15 | 2.54 ± 0.16 | 3.36 ± 0.17 * | 2.71 ± 0.15 # |
| Plasma TC (mmol/L) | 4.09 ± 0.24 | 4.01 ± 0.27 | 6.57 ± 0.37 * | 4.35 ± 0.38 # |
| Plasma HDL-C (mmol/L) | 1.81 ± 0.08 | 1.92 ± 0.08 | 1.41 ± 0.05 * | 1.69 ± 0.08 # |
| Plasma LDL-C (mmol/L) | 2.08 ± 0.12 | 2.05 ± 0.09 | 2.81 ± 0.10 * | 2.30 ± 0.07 # |
| Number | Pathway Name | Input Genes in Pathway | Impact Factor | Corrected Gamma p-Value | Significantly Altered Genes |
|---|---|---|---|---|---|
| 1 | PPAR signaling pathway | 6 | 16.26 | 1.90 × 10−5 | Cd36, Scd1, Cyp7a1, Lpl, Pparg, Pck1 |
| 2 | Biosynthesis of unsaturated fatty acids | 3 | 10.26 | 0.00185 | Acot1, Acot3, Scd1 |
| 3 | Peroxisome | 3 | 10.01 | 0.001974 | Sod2, Prdx5, Prdx1 |
| 4 | Complement and coagulation cascades | 3 | 9.62 | 0.002971 | C4b, Plg, Serpina1a |
| 5 | Renal cell carcinoma | 3 | 8.26 | 0.008088 | Arnt, Jun, Raf1 |
| 6 | Long-term potentiation | 4 | 7.02 | 0.01977 | Araf, pla2g2d, Plcb1, Raf1 |
| 7 | GnRH signaling pathway | 3 | 6.44 | 0.029597 | Jun, Pla2g6, Raf1 |
| 8 | Toll-like receptor signaling pathway | 3 | 5.92 | 0.032924 | Cxcl10, Jun, Spp1 |
| 9 | Insulin signaling pathway | 3 | 5.64 | 0.041843 | Pck1, Raf1, Srebf1 |
| 10 | MAPK signaling pathway | 3 | 5.60 | 0.043097 | Jun, Ntrk2, Pla2g6 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, S.-F.; Le, G.-W.; Wang, P.; Qiu, Y.-Y.; Jiang, Y.-Y.; Tang, X. Regressive Effect of Myricetin on Hepatic Steatosis in Mice Fed a High-Fat Diet. Nutrients 2016, 8, 799. https://0-doi-org.brum.beds.ac.uk/10.3390/nu8120799
Xia S-F, Le G-W, Wang P, Qiu Y-Y, Jiang Y-Y, Tang X. Regressive Effect of Myricetin on Hepatic Steatosis in Mice Fed a High-Fat Diet. Nutrients. 2016; 8(12):799. https://0-doi-org.brum.beds.ac.uk/10.3390/nu8120799
Chicago/Turabian StyleXia, Shu-Fang, Guo-Wei Le, Peng Wang, Yu-Yu Qiu, Yu-Yu Jiang, and Xue Tang. 2016. "Regressive Effect of Myricetin on Hepatic Steatosis in Mice Fed a High-Fat Diet" Nutrients 8, no. 12: 799. https://0-doi-org.brum.beds.ac.uk/10.3390/nu8120799