Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review


1
Department of Chemistry “Giacomo Ciamician”, Alma Mater Studiorum, University of Bologna, 40126 Bologna, Italy
2
Istituto Nazionale Biostrutture e Biosistemi (INBB), 00136 Rome, Italy
3
Centro Interdipartimentale di Ricerca Industriale Energia e Ambiente (CIRI EA), Alma Mater Studiorum, University of Bologna, 47900 Rimini, Italy
4
Department of Medical and Surgical Sciences, Alma Mater Studiorum, University of Bologna, 40138 Bologna, Italy
*
Author to whom correspondence should be addressed.
Nutrients 2017, 9(4), 395; https://0-doi-org.brum.beds.ac.uk/10.3390/nu9040395
Submission received: 26 February 2017
/
Revised: 7 April 2017
/
Accepted: 10 April 2017
/
Published: 18 April 2017
(This article belongs to the Special Issue Dietary Fructose and Glucose: The Multifacetted Aspects of their Metabolism and Implication for Human Health)
{kind=link}
{kind=link}
Abstract
:There is a direct relationship between fructose intake and serum levels of uric acid (UA), which is the final product of purine metabolism. Recent preclinical and clinical evidence suggests that chronic hyperuricemia is an independent risk factor for hypertension, metabolic syndrome, and cardiovascular disease. It is probably also an independent risk factor for chronic kidney disease, Type 2 diabetes, and cognitive decline. These relationships have been observed for high serum UA levels (>5.5 mg/dL in women and >6 mg/dL in men), but also for normal to high serum UA levels (5–6 mg/dL). In this regard, blood UA levels are much higher in industrialized countries than in the rest of the world. Xanthine-oxidase inhibitors can reduce UA and seem to minimize its negative effects on vascular health. Other dietary and pathophysiological factors are also related to UA production. However, the role of fructose-derived UA in the pathogenesis of cardiometabolic disorders has not yet been fully clarified. Here, we critically review recent research on the biochemistry of UA production, the relationship between fructose intake and UA production, and how this relationship is linked to cardiometabolic disorders.
1. Introduction
Uric acid (UA) is the final product of purine metabolism. It is a well-known risk factor for gout [1]. Moreover, a growing body of evidence suggests that high levels of serum UA are also biomarkers for cardiovascular disease (CVD) morbidity and mortality [2].
The increased incidence of gout among rich people in the 18th and 19th centuries was principally due to high consumption of purine-containing meat. However, UA levels are rising in the 21st century too, with mean levels of >5.5 mg/dL in women and >6.0 mg/dL in men [3]. This can partly be explained by a remarkable increase in added sugars in the Western diet, especially fructose [4,5,6]. Accordingly, blood UA levels are higher in Western countries than in the rest of the world. In non-Western countries, hyperuricemia is relatively rare in rural communities. However, there is increased migration from rural areas to cities or communities where the Western diet is dominant and hyperuricemia is more prevalent [7].
Recent research suggests that hyperuricemia may be caused by elevated activity of the enzyme xanthine oxidase (XO) [8]. Xanthine oxidase inhibitors (XOI) have thus been proposed as a strategy for reducing UA and oxidative stress. Both are risk factors for gout, chronic kidney disease (CKD), CVD, obesity, insulin resistance, and metabolic syndrome. Humans and great apes produce UA via XO-catalyzed oxidation of purines. Unlike other mammals, humans and great apes cannot synthesize the uricase enzyme (urate oxidase) and so cannot metabolize UA to allantoin. As a result, UA blood concentrations in humans and great apes are at least 10 times higher than in other mammals, with the consequent risk of developing hyperuricemia [9].
High UA levels favor adipose tissue formation, which was originally an evolutionary advantage for humans [7]. Nowadays, however, excess adipose tissue is considered a predisposing factor for insulin resistance, obesity, and hypertension [10]. This excessive fat storage may be due to increased consumption of fructose-enriched food and drink, which raises serum UA levels [11]. Indeed, several clinical studies have shown that the administration of allopurinol, a competitive antagonist of XO, can significantly improve endothelial function and the circulating markers of oxidative stress in patients with, or at risk of, CVD [12].
Here, we review the most relevant discoveries in the field, focusing on (i) the role of UA in cardiometabolic disorders; and (ii) the link between fructose consumption, high blood UA levels, and associated disorders, particularly CVD.
2. Search Strategy (Methods)
We conducted a literature search of different scientific databases (including Scopus, Google Scholar, PubMed, and Web of Science) for peer-reviewed studies focusing on XO, hyperuricemia, fructose, and CVD. The search strategy was designed to retrieve studies published in English from journal inception to 2016. We used an assessment framework to appraise the quality of basic research studies, prognostic studies, and methodological considerations in the analysis and publication of observational studies. The screening, study selection, and data extraction was undertaken by three independent authors. Disagreement was resolved by discussion and, if required, by a fourth independent author. We have assessed the clinical and methodological heterogeneity across the studies and, where available, included meta-analyses whenever these have been performed.
3. Purine Metabolism and Uric Acid Physiology
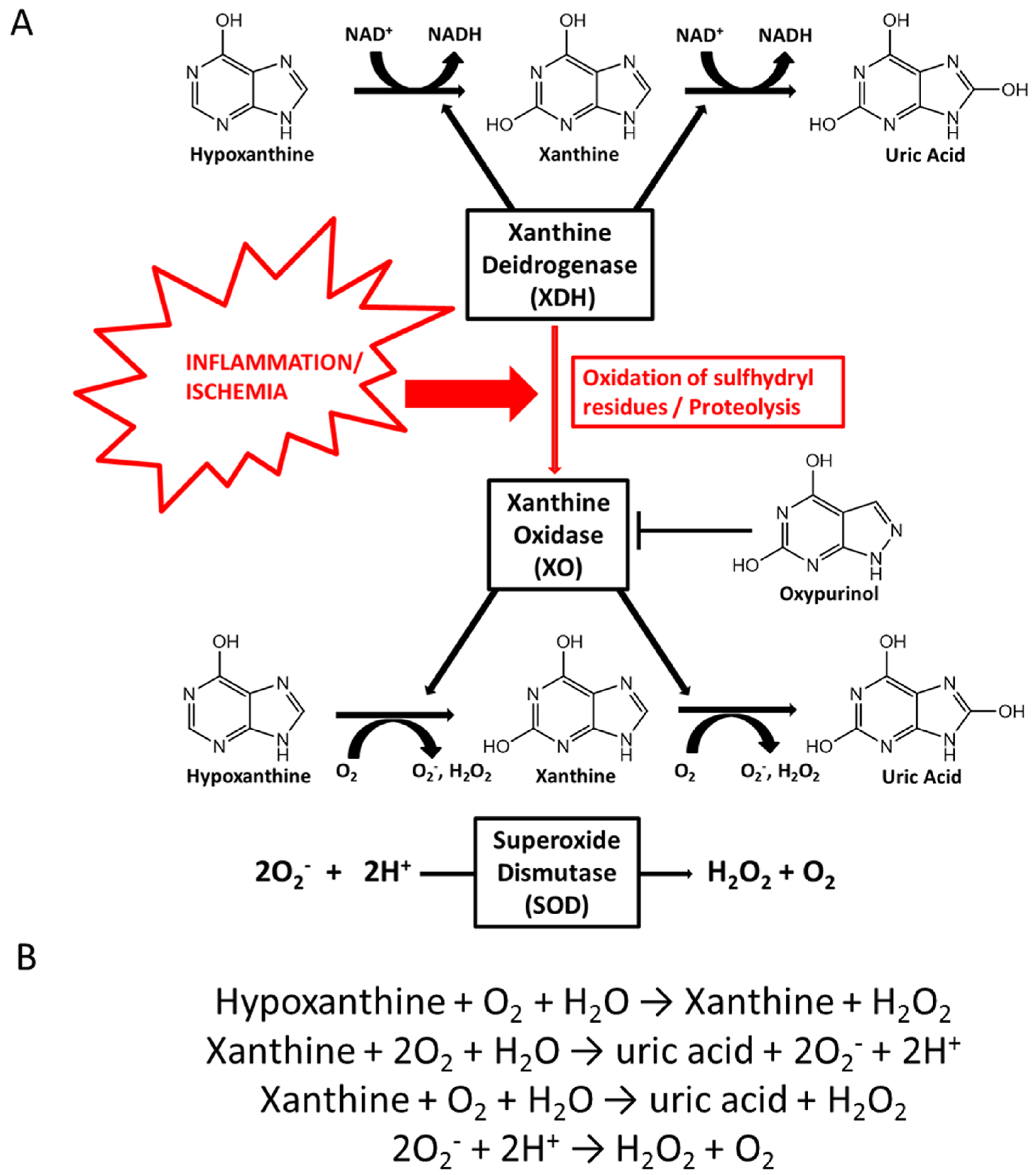
Purines are generated through two pathways. First, there is de novo synthesis from non-purine compounds, such as amino acids and bicarbonate, regulated by phosphoribosyl-pyrophosphate synthetase (PRPP). Second, there is the purine salvage pathway, which economizes the intracellular energy expenditure and is regulated by hypoxanthine-guanine phosphoribosyltransferase (HG-PRTase) [13]. Catabolism of purines is regulated by xanthine-oxidoreductase (XOR), coding for two distinct enzymatic forms: xanthine dehydrogenase (XDH) and XO [14]. XDH and XO catalyze the oxidation of hypoxanthine to xanthine and subsequently to UA, which is the hepatic and intestinal metabolite of purine [15,16]. The main difference between XDH and XO is that XDH-FAD reacts predominantly with NAD+, whereas XO-FAD has higher reactivity for molecular oxygen, producing higher levels of hydrogen peroxide (H2O2) and the superoxide anion (O2−). O2− is then converted into oxygen and H2O2, either spontaneously or catalyzed by the superoxide dismutase (SOD) enzyme, (Figure 1A,B) [14]. XO is the form that is most abundant in inflamed vascular and ischemic tissues. XDH can be converted into XO via the oxidation of sulfhydryl residues or the proteolysis of XDH (Figure 1A) [17]. Allopurinol and its active metabolites oxypurinol act as competitive antagonists of XO and can lower UA levels. In addition, allopurinol has consistently been reported to prevent H2O2 production [12,18] (Figure 1B).
Most UA is filtered by the kidneys and eliminated in urine. The rest passes through the gut and is cleaved by bacteria into waste substances, which are eliminated in faeces. Inefficient renal excretion of UA is the main cause of both primary and secondary hyperuricemia [19]. Renal UA excretion is regulated by several transporters. Renal UA reabsorption is mediated by urate transporter 1 (URAT1, also known as SLC22A12) and glucose transporter 9 (GLUT9, also known as SLC2A9) [20,21]. Renal UA secretion is mediated by sodium phosphate transporter (NPT1, also known as SLC17A1) [22,23]. UA concentration can be decreased by several drugs (benzbromarone, losartan, probenecid, sulfinpyrazon). These drugs mostly work by URAT1 inhibition, which explains their uricosuric effect [24]. UA excretion is also regulated by breast cancer resistance protein (BCRP, also known as ABCG2), which belongs to the superfamily of ATP-binding cassette (ABC) transporters [25,26,27]. Unlike ABC transporters, however, BCRP has only one N-terminal ATP-binding domain [28,29,30]. Reduced intestinal excretion of UA is often associated with polymorphisms of the BCRP gene [26] or with a lack of BCRP dimerization, which is due to oxidative stress [31,32].
As noted above, most mammals can degrade UA to allantoin in a reaction catalyzed by uricase, an enzyme present in peroxisomes. Allantoin is subsequently degraded to urea for excretion [33,34]. Most mammals thus have relatively low UA circulation levels (0.5 to 2.0 mg/dL). However, humans and great apes cannot synthesize functional uricase and therefore have much higher blood UA levels.
According to Neel’s hypothesis, our closest evolutionary ancestors underwent functional genetic mutations, which silenced genes involved in the degradation of UA and the synthesis of vitamin C [10], which is the major water-soluble intracellular free-radical scavenger in human plasma [35]. These mutations increased de novo lipogenesis and weight gain [36]. This is because vitamin C competes with UA for renal resorption in the proximal tubule [37,38,39], producing a uricosuric effect. UA causes endoplasmic reticulum stress; this, in turn, activates SREBP-1c, which stimulates fat accumulation in the liver [40]. Indeed, Cheung et al. recently reported that XOR-knockout mice have a central defect in adipogenesis and fail to gain fat [41,42,43]. Thus, there is a positive correlation between circulating UA and obesity, especially visceral obesity [44]. Accordingly, although hyperuricemia is often considered to be a secondary phenomenon in metabolic syndrome, it is also an independent predictive factor for obesity and hyperinsulinemia [4]. In themselves, UA accumulation and lack of vitamin C do not cause obesity. Rather, they increase susceptibility to obesity and diabetes as a result of an interaction between genetic factors (mostly a polygenic contribution) and environmental factors such as lifestyle, social influences, and fetal surroundings [11,45].
4. Fructose Metabolism and the Mechanisms by Which Fructose May Contribute to Uric Acid Production
In the past 100 years, there has been a progressive rise in blood UA levels, especially in Western countries. This is associated, at least in part, with a rise in the number of people consuming a Western diet. In particular, there has been increased consumption of fructose-containing sugars [46,47], sucrose, high-fructose corn syrup, soft drinks, and fruit juices [11,48].
Fructose is present as a monosaccharide in fruits and vegetables [49], as a disaccharide in sucrose (with d-glucose), and as oligo- and polysaccharides (fructans) in many plants. It is also used as an added sweetener for food and drink, and as an excipient in pharmaceutical preparations, syrups, and solutions [50]. Following fructose ingestion, blood glucose levels are lower (GI 32) than after ingesting a similar amount of glucose (GI 100) or sucrose (GI 68). Similarly, insulin levels do not increase significantly after fructose ingestion. Moreover, fructose has a greater sweetening power than sucrose, so smaller quantities of fructose can be used to sweeten foods. Finally, its calorific value is 3.75 kcal/g, slightly lower than that of sucrose (3.92 kcal/g) [51]. It is not yet clear if a moderate intake of fructose significantly promotes the formation of advanced glycation products (AGE), which damage tissues and thus contribute to ageing and metabolic disorders [52]. Several studies suggest that a fructose-rich diet has negative metabolic consequences, including AGE formation [52,53,54]. However, the effect of a long-term fructose intake on AGE accumulation in tissues has not yet been studied in healthy volunteers.
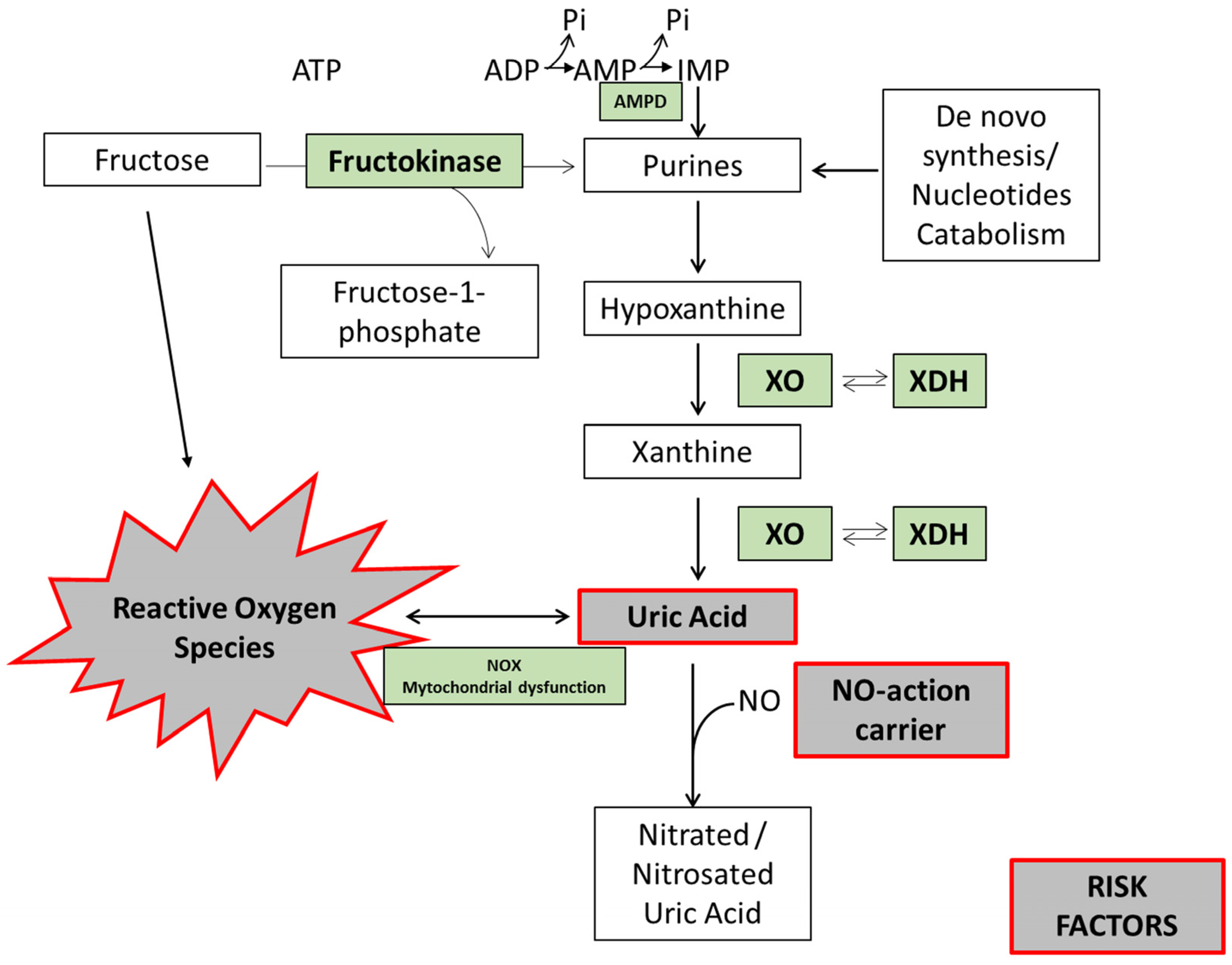
Hyperuricemia is caused by the overproduction and/or underexcretion of UA. It has been reported that metabolism of fructose stimulates UA production, since transient ATP depletion commonly occurs with the generation of AMP [55] and reduced UA excretion [56]. Briefly, during fructose metabolism, fructose is phosphorylated into fructose 1-phosphate in a reaction catalyzed by fructokinase primarily in the liver. This reaction is rapid, has no negative feedback, and hugely decreases the levels of intracellular phosphate and ATP [57]. Next, the enzyme fructose-1-p aldolase (also known as aldolase B) breaks fructose 1-phosphate into dihydroxyacetone phosphate (DHAP) and d-glyceraldehyde. When there is a high intake of fructose, phosphorylation into fructose 1-phosphate is fast, but the reaction with aldolase is slow (Figure 2). Hence, fructose 1-phosphate accumulates, and intracellular phosphate decreases. This decrease stimulates AMP deaminase (AMPD), which catalyzes the degradation of AMP to inosine monophosphate, increasing the rate of purine degradation [58] (Figure 2). The purine degradation produces UA [59] and generates mitochondrial oxidants [36] (Figure 2). Mitochondrial oxidative stress then induces aconitase inhibition in the Krebs cycle, with accumulation of citrate and stimulation of ATP citrate lyase and fatty acid synthase. The result is de novo lipogenesis and hepatic fat accumulation [36].
Physiologically, the increase in intracellular UA is followed by an acute rise in circulating levels of UA, which is likely due to its release from the liver [57,60]. Fructose also stimulates UA synthesis from amino acid precursors such as glycine [61]. Moreover, long-term fructose administration suppresses renal excretion of UA, resulting in elevated serum UA levels [19]. Kaneko and colleagues found that a single administration of fructose affects the excretion of UA to the intestinal lumen, inducing the suppression of BCRP dimerization by reactive oxygen species (ROS)-derived production of dinucleotide phosphate (NADPH) oxidase (NOX) [56].
5. Mechanisms by Which Fructose-Uric Acid May Contribute to Cardiometabolic Disorders
UA has been described as a “paradox molecule” with opposing roles. At physiological concentrations (much higher than ascorbate concentrations in plasma), it is a powerful oxygen radical scavenger in extracellular hydrophilic environments such as human plasma. It may protect the erythrocyte membrane from lipid peroxidation [35]. UA can react with O2−, H2O2, hydroxyl radical (OH−), and particularly peroxynitrite (OONO−) [35,62,63]. However, several authors suggest that UA also acts as a prooxidant inside the cell under certain inflammatory conditions, such as atheromatous plaque formation [35,64]. Recent studies have shown that UA can induce intracellular oxidative stress and proinflammatory effects in various cell types [65,66]. It does this by stimulating NOX [67,68] and by altering mitochondrial function with the consequent alteration in fat synthesis [36]. Fructose also induces NOX activation [69,70].
Oxidative stress significantly contributes to the development of insulin resistance and imbalance in vascular homeostasis, including endothelial cell dysfunction, atherosclerosis, vascular calcification, and impaired myocardial energetics, stimulating the production of interleukin-1 (IL-1), interleukin-6 (IL-6) and tumor necrosis factor α (TNFα) [71,72]. Heme oxygenase 1 (HO1), a potent antioxidant, decreases UA levels and adipocyte dysfunction by decreasing levels of ROS and XO [73]. XO is one of the major endothelial sources of O2− and H2O2. XO levels are substantially elevated in patients with coronary disease or carotid stenosis, and there is an inverse relationship between XO levels and endothelium-dependent vasodilation [74]. Accordingly, UA inhibits the bioavailability of nitric oxide (NO), which is a vasodilator [65,75]. Extracellular UA can also enter endothelial cells and vascular smooth muscle cells through URAT1, GLUT9, and potentially other transporters [76,77] activating the NF-κB axis, which leads to an increase in MCP-1, IL-8, VCAM-1, and ICAM-1 [78]. As a result, it has been hypothesized that intracellular XO activity and increased ROS production might be factors in endothelial dysfunction, which leads to the development of essential hypertension [79]. In this regard, ROS generation and vascular endothelial dysfunction can be reduced by drugs such as allopurinol and febuxostat, which inhibit XO activity and consequently reduce UA production [80].
OONO− is a potent non-radical oxidant species formed by the reaction between NO and O2−, which commonly occurs in the vascular endothelium [81]. NO is usually generated by NO synthase (NOS), However, under hypoxic conditions, XO can also modulate NO concentrations [82,83]. Since reducing NO bioavailability induces endothelial dysfunction and oxidative stress, XO inhibition could prevent oxidative damage, at least in part, by restoring NO generation [84,85]. Several studies have shown that, under aerobic conditions, UA can react with NO but not with OONO−, producing a nitrosated derivative [35,86,87]. Therefore, despite the general belief that survival is associated with increased antioxidant capacity, the opposite appears to be true: the ability to increase oxidative stress may have been associated with survival among early hominoids.
6. Evidence from Clinical Studies, Relevance for Humans
Physiologically, circulating UA levels increase with age. UA levels are lower in women of childbearing age, rising after menopause to levels similar to those in men [24]. There is still no clear threshold, above which uricemia becomes “abnormal”. The pathophysiological approach uses the supersaturation concentration of UA, 6.8 mg/dL at 37 °C, as a cut-off value, as indicated by guidelines for gout management [88]. However, recent findings suggest that this should be revised with reference to rheumatic, cardiovascular, and renal risk. Hyperuricemia is increasing in prevalence, as are its associated pathological conditions, such as metabolic syndrome, CKD, and CVD [8,89]. The average serum UA levels in the general population are rising [90] due to dietary changes, rising body mass indexes (BMI), and improved life expectancy.
Several in vitro and in vivo studies have shown that high fructose consumption increases blood UA levels [4,55,56,91]. In addition, consuming fructose over several days [70] and intravenous fructose administration [92] are both associated with an increase in fasting serum UA levels. It is not yet clear whether circulating UA levels increase when fructose is taken in small doses over several days or when it is taken as a single large dose. It is also unclear whether a high-fructose diet (HFrD) alters the renal clearance of UA or the fractional excretion of UA (UAFE) [93].
A relatively old meta-analysis of 21 controlled feeding trials of at least seven days showed that isocaloric exchange of fructose for other carbohydrates did not affect serum UA. However, in nondiabetic participants, hypercaloric supplementation of control diets with fructose (+35% excess energy) at extreme doses (213–219 g/day) significantly increased serum UA compared with the control diets alone (MD = 31.0 mmol/L (95% CI: 15.4, 46.5)) [94]. This meta-analysis was conducted before the publication of more recent trials, which confirmed the relationship between fructose intake and serum UA levels [95,96,97]. Thus, the available evidence is yet partly conflicting.
Notably, the fructose-induced increase in serum UA may cause acute damage (post-assumption). By excluding shorter studies, the meta-analysis may have excluded insights into this issue [97]. Finally, a fructose-restricted diet has been associated with a decrease in serum UA [98].
Lecoultre and colleagues performed a study on healthy subjects with two approaches: four to six days on an isoenergetic low-fructose diet (LFrD) and then either 6 days on a hyperenergetic HFrD with 34% excess energy as fructose or four days on a weight-maintenance HFrD in which 30% starch was substituted with fructose [42]. There was no difference in urinary UA excretion after the HFrD and the LFrD [42]. This suggests that UA production increases mainly when there is abnormally high hepatic metabolism of fructose. These findings also demonstrate that decreased urinary UA excretion may contribute to fructose-induced hyperuricemia. This mechanism could substantially increase the risk of gout in people who consume high amounts of fructose. Moreover, chronic exposure to fructose favours the onset of metabolic syndrome and increases insulin resistance [51].
Many experiments have shown that hyperuricemia may have a potential role in endothelial dysfunction and reduced NO bioavailability. However, several other studies have failed to demonstrate that an increase in blood UA levels can contribute to the onset of coronary and cardiovascular diseases. These studies also found no association between raised serum UA levels and the incidence of cardiovascular events [35,99,100,101,102,103]. It is not yet possible to say whether UA is a causal, compensatory, or coincidental factor for CVD [104]. Generally, gout and hyperuricemia patients also suffer from hypertension, CKD, insulin resistance, and obesity [105]. However, no clear link has yet been demonstrated between a HFrD and CVD. A recent systematic review and meta-analysis of prospective cohort studies was undertaken to quantify the association between intake of fructose-containing sugar (high-fructose corn syrup, sucrose, and fructose) and incidence of hypertension. This review searched MEDLINE, EMBASE, CINAHL, and the Cochrane Library for relevant studies [106]. It demonstrated that there was no association between a high total fructose intake and an increased risk of hypertension in three large prospective cohorts of men and women in the USA. In contrast, the same group conducted another systematic review and meta-analysis of prospective cohort studies to quantify the association between the consumption of fructose-containing sugar-sweetened beverages (SSBs) and the risk of hypertension. This demonstrated that SSBs were associated with a modest risk of developing hypertension in six cohorts [107].
The well-characterized Mediterranean cohort of the Brisighella Heart Study has a high intake of fruit [108]. Here, the increase in blood UA levels in the general population has been associated with increased incidence of hypertension and diabetes [109], LDL oxidation [110], arterial stiffness [111], impaired cognitive function [112], and heart failure [113].
In general, epidemiologists and other scientists have attempted to prove a causal link between dietary fructose intake and metabolic disorders, such as obesity, diabetes, and metabolic syndrome. However, while some studies have found a link, other studies have not [114].
Recent relevant clinical research has sought to verify if XO inhibition could benefit patients with high circulating UA levels, focusing on the XO inhibitor allopurinol and its active metabolite oxypurinol. Allopurinol has displayed beneficial effects on blood pressure in adolescents with newly diagnosed essential hypertension [89], and in patients with heart failure, coronary artery disease, and stroke [115]. Allopurinol may thus improve endothelial function and endothelium-dependent vasodilation in chronic heart failure patients [18].
XO inhibition has also been shown to improve a range of surrogate markers in hyperuricemic patients with CVD [9]. Wu and colleagues have demonstrated that high blood UA levels could be an independent predictor of mortality in patients with severe heart failure [116]. Baldus and colleagues have shown that oxypurinol improves myocardial contractility in patients with ischemic cardiomyopathy [117]. It also improves coronary vascular endothelial dysfunction in patients with coronary artery disease (CAD) [74]. The same group has shown that, without XO inhibition, lowering UA by uricosuric treatment (with benzbromarone) has no beneficial effect for chronic heart failure (CHF) patients [118]. This finding suggests that it is upregulated XO activity, rather than increased UA, that is actively involved in hemodynamic impairment in CHF. However, other studies suggest that elevated serum UA levels may be a risk marker for developing CVD [119,120]. Therefore, the scientific community needs to clarify the role of XO activity in CVD, so that the administration of natural or synthetic XO inhibitors as therapeutic agents for CVD can be properly evaluated [121,122].
7. Conclusions
In summary, a specific causal link between fructose consumption, hyperuricemia, and CVD has not yet been established. There is an association between UA and established cardiovascular risk factors, and there is a limit to how much population-based studies can adjust for confounding variables. As such, it is not yet possible to conclude that fructose intake is the main contributor to an increase in blood UA, and that this detrimentally affects vascular health. Further studies are required to prove or exclude a causal correlation between dietary fructose intake, UA production, and metabolic disorders. This would allow researchers to better understand which patients would obtain the greatest preventative benefit from reducing their UA levels, with diet and/or with XO inhibitors.
An important issue to consider is the bioavailability of XO inhibitors, especially the ability to cross cell plasma membranes. To address this, members of our group recently developed a cell-based biosensor that only measures intracellular XO activity and its inhibition by drugs that cross cell membranes [123]. The scientific community will need to focus on bioanalytical methods for directly monitoring XO activity and UA production. These will be useful tools for predicting the potential effects of new XO inhibitors, which could be used to treat hyperuricemia linked with cardiometabolic disorders.
Acknowledgments
We thank Grace Fox for copyediting and proofreading the manuscript.
Author Contributions
Cristiana Caliceti and Arrigo F. G. Cicero conceived the review. Cristiana Caliceti and Donato Calabria systematically searched and analysed the references. Aldo Roda critically revised the biochemistry related references, while Arrigo F. G. Cicero the epidemiology and clinical ones. Arrigo F. G. Cicero coordinated the authors. All authors contributed to the revision of the paper before publication.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zamudio-Cuevas, Y.; Hernandez-Diaz, C.; Pineda, C.; Reginato, A.M.; Cerna-Cortes, J.F.; Ventura-Rios, L.; Lopez-Reyes, A. Molecular basis of oxidative stress in gouty arthropathy. Clin. Rheumatol. 2015, 34, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Stack, A.G.; Hanley, A.; Casserly, L.F.; Cronin, C.J.; Abdalla, A.A.; Kiernan, T.J.; Murthy, B.V.; Hegarty, A.; Hannigan, A.; Nguyen, H.T. Independent and conjoint associations of gout and hyperuricaemia with total and cardiovascular mortality. QJM 2013, 106, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Church, T.S.; Meriwether, R.A.; Lobelo, F.; Blair, S.N. Uric acid and the development of metabolic syndrome in women and men. Metabolism 2008, 57, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Renal. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef] [PubMed]
- Gersch, M.S.; Mu, W.; Cirillo, P.; Reungjui, S.; Zhang, L.; Roncal, C.; Sautin, Y.Y.; Johnson, R.J.; Nakagawa, T. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2007, 293, F1256–F1261. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.H.; Gersch, M.S.; Benner, S.; Sanchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar] [PubMed]
- Johnson, R.J.; Titte, S.; Cade, J.R.; Rideout, B.A.; Oliver, W.J. Uric acid, evolution and primitive cultures. Semin. Nephrol. 2005, 25, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Borghi, C. The role of uric acid in the development of cardiovascular disease. Curr. Med. Res. Opin. 2015, 31 (Suppl. S2), 1–2. [Google Scholar] [CrossRef] [PubMed]
- Iwama, M.; Kondo, Y.; Shimokado, K.; Maruyama, N.; Ishigami, A. Uric acid levels in tissues and plasma of mice during aging. Biol. Pharm. Bull. 2012, 35, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Andrews, P.; Benner, S.A.; Oliver, W.; Theodore, E. Woodward award. The evolution of obesity: Insights from the mid-Miocene. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 295–305. [Google Scholar] [PubMed]
- Johnson, R.J.; Sautin, Y.Y.; Oliver, W.J.; Roncal, C.; Mu, W.; Gabriela Sanchez-Lozada, L.; Rodriguez-Iturbe, B.; Nakagawa, T.; Benner, S.A. Lessons from comparative physiology: Could uric acid represent a physiologic alarm signal gone awry in western society? J. Comp. Physiol. B 2009, 179, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Higgins, P.; Dawson, J.; Lees, K.R.; McArthur, K.; Quinn, T.J.; Walters, M.R. Xanthine oxidase inhibition for the treatment of cardiovascular disease: A systematic review and meta-analysis. Cardiovasc. Ther. 2012, 30, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, L.B. Mechanism of excessive purine biosynthesis in hypoxanthine-guanine phosphoribosyltransferase deficiency. J. Clin. Investig. 1970, 49, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Pritsos, C.A. Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chem. Biol. Interact. 2000, 129, 195–208. [Google Scholar] [CrossRef]
- Vorbach, C.; Harrison, R.; Capecchi, M.R. Xanthine oxidoreductase is central to the evolution and function of the innate immune system. Trends Immunol. 2003, 24, 512–517. [Google Scholar] [CrossRef]
- Friedl, H.P.; Till, G.O.; Ryan, U.S.; Ward, P.A. Mediator-induced activation of xanthine oxidase in endothelial cells. FASEB J. 1989, 3, 2512–2518. [Google Scholar] [PubMed]
- Doehner, W.; Schoene, N.; Rauchhaus, M.; Leyva-Leon, F.; Pavitt, D.V.; Reaveley, D.A.; Schuler, G.; Coats, A.J.; Anker, S.D.; Hambrecht, R. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: Results from 2 placebo-controlled studies. Circulation 2002, 105, 2619–2624. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ruiz, F.; Calabozo, M.; Erauskin, G.G.; Ruibal, A.; Herrero-Beites, A.M. Renal underexcretion of uric acid is present in patients with apparent high urinary uric acid output. Arthritis Rheum. 2002, 47, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Anzai, N.; Ichida, K.; Jutabha, P.; Kimura, T.; Babu, E.; Jin, C.J.; Srivastava, S.; Kitamura, K.; Hisatome, I.; Endou, H.; et al. Plasma urate level is directly regulated by a voltage-driven urate efflux transporter URATv1 (SLC2A9) in humans. J. Biol. Chem. 2008, 283, 26834–26838. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Iharada, M.; Miyaji, T.; Fujimoto, T.; Hiasa, M.; Anzai, N.; Omote, H.; Moriyama, Y. Type 1 sodium-dependent phosphate transporter (SLC17A1 Protein) is a Cl(-)-dependent urate exporter. J. Biol. Chem. 2010, 285, 26107–26113. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Tamai, I.; Yamashita, K.; Minemoto, Y.; Sai, Y.; Yabuuchi, H.; Miyamoto, K.; Takeda, E.; Tsuji, A. p-Aminohippuric acid transport at renal apical membrane mediated by human inorganic phosphate transporter NPT1. Biochem. Biophys. Res. Commun. 2000, 270, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Richette, P.; Bardin, T. Gout. Lancet 2010, 375, 318–328. [Google Scholar] [CrossRef]
- Hosomi, A.; Nakanishi, T.; Fujita, T.; Tamai, I. Extra-renal elimination of uric acid via intestinal efflux transporter BCRP/ABCG2. PLoS ONE 2012, 7, e30456. [Google Scholar] [CrossRef] [PubMed]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Kottgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Kottgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [PubMed]
- Litman, T.; Jensen, U.; Hansen, A.; Covitz, K.M.; Zhan, Z.; Fetsch, P.; Abati, A.; Hansen, P.R.; Horn, T.; Skovsgaard, T.; et al. Use of peptide antibodies to probe for the mitoxantrone resistance-associated protein MXR/BCRP/ABCP/ABCG2. Biochim. Biophys. Acta 2002, 1565, 6–16. [Google Scholar] [CrossRef]
- Kage, K.; Tsukahara, S.; Sugiyama, T.; Asada, S.; Ishikawa, E.; Tsuruo, T.; Sugimoto, Y. Dominant-negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S-S dependent homodimerization. Int. J. Cancer 2002, 97, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Suzuki, H.; Sugiyama, Y. Characterization of polarized expression of point- or deletion-mutated human BCRP/ABCG2 in LLC-PK1 cells. Pharm. Res. 2005, 22, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Ogura, J.; Kuwayama, K.; Sasaki, S.; Kaneko, C.; Koizumi, T.; Yabe, K.; Tsujimoto, T.; Takeno, R.; Takaya, A.; Kobayashi, M.; et al. Reactive oxygen species derived from xanthine oxidase interrupt dimerization of breast cancer resistance protein, resulting in suppression of uric acid excretion to the intestinal lumen. Biochem. Pharmacol. 2015, 97, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ogura, J.; Kuwayama, K.; Takaya, A.; Terada, Y.; Tsujimoto, T.; Koizumi, T.; Maruyama, H.; Fujikawa, A.; Takahashi, N.; Kobayashi, M.; et al. Intestinal ischemia-reperfusion increases efflux for uric acid via paracellular route in the intestine, but decreases that via transcellular route mediated by BCRP. J. Pharm. Pharm. Sci. 2012, 15, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Hediger, M.A.; Johnson, R.J.; Miyazaki, H.; Endou, H. Molecular physiology of urate transport. Physiology (Bethesda) 2005, 20, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.W.; Muzny, D.M.; Lee, C.C.; Caskey, C.T. Two independent mutational events in the loss of urate oxidase during hominoid evolution. J. Mol. Evol. 1992, 34, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Gao, X.; Curhan, G. Vitamin C intake and the risk of gout in men: A prospective study. Arch. Intern. Med. 2009, 169, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Berger, L.; Gerson, C.D.; Yu, T.F. The effect of ascorbic acid on uric acid excretion with a commentary on the renal handling of ascorbic acid. Am. J. Med. 1977, 62, 71–76. [Google Scholar] [CrossRef]
- Stein, H.B.; Hasan, A.; Fox, I.H. Ascorbic acid-induced uricosuria. A consequency of megavitamin therapy. Ann. Intern. Med. 1976, 84, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Shin, H.S.; Choi, H.S.; Park, J.W.; Jo, I.; Oh, E.S.; Lee, K.Y.; Lee, B.H.; Johnson, R.J.; Kang, D.H. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab. Investig. 2014, 94, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Bedir, A.; Topbas, M.; Tanyeri, F.; Alvur, M.; Arik, N. Leptin might be a regulator of serum uric acid concentrations in humans. Jpn. Heart J. 2003, 44, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Tzameli, I.; Pissios, P.; Rovira, I.; Gavrilova, O.; Ohtsubo, T.; Chen, Z.; Finkel, T.; Flier, J.S.; Friedman, J.M. Xanthine oxidoreductase is a regulator of adipogenesis and PPARgamma activity. Cell Metab. 2007, 5, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Fruehwald-Schultes, B.; Peters, A.; Kern, W.; Beyer, J.; Pfutzner, A. Serum leptin is associated with serum uric acid concentrations in humans. Metabolism 1999, 48, 677–680. [Google Scholar] [CrossRef]
- Matsuura, F.; Yamashita, S.; Nakamura, T.; Nishida, M.; Nozaki, S.; Funahashi, T.; Matsuzawa, Y. Effect of visceral fat accumulation on uric acid metabolism in male obese subjects: Visceral fat obesity is linked more closely to overproduction of uric acid than subcutaneous fat obesity. Metabolism 1998, 47, 929–933. [Google Scholar] [CrossRef]
- Romao, I.; Roth, J. Genetic and environmental interactions in obesity and type 2 diabetes. J. Am. Diet. Assoc. 2008, 108 (4 Suppl. 1S), S24–S28. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Atkinson, K.; Karlson, E.W.; Willett, W.; Curhan, G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N. Engl. J. Med. 2004, 350, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Ford, E.S.; Gao, X.; Choi, H.K. Sugar-sweetened soft drinks, diet soft drinks, and serum uric acid level: The Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2008, 59, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Bleich, S.N.; Gortmaker, S.L. Increasing caloric contribution from sugar-sweetened beverages and 100% fruit juices among US children and adolescents, 1988–2004. Pediatrics 2008, 121, e1604–e1614. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; van Eys, J. Nutritional significance of fructose and sugar alcohols. Annu. Rev. Nutr. 1981, 1, 437–475. [Google Scholar] [CrossRef] [PubMed]
- Hanover, L.M.; White, J.S. Manufacturing, composition, and applications of fructose. Am. J. Clin. Nutr. 1993, 58 (Suppl. S5), 724S–732S. [Google Scholar] [PubMed]
- Lustig, R.H. Fructose: Metabolic, hedonic, and societal parallels with ethanol. J. Am. Diet. Assoc. 2010, 110, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, A.; Niquet-Leridon, C.; Boulanger, E.; Tessier, F.J. How Can Diet Affect the Accumulation of Advanced Glycation End-Products in the Human Body? Foods 2016, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Bunn, H.F.; Higgins, P.J. Reaction of monosaccharides with proteins: Possible evolutionary significance. Science 1981, 213, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Suarez, G.; Rajaram, R.; Oronsky, A.L.; Gawinowicz, M.A. Nonenzymatic glycation of bovine serum albumin by fructose (fructation). Comparison with the Maillard reaction initiated by glucose. J. Biol. Chem. 1989, 264, 3674–3679. [Google Scholar] [PubMed]
- Hallfrisch, J. Metabolic effects of dietary fructose. FASEB J. 1990, 4, 2652–2660. [Google Scholar] [PubMed]
- Kaneko, C.; Ogura, J.; Sasaki, S.; Okamoto, K.; Kobayashi, M.; Kuwayama, K.; Narumi, K.; Iseki, K. Fructose suppresses uric acid excretion to the intestinal lumen as a result of the induction of oxidative stress by NADPH oxidase activation. Biochim. Biophys. Acta 2017, 1861, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, G.; Bronfman, M.; Vanneste, R.; Hers, H.G. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem. J. 1977, 162, 601–609. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.P.; Burini, R.C. High plasma uric acid concentration: Causes and consequences. Diabetol. Metab. Syndr. 2012, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Brosh, S.; Boer, P.; Sperling, O. Effects of fructose on synthesis and degradation of purine nucleotides in isolated rat hepatocytes. Biochim. Biophys. Acta 1982, 717, 459–464. [Google Scholar] [CrossRef]
- Emmerson, B.T. Effect of oral fructose on urate production. Ann. Rheum. Dis. 1974, 33, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite with uric acid in the presence of ascorbate and thiols: Implications for uncoupling endothelial nitric oxide synthase. Biochem. Pharmacol. 2005, 70, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Morre, J.T.; Beckman, J.S. Triuret: A novel product of peroxynitrite-mediated oxidation of urate. Arch Biochem. Biophys. 2004, 423, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Holme, I.; Aastveit, A.H.; Hammar, N.; Jungner, I.; Walldius, G. Uric acid and risk of myocardial infarction, stroke and congestive heart failure in 417,734 men and women in the Apolipoprotein MOrtality RISk study (AMORIS). J. Intern. Med. 2009, 266, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Park, S.K.; Lee, I.K.; Johnson, R.J. Uric acid-induced C-reactive protein expression: Implication on cell proliferation and nitric oxide production of human vascular cells. J. Am. Soc. Nephrol. 2005, 16, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.A.; Sanchez-Lozada, L.G.; Johnson, R.J.; Kang, D.H. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Ratto, E.; Villaggio, B.; Parodi, E.L.; Pontremoli, R.; Garibotto, G.; Viazzi, F. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS ONE 2014, 9, e115210. [Google Scholar] [CrossRef] [PubMed]
- De Souto Padron, F.A.; Salmon, A.B.; Bruno, F.; Jimenez, F.; Martinez, H.G.; Halade, G.V.; Ahuja, S.S.; Clark, R.A.; DeFronzo, R.A.; Abboud, H.E.; et al. Nox2 mediates skeletal muscle insulin resistance induced by a high fat diet. J. Biol. Chem. 2015, 290, 13427–13439. [Google Scholar] [CrossRef] [PubMed]
- Streeter, J.; Thiel, W.; Brieger, K.; Miller, F.J., Jr. Opportunity nox: The future of NADPH oxidases as therapeutic targets in cardiovascular disease. Cardiovasc. Ther. 2013, 31, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Kanellis, J.; Kang, D.H. Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Semin. Nephrol. 2005, 25, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Prata, C.; Grasso, C.; Loizzo, S.; Vieceli Dalla Sega, F.; Caliceti, C.; Zambonin, L.; Fiorentini, D.; Hakim, G.; Berridge, M.V.; Landi, L. Inhibition of trans-plasma membrane electron transport: A potential anti-leukemic strategy. Leuk. Res. 2010, 34, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Hilgefort, J.; Banks, G.; Gilliam, C.; Stevens, S.; Ansinelli, H.A.; Getty, M.; Abraham, N.G.; Shapiro, J.I.; Khitan, Z. Uric Acid-Induced Adipocyte Dysfunction Is Attenuated by HO-1 Upregulation: Potential Role of Antioxidant Therapy to Target Obesity. Stem Cells Int. 2016, 2016, 8197325. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.; Koster, R.; Chumley, P.; Heitzer, T.; Rudolph, V.; Ostad, M.A.; Warnholtz, A.; Staude, H.J.; Thuneke, F.; Koss, K.; et al. Oxypurinol improves coronary and peripheral endothelial function in patients with coronary artery disease. Free Radic. Biol. Med. 2005, 39, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Price, K.L.; Sautin, Y.Y.; Long, D.A.; Zhang, L.; Miyazaki, H.; Mu, W.; Endou, H.; Johnson, R.J. Human vascular smooth muscle cells express a urate transporter. J. Am. Soc. Nephrol. 2006, 17, 1791–1795. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, S.; Hisatome, I.; Kuwabara, M.; Niwa, K.; Maharani, N.; Kato, M.; Ogino, K.; Hamada, T.; Ninomiya, H.; Higashi, Y.; et al. Depletion of Uric Acid Due to SLC22A12 (URAT1) Loss-of-Function Mutation Causes Endothelial Dysfunction in Hypouricemia. Circ. J. 2015, 79, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.Y.; Zhu, X.Y.; Zhang, J.W.; Feng, X.R.; Wang, Y.C.; Liu, M.L. Uric acid promotes chemokine and adhesion molecule production in vascular endothelium via nuclear factor-kappa B signaling. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, B.; Brand, S.M. Uric acid and essential hypertension: The endothelial connection. J. Hypertens. 2016, 34, 2138–2139. [Google Scholar] [CrossRef] [PubMed]
- Puddu, P.; Puddu, G.M.; Cravero, E.; Vizioli, L.; Muscari, A. Relationships among hyperuricemia, endothelial dysfunction and cardiovascular disease: Molecular mechanisms and clinical implications. J. Cardiol. 2012, 59, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Godber, B.L.; Doel, J.J.; Sapkota, G.P.; Blake, D.R.; Stevens, C.R.; Eisenthal, R.; Harrison, R. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J. Biol. Chem. 2000, 275, 7757–7763. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Samouilov, A.; Liu, X.; Zweier, J.L. Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrate reduction: Evaluation of its role in nitrite and nitric oxide generation in anoxic tissues. Biochemistry 2003, 42, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Millar, T.M.; Stevens, C.R.; Benjamin, N.; Eisenthal, R.; Harrison, R.; Blake, D.R. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett. 1998, 427, 225–228. [Google Scholar] [CrossRef]
- Hink, H.U.; Santanam, N.; Dikalov, S.; McCann, L.; Nguyen, A.D.; Parthasarathy, S.; Harrison, D.G.; Fukai, T. Peroxidase properties of extracellular superoxide dismutase: Role of uric acid in modulating in vivo activity. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Suliman, H.B.; Piantadosi, C.A. Extracellular superoxide dismutase. Int. J. Biochem. Cell Biol. 2005, 37, 2466–2471. [Google Scholar] [CrossRef] [PubMed]
- Nieto, F.J.; Iribarren, C.; Gross, M.D.; Comstock, G.W.; Cutler, R.G. Uric acid and serum antioxidant capacity: A reaction to atherosclerosis? Atherosclerosis 2000, 148, 131–139. [Google Scholar] [CrossRef]
- Suzuki, T. Nitrosation of uric acid induced by nitric oxide under aerobic conditions. Nitric Oxide 2007, 16, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Fitzgerald, J.D.; Khanna, P.P.; Bae, S.; Singh, M.K.; Neogi, T.; Pillinger, M.H.; Merill, J.; Lee, S.; Prakash, S.; et al. 2012 American College of Rheumatology guidelines for management of gout. Part 1: Systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. (Hoboken) 2012, 64, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Feig, D.I.; Soletsky, B.; Johnson, R.J. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: A randomized trial. JAMA 2008, 300, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pandya, B.J.; Choi, H.K. Prevalence of gout and hyperuricemia in the US general population: The National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011, 63, 3136–3141. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Bremer, A.A.; Berglund, L.; McGahan, J.P.; Keim, N.L.; et al. Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr. Metab. 2012, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Lazo, M.; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; Clark, J.M. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.H.; Wang, C.; Li, J.M.; Zhang, D.M.; Kong, L.D. Allopurinol, rutin, and quercetin attenuate hyperuricemia and renal dysfunction in rats induced by fructose intake: Renal organic ion transporter involvement. Am. J. Physiol. Renal. Physiol. 2009, 297, F1080–F1091. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Sievenpiper, J.L.; de Souza, R.J.; Chiavaroli, L.; Ha, V.; Cozma, A.I.; Mirrahimi, A.; Yu, M.E.; Carleton, A.J.; Di Buono, M.; et al. The effects of fructose intake on serum uric acid vary among controlled dietary trials. J. Nutr. 2012, 142, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Bruun, J.M.; Maersk, M.; Belza, A.; Astrup, A.; Richelsen, B. Consumption of sucrose-sweetened soft drinks increases plasma levels of uric acid in overweight and obese subjects: A 6-month randomised controlled trial. Eur. J. Clin. Nutr. 2015, 69, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Medici, V.; Bremer, A.A.; Lee, V.; Lam, H.D.; Nunez, M.V.; Chen, G.X.; Keim, N.L.; Havel, P.J. A dose-response study of consuming high-fructose corn syrup-sweetened beverages on lipid/lipoprotein risk factors for cardiovascular disease in young adults. Am. J. Clin. Nutr. 2015, 101, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.; Frye, R.F.; Rivard, C.J.; Cheng, J.; McFann, K.K.; Segal, M.S.; Johnson, R.J.; Johnson, J.A. Effects of high-fructose corn syrup and sucrose on the pharmacokinetics of fructose and acute metabolic and hemodynamic responses in healthy subjects. Metabolism 2012, 61, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Madero, M.; Rodríguez Castellanos, F.E.; Jalal, D.; Villalobos-Martín, M.; Salazar, J.; Vazquez-Rangel, A.; Johnson, R.J.; Sanchez-Lozada, L.G. A pilot study on the impact of a low fructose diet and allopurinol on clinic blood pressure among overweight and prehypertensive subjects: A randomized placebo controlled trial. J. Am. Soc. Hypertens. 2015, 9, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Moriarity, J.T.; Folsom, A.R.; Iribarren, C.; Nieto, F.J.; Rosamond, W.D. Serum uric acid and risk of coronary heart disease: Atherosclerosis Risk in Communities (ARIC) Study. Ann. Epidemiol. 2000, 10, 136–143. [Google Scholar] [CrossRef]
- Sakata, K.; Hashimoto, T.; Ueshima, H.; Okayama, A. Absence of an association between serum uric acid and mortality from cardiovascular disease: NIPPON DATA 80, 1980–1994. National Integrated Projects for Prospective Observation of Non-communicable Diseases and its Trend in the Aged. Eur. J. Epidemiol. 2001, 17, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Wannamethee, S.G.; Shaper, A.G.; Whincup, P.H. Serum urate and the risk of major coronary heart disease events. Heart 1997, 78, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.G.; Juzwishin, K.D.; Eiriksdottir, G.; Gudnason, V.; Danesh, J. Serum uric acid and coronary heart disease in 9,458 incident cases and 155,084 controls: Prospective study and meta-analysis. PLoS Med. 2005, 2, e76. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.D.; Brand, F.N.; Kannel, W.B.; Castelli, W.P. Gout and coronary heart disease: The Framingham Study. J. Clin. Epidemiol. 1988, 41, 237–242. [Google Scholar] [CrossRef]
- Waring, W.S.; Webb, D.J.; Maxwell, S.R. Uric acid as a risk factor for cardiovascular disease. QJM 2000, 93, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Pillinger, M.H.; Goldfarb, D.S.; Keenan, R.T. Gout and its comorbidities. Bull. NYU Hosp. Jt. Dis. 2010, 68, 199–203. [Google Scholar] [PubMed]
- Jayalath, V.H.; Sievenpiper, J.L.; de Souza, R.J.; Ha, V.; Mirrahimi, A.; Santaren, I.D.; Blanco, M.S.; Di, B.M.; Jenkins, A.L.; Leiter, L.A.; et al. Total fructose intake and risk of hypertension: A systematic review and meta-analysis of prospective cohorts. J. Am. Coll. Nutr. 2014, 33, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Jayalath, V.H.; de Souza, R.J.; Ha, V.; Mirrahimi, A.; Blanco-Mejia, S.; Di, B.M.; Jenkins, A.L.; Leiter, L.A.; Wolever, T.M.; Beyene, J.; et al. Sugar-sweetened beverage consumption and incident hypertension: A systematic review and meta-analysis of prospective cohorts. Am. J. Clin. Nutr. 2015, 102, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Dormi, A.; D’Addato, S.; Gaddi, A.V.; Borghi, C. Long-term effect of a dietary education program on postmenopausal cardiovascular risk and metabolic syndrome: The Brisighella Heart Study. J. Womens Health 2010, 19, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Rosticci, M.; Bove, M.; Fogacci, F.; Giovannini, M.; Urso, R.; D’Addato, S.; Borghi, C. Serum uric acid change and modification of blood pressure and fasting plasma glucose in an overall healthy population sample: Data from the Brisighella heart study. Ann. Med. 2017, 49, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Rosticci, M.; Cagnati, M.; Urso, R.; Scapagnini, G.; Morbini, M.; Grandi, E.; D’Addato, S.; Borghi, C. Serum uric acid and markers of low-density lipoprotein oxidation in nonsmoking healthy subjects: Data from the Brisighella Heart Study. Pol. Arch. Med. Wewn. 2014, 124, 661–668. [Google Scholar] [PubMed]
- Cicero, A.F.; Rosticci, M.; Fogacci, F.; Grandi, E.; D’Addato, S.; Borghi, C. High serum uric acid is associated to poorly controlled blood pressure and higher arterial stiffness in hypertensive subjects. Eur. J. Intern. Med. 2017, 37, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Desideri, G.; Grossi, G.; Urso, R.; Rosticci, M.; D’Addato, S.; Borghi, C. Serum uric acid and impaired cognitive function in a cohort of healthy young elderly: Data from the Brisighella Study. Intern. Emerg Med. 2015, 10, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Rosticci, M.; Parini, A.; Baronio, C.; D’Addato, S.; Borghi, C. Serum uric acid is inversely proportional to estimated stroke volume and cardiac output in a large sample of pharmacologically untreated subjects: Data from the Brisighella Heart Study. Intern. Emerg Med. 2014, 9, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Z.; Anderson, G.H.; Flickinger, B.D.; Williamson-Hughes, P.S.; Empie, M.W. Fructose and non-fructose sugar intakes in the US population and their associations with indicators of metabolic syndrome. Food Chem. Toxicol. 2011, 49, 2875–2882. [Google Scholar] [CrossRef] [PubMed]
- Okafor, O.N.; Farrington, K.; Gorog, D.A. Allopurinol as a therapeutic option in cardiovascular disease. Pharmacol. Ther. 2016, 172, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.; Ghali, J.K.; Neuberg, G.W.; O’Connor, C.M.; Carson, P.E.; Levy, W.C. Uric acid level and allopurinol use as risk markers of mortality and morbidity in systolic heart failure. Am. Heart J. 2010, 160, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.; Mullerleile, K.; Chumley, P.; Steven, D.; Rudolph, V.; Lund, G.K.; Staude, H.J.; Stork, A.; Koster, R.; Kahler, J.; et al. Inhibition of xanthine oxidase improves myocardial contractility in patients with ischemic cardiomyopathy. Free Radic. Biol. Med. 2006, 41, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Ogino, K.; Kato, M.; Furuse, Y.; Kinugasa, Y.; Ishida, K.; Osaki, S.; Kinugawa, T.; Igawa, O.; Hisatome, I.; Shigemasa, C.; et al. Uric acid-lowering treatment with benzbromarone in patients with heart failure: A double-blind placebo-controlled crossover preliminary study. Circ. Heart Fail. 2010, 3, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, C.; Folsom, A.R.; Eckfeldt, J.H.; McGovern, P.G.; Nieto, F.J. Correlates of uric acid and its association with asymptomatic carotid atherosclerosis: The ARIC Study. Atherosclerosis Risk in Communities. Ann. Epidemiol. 1996, 6, 331–340. [Google Scholar] [CrossRef]
- Johnson, R.J.; Kang, D.H.; Feig, D.; Kivlighn, S.; Kanellis, J.; Watanabe, S.; Tuttle, K.R.; Rodriguez-Iturbe, B.; Herrera-Acosta, J.; Mazzali, M. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 2003, 41, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Borghi, C.; Cicero, A.F. Serum Uric Acid and Cardiometabolic Disease: Another Brick in the Wall? Hypertension 2017. [Google Scholar] [CrossRef] [PubMed]
- Bove, M.; Cicero, A.F.; Veronesi, M.; Borghi, C. An evidence-based review on urate-lowering treatments: Implications for optimal treatment of chronic hyperuricemia. Vasc. Health Risk Manag. 2017, 13, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Caliceti, C.; Calabria, D.; Roda, A. A new sensitive and quantitative chemiluminescent assay to monitor intracellular xanthine oxidase activity for rapid screening of inhibitors in living endothelial cells. Anal. Bioanal. Chem. 2016, 408, 8755–8760. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Uric acid formation through xanthine oxidase activity. (A) Under ischemic or inflammatory conditions, xanthine dehydrogenase (XDH) is converted to xanthine oxidase (XO) via the oxidation of sulfhydryl residues or proteolysis of XDH. In the presence of oxygen, XO catalyses the oxidation of hypoxanthine to xanthine and then to uric acid (UA), with consequent production of the superoxide anion (O2−) and hydrogen peroxide (H2O2). The competitive antagonist allopurinol is converted in the active form, oxypurinol, via XO activity, acting as an XO inhibitor; (B) During hypoxanthine conversion to xanthine and then UA, high levels of H2O2 and O2− are produced and converted to O2 and H2O2, spontaneously or in a reaction catalyzed by the enzyme superoxide dismutase (SOD).
Figure 1.
Uric acid formation through xanthine oxidase activity. (A) Under ischemic or inflammatory conditions, xanthine dehydrogenase (XDH) is converted to xanthine oxidase (XO) via the oxidation of sulfhydryl residues or proteolysis of XDH. In the presence of oxygen, XO catalyses the oxidation of hypoxanthine to xanthine and then to uric acid (UA), with consequent production of the superoxide anion (O2−) and hydrogen peroxide (H2O2). The competitive antagonist allopurinol is converted in the active form, oxypurinol, via XO activity, acting as an XO inhibitor; (B) During hypoxanthine conversion to xanthine and then UA, high levels of H2O2 and O2− are produced and converted to O2 and H2O2, spontaneously or in a reaction catalyzed by the enzyme superoxide dismutase (SOD).

Figure 2.
Fructose-induced uric acid formation: risk factor biomarkers. In hepatocytes, fructokinase catalyzes the rapid phosphorylation of fructose to fructose-1-phosphate, using ATP as a phosphate donor. Intracellular phosphate (Pi) levels decrease, stimulating the activity of AMP deaminase (AMPD). AMPD converts AMP to inosine monophosphate (IMP). IMP is metabolized to inosine, which is further degraded to xanthine and hypoxanthine by xanthine oxidase (XO), ultimately generating uric acid (UA). UA can react with nitric oxide (NO), reducing NO bioavailability and inducing dinucleotide phosphate oxidase (NOX) activation and mitochondrial dysfunction. In turn, this promotes oxidative stress and endothelial dysfunction. Fructose per se can also induce oxidative stress.
Figure 2.
Fructose-induced uric acid formation: risk factor biomarkers. In hepatocytes, fructokinase catalyzes the rapid phosphorylation of fructose to fructose-1-phosphate, using ATP as a phosphate donor. Intracellular phosphate (Pi) levels decrease, stimulating the activity of AMP deaminase (AMPD). AMPD converts AMP to inosine monophosphate (IMP). IMP is metabolized to inosine, which is further degraded to xanthine and hypoxanthine by xanthine oxidase (XO), ultimately generating uric acid (UA). UA can react with nitric oxide (NO), reducing NO bioavailability and inducing dinucleotide phosphate oxidase (NOX) activation and mitochondrial dysfunction. In turn, this promotes oxidative stress and endothelial dysfunction. Fructose per se can also induce oxidative stress.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Caliceti, C.; Calabria, D.; Roda, A.; Cicero, A.F.G. Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review. Nutrients 2017, 9, 395. https://0-doi-org.brum.beds.ac.uk/10.3390/nu9040395
AMA Style
Caliceti C, Calabria D, Roda A, Cicero AFG. Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review. Nutrients. 2017; 9(4):395. https://0-doi-org.brum.beds.ac.uk/10.3390/nu9040395
Chicago/Turabian StyleCaliceti, Cristiana, Donato Calabria, Aldo Roda, and Arrigo F. G. Cicero. 2017. "Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review" Nutrients 9, no. 4: 395. https://0-doi-org.brum.beds.ac.uk/10.3390/nu9040395
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.