Uremia Impacts VE-Cadherin and ZO-1 Expression in Human Endothelial Cell-to-Cell Junctions

 , ,
, , 
Abstract
:
1. Introduction
2. Results
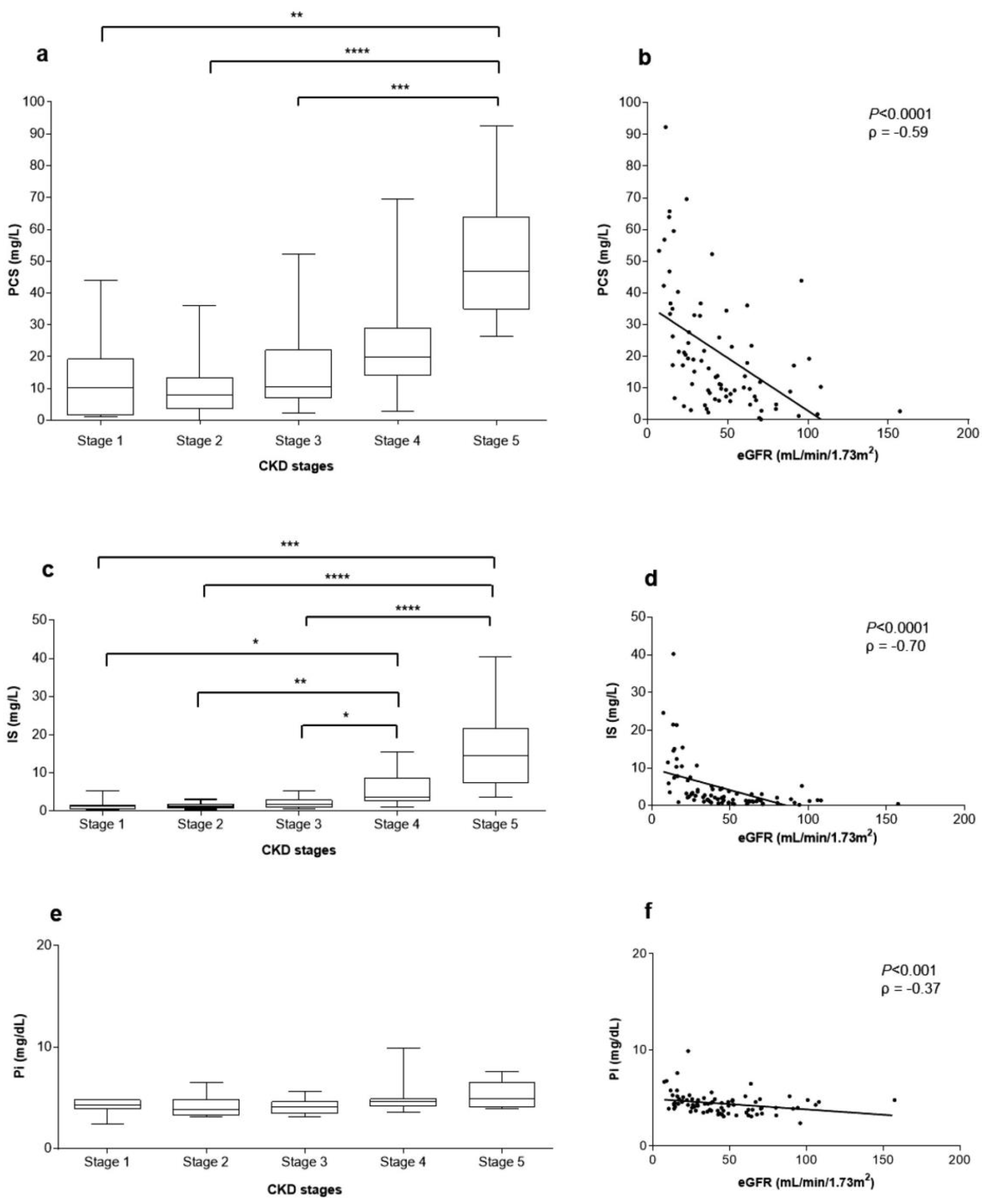
2.1. Clinical and Laboratory Characteristics of the Study Population
2.2. Clinical and Biochemical Characteristics of Each Uremic Pool
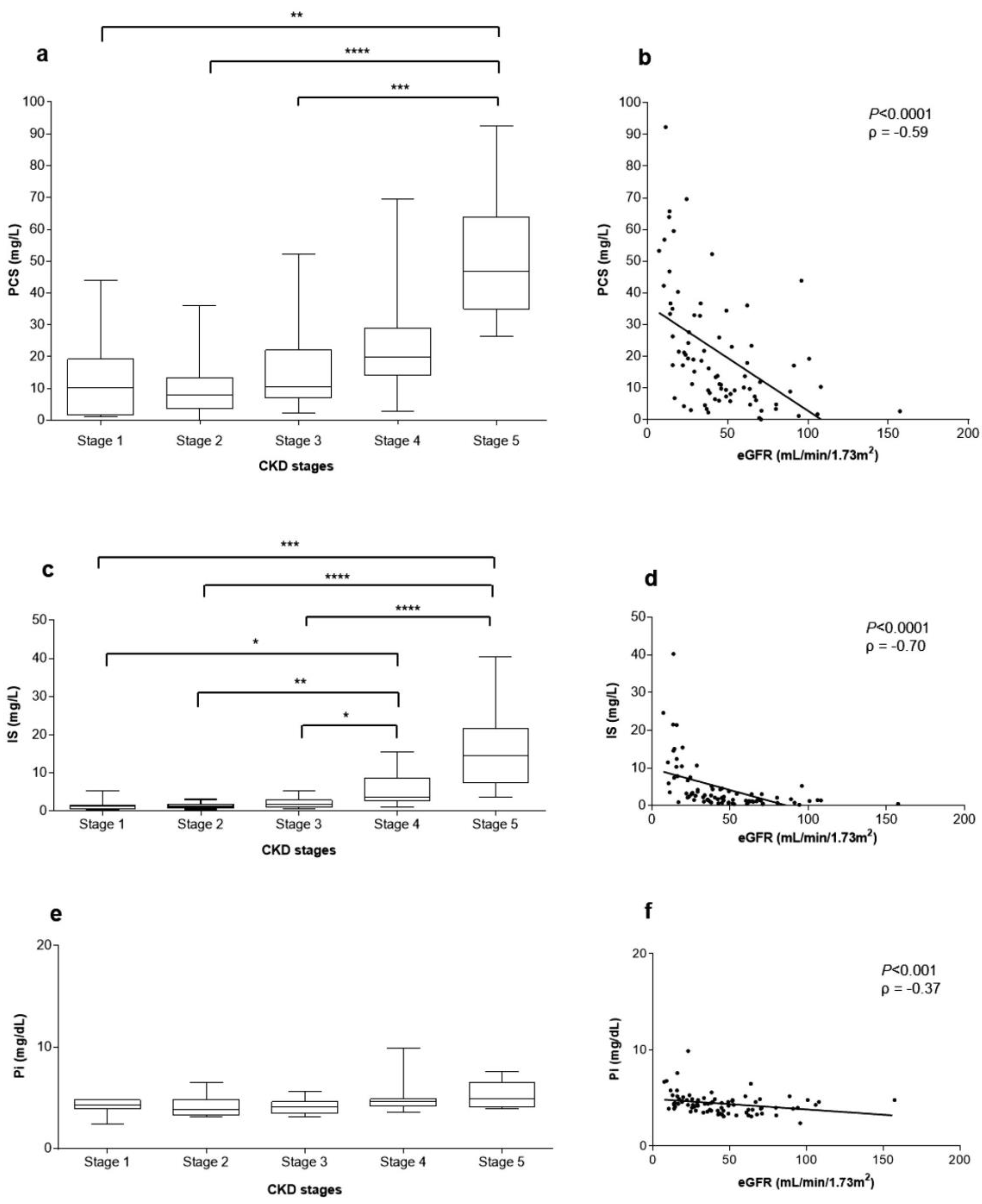
2.3. Concentration of Systemic and Vascular Inflammatory Biomarkers
2.4. Multivariate Analysis of Independent Determinants of Chemokines, Adhesion Molecules
2.5. Correlations between Uremic Toxins Serum Concentration and eGFR
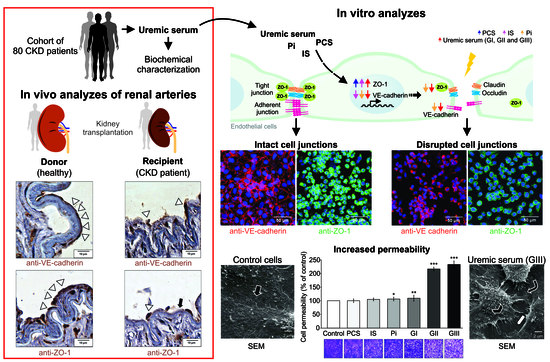
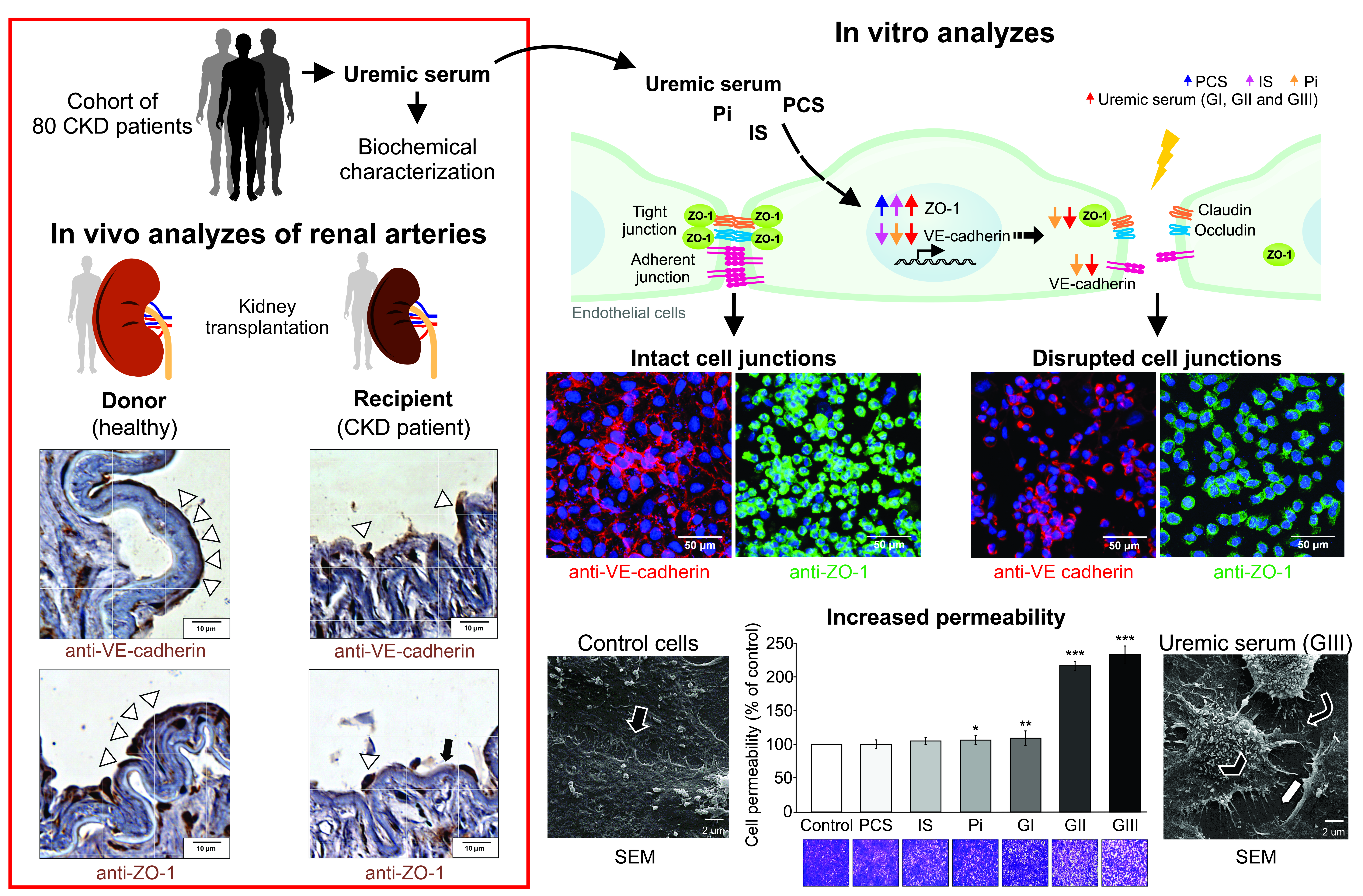
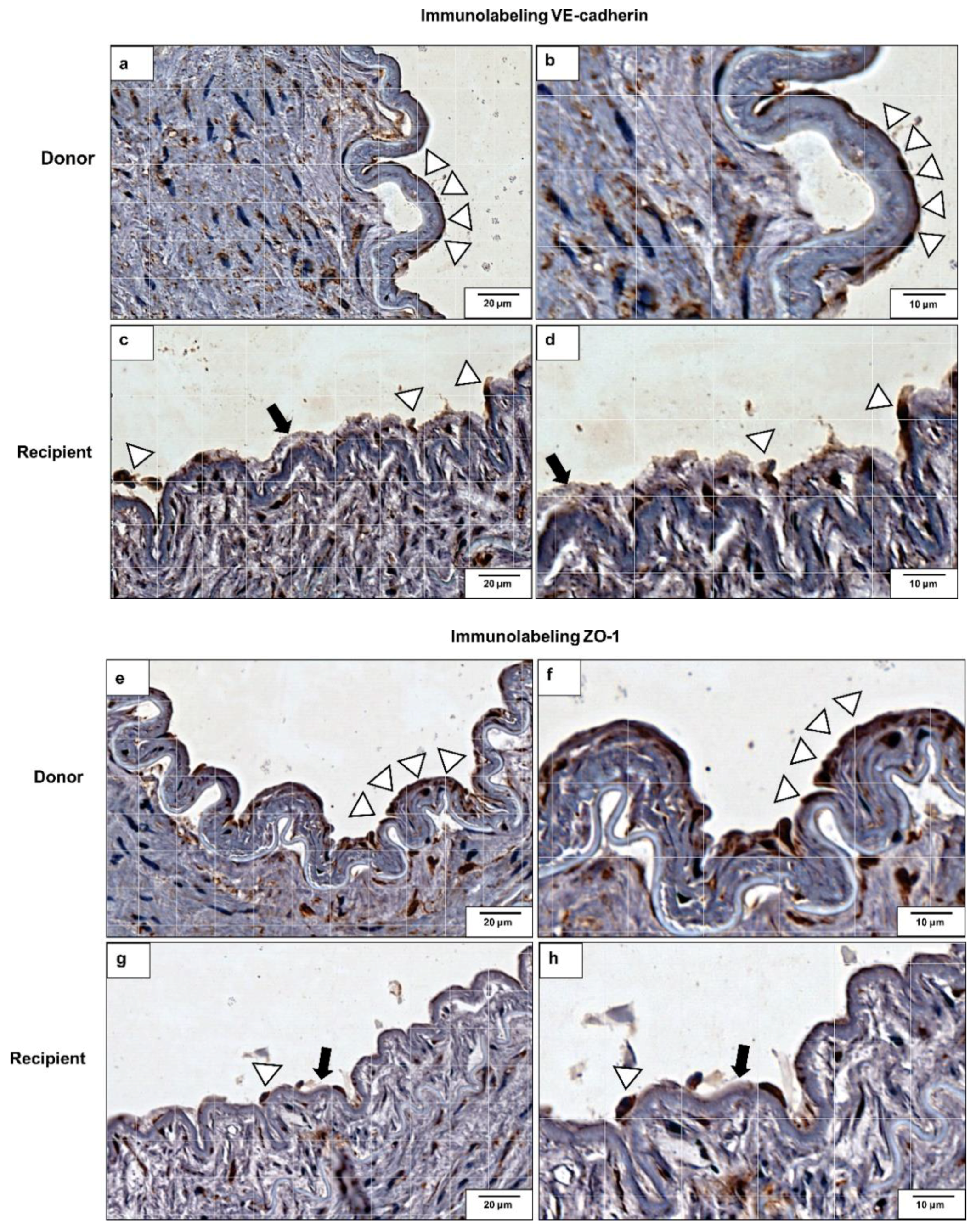
2.6. VE-Cadherin and ZO-1 Expression Increased in CKD Iliac and Renal Arteries
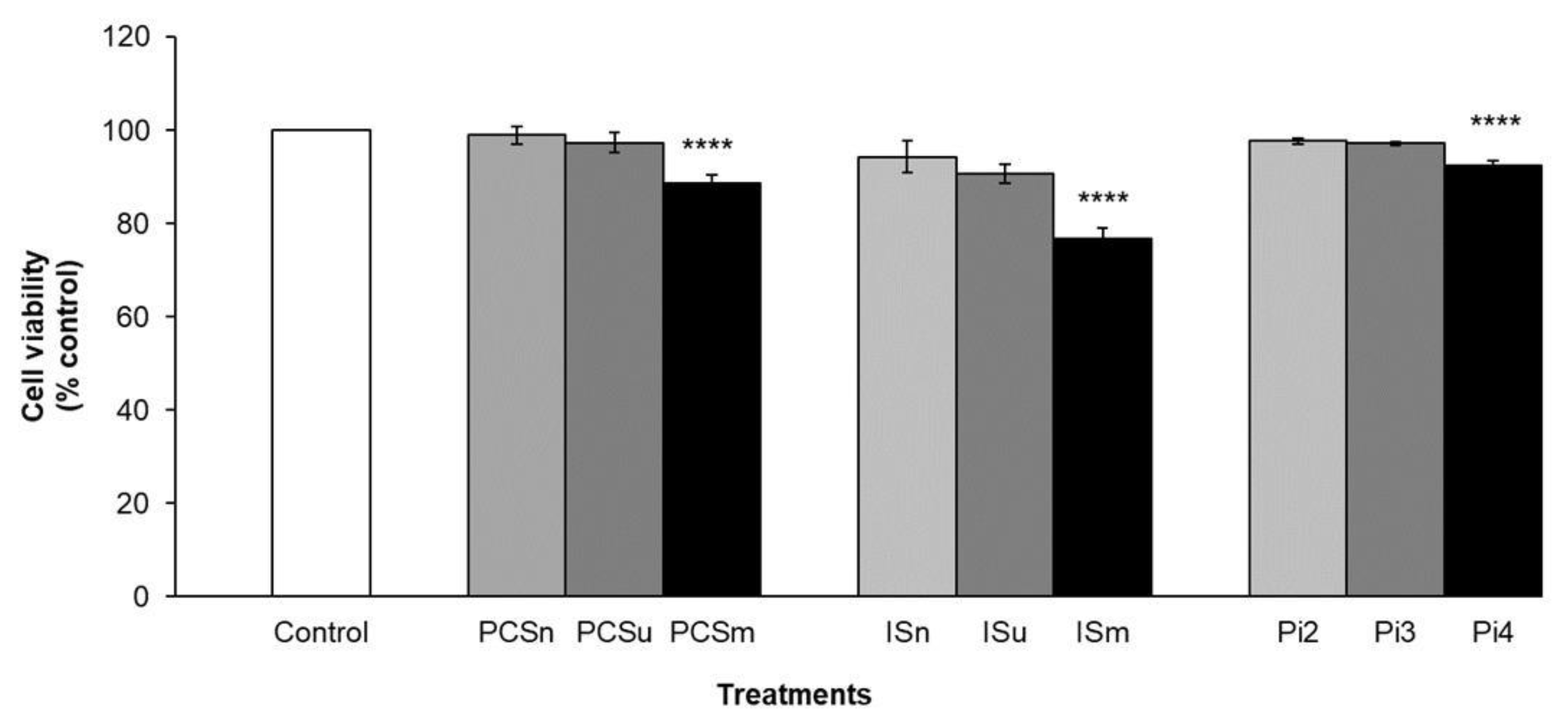
2.7. Cell Viability
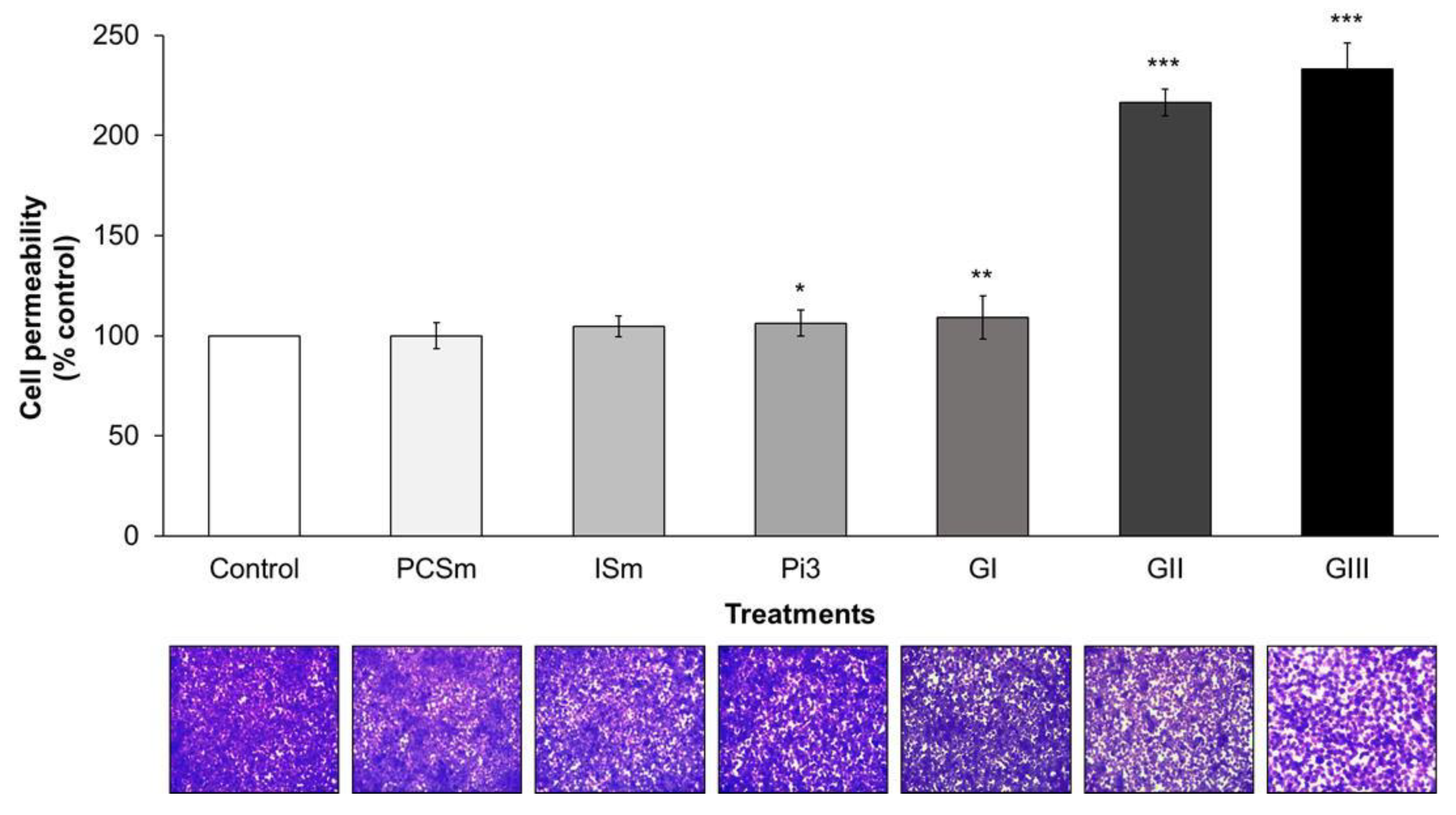
2.8. Uremic Milieu Increases Endothelial Cell Permeability
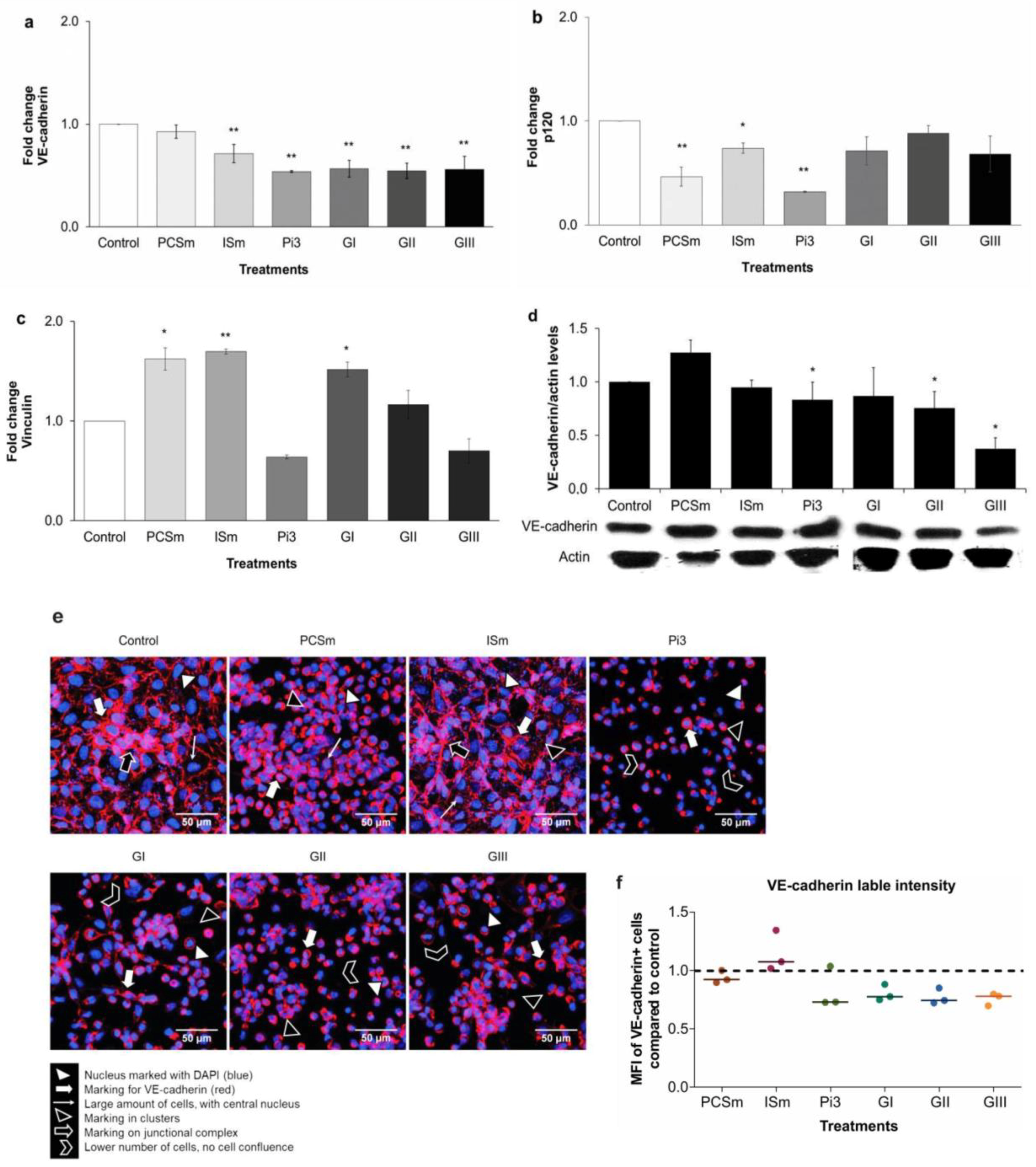
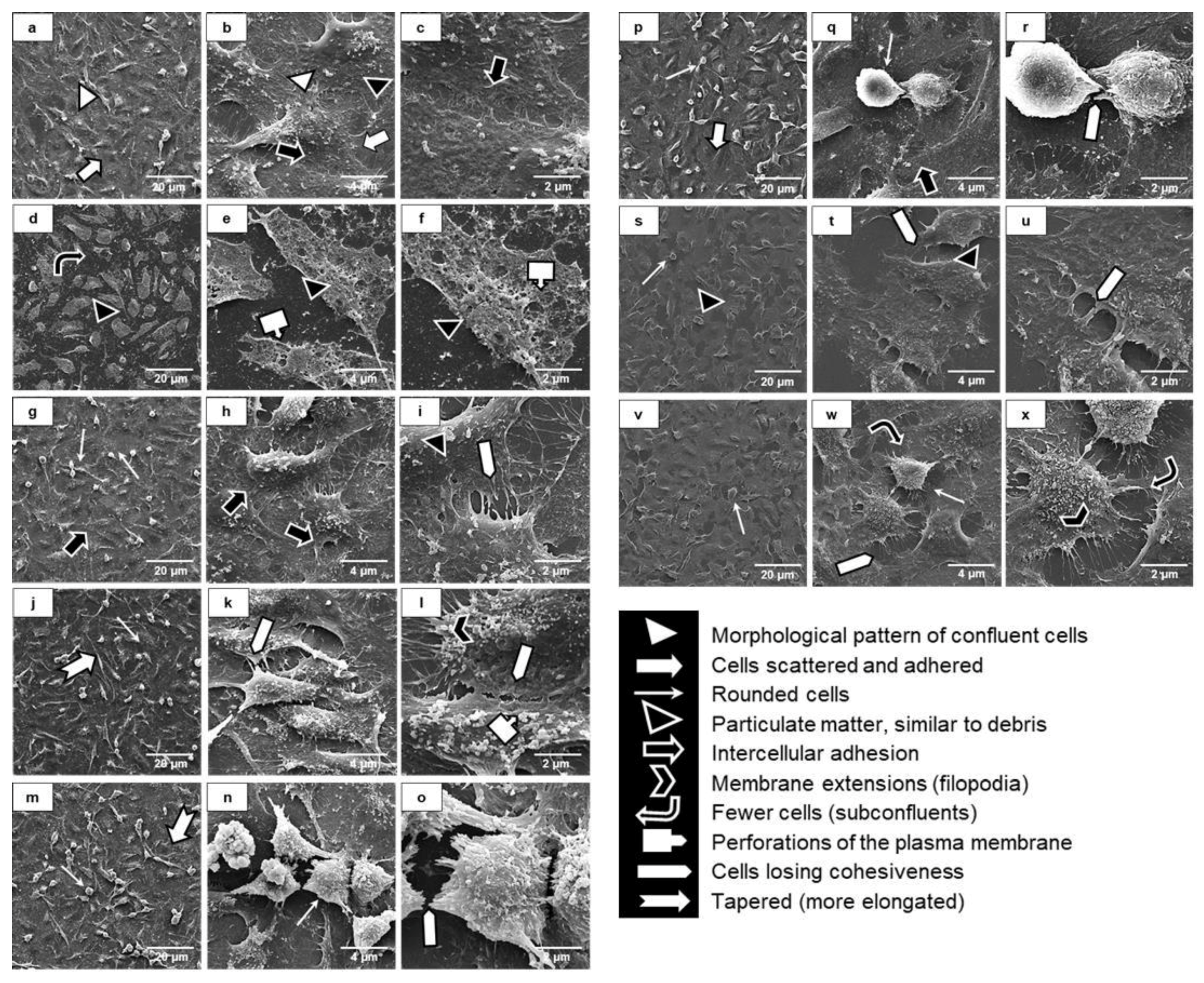
2.9. Uremia Impacts the Intercellular Adhesion and the Endothelial Cell Phenotype
2.10. Uremic Environment Modifies the Endothelial Cell Cytoskeleton
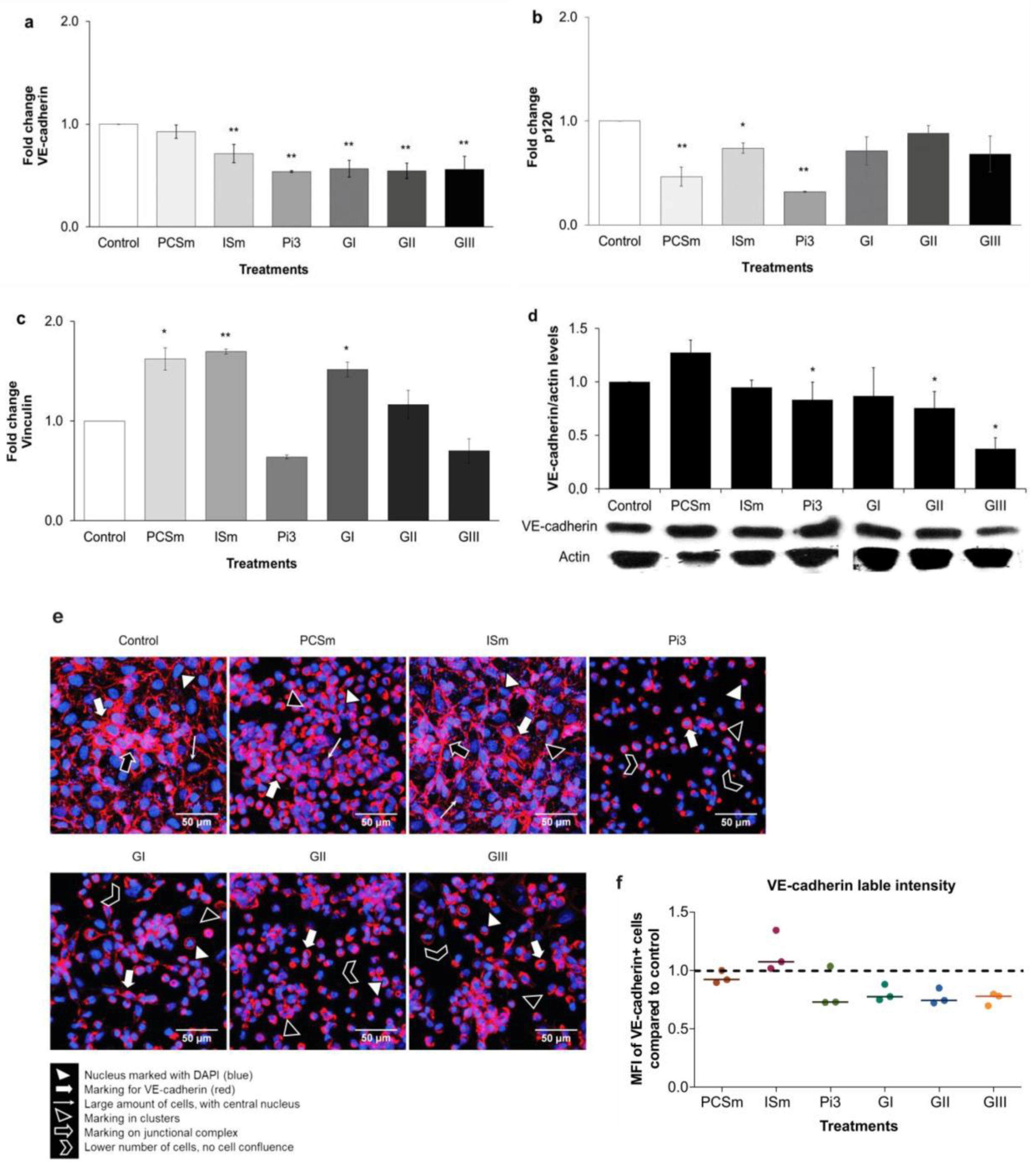
2.11. Uremic Environment Impacts Endothelial Cell Adherent Junction and VE-Cadherin Expression
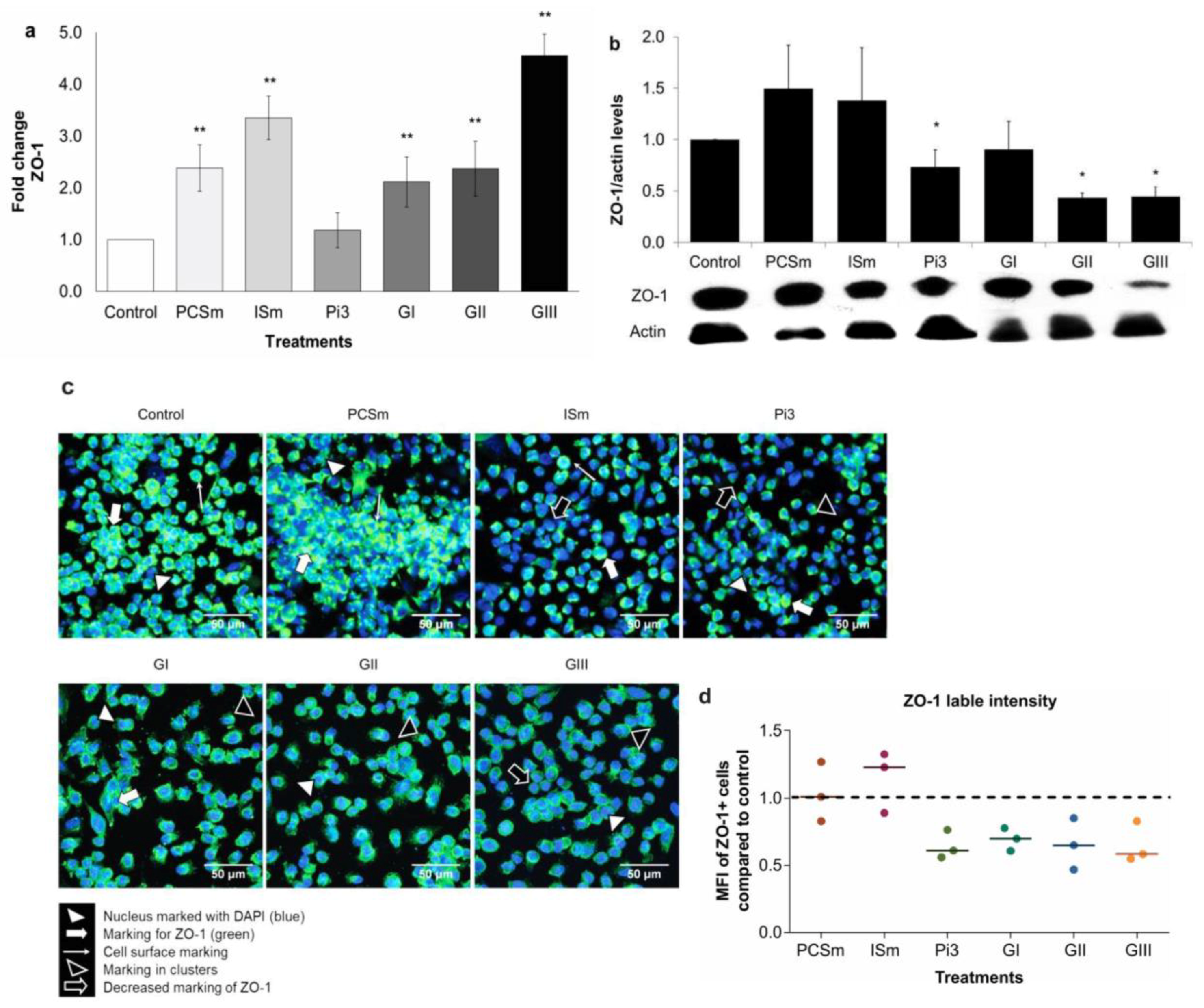
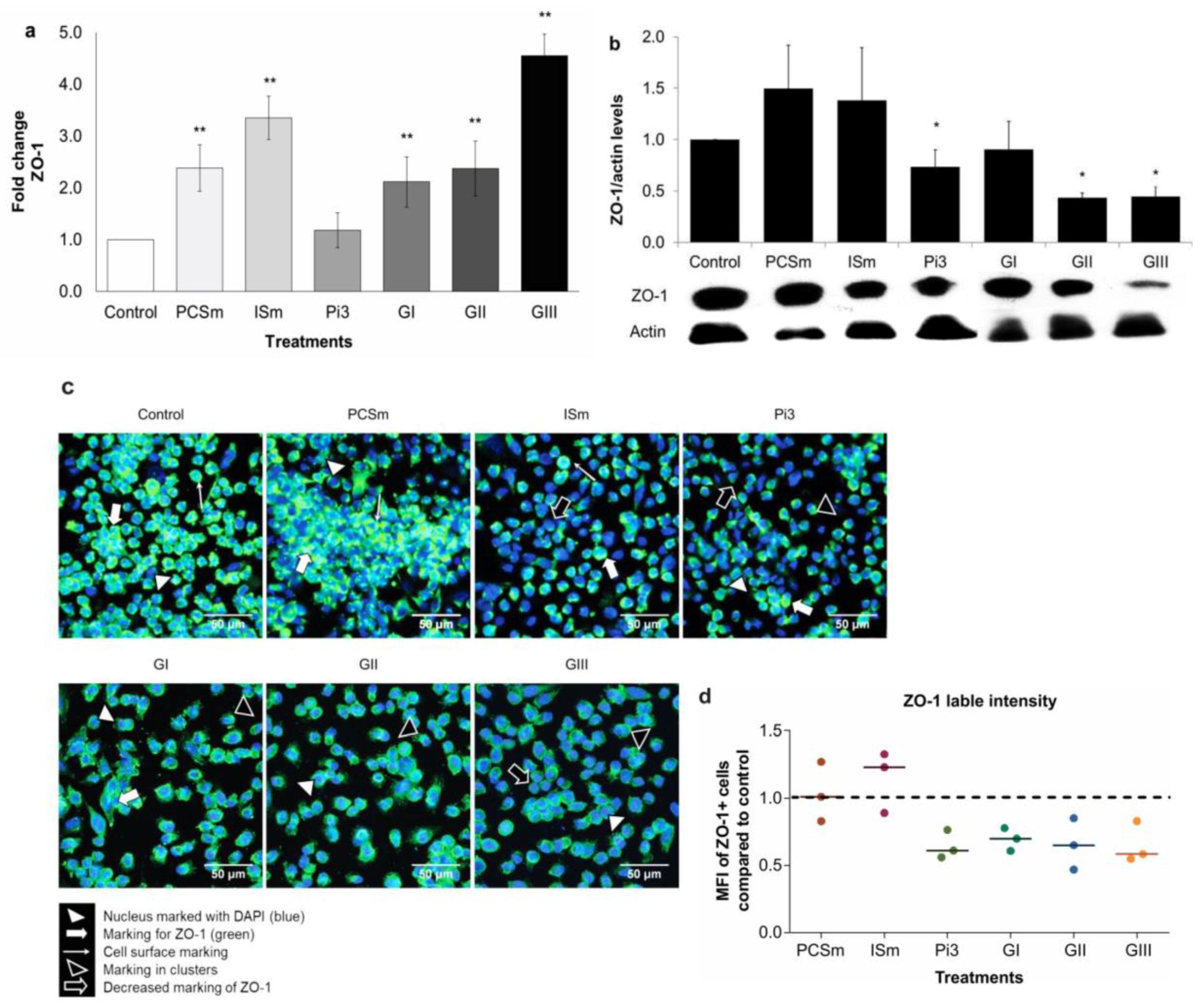
2.12. Uremic Environment Differently Modulates ZO-1 Gene and Protein Expression
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Patients
5.1.1. Patients’ Samples Collection and Processing for In Vivo and In Vitro Assays
5.1.2. Clinical and Biochemical Characteristics of the Patients
5.2. Materials
5.3. Uremic Toxins’ Preparation
5.4. PCS and IS Serum Measurement
5.5. Measurement of MCP-1, IL-8, sVCAM-1, and sICAM-1 Serum Concentrations
5.6. Endothelial Cell Culture and Treatment
5.7. MTT Cell Viability Assay
5.8. Cell Permeability Assay
5.9. Scanning Electron Microscopy (SEM)
5.10. F-actin Staining by Fluorescence Microscopy
5.11. Immunochemical Analysis of VE-Cadherin and ZO-1 On Arteries
5.12. VE-Cadherin and ZO-1 Gene Expression
5.13. VE-Cadherin and ZO-1 Western Blot Analysis
5.14. VE-Cadherin and ZO-1 Immunofluorescence Analysis
5.15. VE-Cadherin and ZO-1 Flow Cytometry Analysis
5.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Suliman, M.E.; Qureshi, A.R.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P. Soluble adhesion molecules in end-stage renal disease: A predictor of outcome. Nephrol. Dial. Transplant. 2006, 21, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Maciel, R.A.P.; Rempel, L.C.T.; Bosquetti, B.; Finco, A.B.; Pecoits-Filho, R.; de Souza, W.M.; Stinghen, A.E.M. p-cresol but not p-cresyl sulfate stimulate MCP-1 production via NF-κB p65 in human vascular smooth muscle cells. J. Bras. Nefrol. 2016, 38, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.M.; Gonçalves, S.M.; Martines, E.G.; Nakao, L.S.; Riella, M.C.; Aita, C.A.; Pecoits-Filho, R. Increased plasma and endothelial cell expression of chemokines and adhesion molecules in chronic kidney disease. Nephron Clin. Pract. 2009, 111, c117–c126. [Google Scholar] [CrossRef] [PubMed]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; De Loor, H.; Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-cresyl sulfate and indoxyl sulfate in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y. Sen; Lin, Y.T.; Chen, Y.; Hung, K.Y.; Wang, S.M. Effects of indoxyl sulfate on adherens junctions of endothelial cells and the underlying signaling mechanism. J. Cell. Biochem. 2012, 113, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y. Sen; Ding, H.C.; Lin, Y.T.; Syu, J.P.; Chen, Y.; Wang, S.M. Uremic toxin p-cresol induces disassembly of gap junctions of cardiomyocytes. Toxicology 2012, 302, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [PubMed]
- Boda-Heggemann, J.; Régnier-Vigouroux, A.; Franke, W.W. Beyond vessels: Occurrence and regional clustering of vascular endothelial (VE-) cadherin-containing junctions in non-endothelial cells. Cell Tissue Res. 2009, 335, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G.; Dejana, E. Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb. Res. 2007, 120, S1–S6. [Google Scholar] [CrossRef]
- Kourtidis, A.; Ngok, S.P.; Anastasiadis, P.Z. P120 catenin: An essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Prog. Mol. Biol. Transl. Sci. 2013, 116, 409–432. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Nyqvist, D.; Orsenigo, F.; Caprini, A.; Giampietro, C.; Taketo, M.M.; Iruela-Arispe, M.L.; Adams, R.H.; Dejana, E. The Wnt/β-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/notch signaling. Dev. Cell 2010, 18, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Huveneers, S.; Oldenburg, J.; Spanjaard, E.; van der Krogt, G.; Grigoriev, I.; Akhmanova, A.; Rehmann, H.; de Rooij, J. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J. Cell Biol. 2012, 196, 641–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umeda, K.; Matsui, T.; Nakayama, M.; Furuse, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Establishment and characterization of cultured epithelial cells lacking expression of ZO-1. J. Biol. Chem. 2004, 279, 44785–44794. [Google Scholar] [CrossRef] [PubMed]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.B.; Azevedo, I.R.F.; Gonçalves, S.; Stinghen, A.; Aita, C.; Pecoits-Filho, R. Sevelamer carbonate reduces inflammation and endotoxemia in an animal model of uremia. Blood Purif. 2010, 30, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Rempel, L.C.T.; Finco, A.B.; Maciel, R.A.P.; Bosquetti, B.; Alvarenga, L.M.; Souza, W.M.; Pecoits-Filho, R.; Stinghen, A.E.M. Effect of PKC-β signaling pathway on expression of MCP-1 and VCAM-1 in different cell models in response to advanced glycation end products (AGEs). Toxins 2015, 7, 1722–1737. [Google Scholar] [CrossRef] [PubMed]
- Aznar-Salatti, J.; Escolar, G.; Cases, A.; Gomez-Ortiz, G.; Anton, P.; Castillo, R.; Revert, L.; Ordinas, A. Uraemic medium causes endothelial cell dysfunction characterized by an alteration of the properties of its subendothelial matrix. Nephrol. Dial. Transplant. 1995, 10, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Selzman, C.H.; Miller, S.A.; Zimmerman, M.A.; Gamboni-Robertson, F.; Harken, A.H.; Banerjee, A. Monocyte chemotactic protein-1 directly induces human vascular smooth muscle proliferation. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1455–H1461. [Google Scholar] [CrossRef] [PubMed]
- Gregório, P.C.; Favretto, G.; Sassaki, G.L.; Cunha, R.S.; Becker-Finco, A.; Pecoits-Filho, R.; Souza, W.M.; Barreto, F.C.; Stinghen, A.E.M. Sevelamer reduces endothelial inflammatory response to advanced glycation end products. Clin. Kidney J. 2018, 11, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z. a Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Gross, P.; Massy, Z.A.; Henaut, L.; Boudot, C.; Cagnard, J.; March, C.; Kamel, S.; Drueke, T.B.; Six, I. Para-Cresyl Sulfate Acutely Impairs Vascular Reactivity and Induces Vascular Remodeling. J. Cell. Physiol. 2015, 230, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-A.; Lu, L.-F.; Yu, T.-H.; Hung, W.-C.; Chung, F.-M.; Tsai, I.-T.; Yang, C.-Y.; Hsu, C.-C.; Lu, Y.-C.; Wang, C.-P.; et al. Increased levels of total P-Cresylsulphate and indoxyl sulphate are associated with coronary artery disease in patients with diabetic nephropathy. Rev. Diabet. Stud. 2010, 7, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Evenepoel, P.; Keuleers, H.; Verbeke, K.; Vanrenterghem, Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006, 69, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B. K. I.; Bammens, B.; De Moor, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. 2008, 73, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.; Chillon, J.-M.; Massy, Z.; Boullier, A. Differential Effects of Indoxyl Sulfate and Inorganic Phosphate in a Murine Cerebral Endothelial Cell Line (bEnd.3). Toxins 2014, 6, 1742–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Six, I.; Gross, P.; Rémond, M. C.; Chillon, J.M.; Poirot, S.; Drueke, T.B.; Massy, Z.A. Deleterious vascular effects of indoxyl sulfate and reversal by oral adsorbent AST-120. Atherosclerosis 2015, 243, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transplant. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzolino, M.; Pasho, S.; Fallabrino, G.; Olivi, L.; Gallieni, M.; Brancaccio, D. Pathogenesis of secondary hyperparathyroidism. Int. J. Artif. Organs 2009, 32, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M. Fibroblast growth factor 23 and the future of phosphorus management. Curr. Opin. Nephrol. Hypertens. 2009, 18, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Román-García, P.; Carrillo-López, N.; Cannata-Andía, J.B. Pathogenesis of bone and mineral related disorders in chronic kidney disease: Key role of hyperphosphatemia. J. Ren. Care 2009, 35, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Escolar, G.; Díaz-Ricart, M.; Cases, A.; Castillo, R.; Ordinas, A.; White, J.G. Abnormal Cytoskeletal Assembly in Platelets from Uremic Patients. Am. J. Pathol. 1993, 143, 823–831. [Google Scholar] [PubMed]
- Madsen, M.; Aarup, A.; Albinsson, S.; Hartvigsen, K.; Sørensen, C.M.; Turczynska, K.; Nielsen, L.B.; Pedersen, T.X. Uremia modulates the phenotype of aortic smooth muscle cells. Atherosclerosis 2017, 257, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Monroy, M.A.; Fang, J.; Li, S.; Ferrer, L.; Birkenbach, M.P.; Lee, I.J.; Wang, H.; Yang, X.F.; Choi, E.T. Chronic kidney disease alters vascular smooth muscle cell phenotype. Front. Biosci. (Landmark Ed) 2015, 20, 784–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzoni, G. Pathobiology of junctional adhesion molecules. Antioxid. Redox Signal. 2011, 15, 1221–1234. [Google Scholar] [CrossRef] [PubMed]
- Crosby, C.V.; Fleming, P.A.; Argraves, W.S.; Corada, M.; Zanetta, L.; Dejana, E.; Drake, C.J. VE-cadherin is not required for the formation of nascent blood vessels but acts to prevent their disassembly. Blood 2005, 105, 2771–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Lampugnani, M.G.; Moons, L.; Breviario, F.; Compernolle, V.; Bono, F.; Balconi, G.; Spagnuolo, R.; Oosthuyse, B.; Dewerchin, M.; et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 1999, 98, 147–157. [Google Scholar] [CrossRef]
- Yan, Y.; Chang, Q.; Li, Q.; Li, L.; Wang, S.; Du, R.; Hu, X. Identification of plasma vascular endothelia-cadherin as a biomarker for coronary artery disease in type 2 diabetes mellitus patients. Int. J. Clin. Exp. Med. 2015, 8, 19466–19470. [Google Scholar] [PubMed]
- Yuan, J.; Guo, Q.; Qureshi, A.R.; Anderstam, B.; Eriksson, M.; Heimbürger, O.; Bárány, P.; Stenvinkel, P.; Lindholm, B. Circulating vascular endothelial growth factor (VEGF) and its soluble receptor 1 (sVEGFR-1) are associated with inflammation and mortality in incident dialysis patients. Nephrol. Dial. Transplant. 2013, 28, 2356–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Goshtasbi, N.; Yuan, J.; Jellbauer, S.; Moradi, H.; Raffatellu, M.; Kalantar-Zadeh, K. Uremic plasma impairs barrier function and depletes the tight junction protein constituents of intestinal epithelium. Am. J. Nephrol. 2012, 36, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Yuan, J.; Khazaeli, M.; Masuda, Y.; Ichii, H.; Liu, S. Oral activated charcoal adsorbent (AST-120) ameliorates chronic kidney disease-induced intestinal epithelial barrier disruption. Am. J. Nephrol. 2013, 37, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Yuan, J.; Rahimi, A.; Ni, Z.; Said, H.; Subramanian, V.S. Disintegration of colonic epithelial tight junction in uremia: A likely cause of CKD-associated inflammation. Nephrol. Dial. Transplant. 2012, 27, 2686–2693. [Google Scholar] [CrossRef] [PubMed]
- Umeda, K.; Ikenouchi, J.; Katahira-Tayama, S.; Furuse, K.; Sasaki, H.; Nakayama, M.; Matsui, T.; Tsukita, S.; Furuse, M.; Tsukita, S. ZO-1 and ZO-2 Independently Determine Where Claudins Are Polymerized in Tight-Junction Strand Formation. Cell 2006, 126, 741–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, H.; Zweimueller-Mayer, J.; Steinbacher, P.; Lametschwandtner, A.; Bauer, H.C. The dual role of zonula occludens (ZO) proteins. J. Biomed. Biotechnol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F.; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Inker, L.A.; Astor, B.C.; Fox, C.H.; Isakova, T.; Lash, J.P.; Peralta, C.A.; Kurella Tamura, M.; Feldman, H.I. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am. J. Kidney Dis. 2014, 63, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Feigenbaum, J.; Neuberg, C.A. Simplified Method for the preparation of aromatic sulfuric acid esters. Notes 1941, 63, 3529–3530. [Google Scholar] [CrossRef]
- Favretto, G.; Souza, L.M.; Gregório, P.C.; Cunha, R.S.; MacIel, R.A.P.; Sassaki, G.L.; Toledo, M.G.; Pecoits-Filho, R.; Souza, W.M.; Stinghen, A.E.M. Role of Organic Anion Transporters in the Uptake of Protein-Bound Uremic Toxins by Human Endothelial Cells and Monocyte Chemoattractant Protein-1 Expression. J. Vasc. Res. 2017, 54, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Ise, M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J. Lab. Clin. Med. 1994, 124, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Mazière, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockler-Pinto, M.B.; Soulage, C.O.; Borges, N.A.; Cardozo, L.F.M.F.; Dolenga, C.J.; Nakao, L.S.; Pecoits-Filho, R.; Fouque, D.; Mafra, D. From bench to the hemodialysis clinic: Protein-bound uremic toxins modulate NF-κB/Nrf2 expression. Int. Urol. Nephrol. 2018, 50, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Meert, N.; Schepers, E.; Glorieux, G.; Van Landschoot, M.; Goeman, J.L.; Waterloos, M.-A.; Dhondt, A.; Van der Eycken, J.; Vanholder, R. Novel method for simultaneous determination of p-cresylsulphate and p-cresylglucuronide: Clinical data and pathophysiological implications. Nephrol. Dial. Transplant. 2012, 27, 2388–2396. [Google Scholar] [CrossRef] [PubMed]
- de Loor, H.; Meijers, B.K.I.; Meyer, T.W.; Bammens, B.; Verbeke, K.; Dehaen, W.; Evenepoel, P. Sodium octanoate to reverse indoxyl sulfate and p-cresyl sulfate albumin binding in uremic and normal serum during sample preparation followed by fluorescence liquid chromatography. J. Chromatogr. A 2009, 1216, 4684–4688. [Google Scholar] [CrossRef] [PubMed]
- Calaf, R.; Cerini, C.; Génovésio, C.; Verhaeghe, P.; Jourde-Chiche, N.; Bergé-Lefranc, D.; Gondouin, D.; Dou, L.; Morange, S.; Argilésg, A.; et al. Determination of uremic solutes in biological fluids of chronic kidney disease patients by HPLC assay. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Edgell, C.J.; McDonald, C.C.; Graham, J.B. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc. Natl. Acad. Sci. USA 1983, 80, 3734–3737. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ma, C.; Wang, X.F. In vitro assays for the extracellular matrix protein-regulated extravasation process. Cold Spring Harb. Protoc. 2008, 8, pdb–prot5034. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, F.S.F.; Abud, A.P.R.; Oliveira, S.M.; Oliveira, C.C.; César, B.; Andrade, L.F.; Donatti, L.; Gabardo, J.; Trindade, E.S.; Buchi, D.F. Stimulation of lymphocyte anti-melanoma activity by co-cultured macrophages activated by complex homeopathic medication. BMC Cancer 2009, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- Guilgen, G.; Werneck, M.L.; De Noronha, L.; Martins, A.P.C.; Varela, A.M.; Nakao, L.S.; Pecoits-Filho, R. Increased calcification and protein nitration in arteries of chronic kidney disease patients. Blood Purif. 2011, 32, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: M inimum I nformation for Publication of Q uantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The digital MIQE guidelines: Minimum information for publication of quantitative digital PCR experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Chang, H.M.; Leung, P.C.K. Transforming growth factor-β1 inhibits trophoblast cell invasion by inducing snail-mediated down-regulation of vascular endothelial-cadherin protein. J. Biol. Chem. 2013, 288, 33181–33192. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.E.; Torr, E.E.; Mohd Jamili, N.H.; Bosquillon, C.; Sayers, I. Evaluation of Differentiated Human Bronchial Epithelial Cell Culture Systems for Asthma Research. J. Allergy 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, J.; Flozak, A.S.; Albuquerque, M.L.C. Laminar Shear Stress Differentially Modulates Gene Expression of p120 Catenin, Kaiso Transcription Factor, and Vascular Endothelial Cadherin in Human Coronary Artery Endothelial Cells. J. Biol. Chem. 2004, 279, 11417–11424. [Google Scholar] [CrossRef] [PubMed]
- Stein, U.; Arlt, F.; Smith, J.; Sack, U.; Herrmann, P.; Walther, W.; Lemm, M.; Fichtner, I.; Shoemaker, R.H.; Schlag, P.M. Intervening in β-Catenin Signaling by Sulindac Inhibits S100A4-Dependent Colon Cancer Metastasis. Neoplasia 2011, 13, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Mayanagi, T.; Morita, T.; Hayashi, K.; Fukumoto, K.; Sobue, K. Glucocorticoid receptor-mediated expression of caldesmon regulates cell migration via the reorganization of the actin cytoskeleton. J. Biol. Chem. 2008, 283, 31183–31196. [Google Scholar] [CrossRef] [PubMed]
- Figueira, R.C.S.; Gomes, L.R.; Neto, J.S.; Silva, F.C.; Silva, I.D.C.G.; Sogayar, M.C. Correlation between MMPs and their inhibitors in breast cancer tumor tissue specimens and in cell lines with different metastatic potential. BMC Cancer 2009, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyzed Parameters | Mean Or Percent |
|---|---|
| Patients (n) | 80 |
| Traditional risk factors | |
| Mean age ± SEM, years | 62.7 ± 1.3 |
| Gender, % male | 61.0 |
| Race, % caucasians | 86.0 |
| Smoking, % | 33.0 |
| Alcoholism, % | 14.0 |
| Diabetes mellitus, % | 43.0 |
| Hypertension, % | 79.0 |
| Dyslipidemia, % | 56.3 |
| Primary kidney disease | |
| Nephrosclerosis, % | 30.0 |
| Diabetic nephropathy, % | 21.3 |
| Chronic glomerulonephritis, % | 10.0 |
| Polycystic kidney disease, % | 12.5 |
| Others and unknown, % | 26.2 |
| Analyzed Parameters | Media ± SEM | Range |
|---|---|---|
| Biochemical characterization | ||
| Uric acid, mg/dL | 7.0 ± 2.0 | 1.7–15.6 |
| Albumin, g/dL | 4.2 ± 0.6 | 2.2–5.7 |
| Calcium, mg/dL | 8.6 ± 3.0 | 1.0–12.7 |
| Creatinine, mg/dL | 2.3 ± 1.5 | 0.5–7.6 |
| Glucose, mg/dL | 116.0 ± 52.1 | 53.0–284.0 |
| Potassium, mmol/L | 5.0 ± 0.6 | 3.5–7.0 |
| Sodium, mmol/L | 141.0 ± 4.5 | 132.0–158.0 |
| Urea, mg/dL | 77.0 ± 40.6 | 13.0–189.0 |
| PCS, mg/L | 20.5 ± 19.0 | 0.01–92.35 |
| IS, mg/L | 4.6 ± 6.5 | 0.25–40.26 |
| Pi, mg/dL | 4.4 ± 1.1 | 2.4–9.9 |
| Analyzed Parameters | GI (n = 7) Latent CKD | GII (n = 42) Mild CKD | GIII (n = 31) Severe CKD |
|---|---|---|---|
| Traditional risk factors | |||
| Smoking, % | 52 | 36 | 45 |
| Alcoholism, % | 22 | 12 | 26 |
| Diabetes mellitus, % | 43 | 32 | 58 |
| Hypertension, % | 78 | 100 | 94 |
| Dyslipidemia, % | 57 | 56 | 58 |
| CVD, % | 43 | 36 | 55 |
| Biochemical characterization | |||
| Uric acid (mg/dL) | 6.1 | 7.0 | 7.7 |
| Albumin (g/dL) | 4.5 | 4.2 | 4.0 |
| Calcium (mg/dL) | 9.7 | 7.7 | 8.6 |
| Creatinine (mg/dL) | 1.0 | 1.6 | 3.7 |
| Glucose (mg/dL) | 137.1 | 95.3 | 120.8 |
| hs-CRP (mg/L) | 5.1 | 5.9 | 7.4 |
| Potassium (mmol/L) | 5.0 | 5.0 | 5.0 |
| Sodium (mmol/L) | 141.0 | 142.0 | 139.0 |
| Urea (mg/dL) | 42.3 | 63.9 | 114.0 |
| PCS (mg/L) | 11.1 | 15.4 | 33.9 |
| IS (mg/L) | 1.5 | 2.1 | 9.6 |
| Pi (mg/dL) | 4.1 | 4.1 | 5.0 |
| Target Gene | Primers | Amplicon |
|---|---|---|
| VE-cadherin | 5’-CAGCCCAAAGTGTGTGAGAA-3’ (F) 5’-CGGTCAAACTGCCCATACTT-3’ (R) | 185 pb |
| ZO-1 | 5’-GCGGTCAGAGCCTTCTGATC-3’ (F) 5’-CATGCTTTACAGGAGTTGAGACAG-3’ (R) | 122 pb |
| p120 | 5’-GATGCTGTCAAGTCCAATGCAG-3’ (F) 5’-AGTACTGGGATGCCCTTGAGC-3’ (R) | 101 pb |
| β-catenin | 5’-GTGCTATCTGTCTGCTCTAGTA-3’ (F) 5’-CTTCCTGTTTAGTTGCAGCATC-3’ (R) | 154 pb |
| Vinculin | 5’-TCAGATGAGGTGACTCGGTTGG-3’ (F) 5’-GGGTGCTTATGGTTGGGATTCG-3’ (R) | 109 pb |
| HPRT | 5’-GAACGTCTTGCTCGAGATGTGA-3’ (F) 5’-TCCAGCAGGTCAGCAAAGAAT-3’ (R) | 101 pb |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maciel, R.A.P.; Cunha, R.S.; Busato, V.; Franco, C.R.C.; Gregório, P.C.; Dolenga, C.J.R.; Nakao, L.S.; Massy, Z.A.; Boullier, A.; Pecoits-Filho, R.; et al. Uremia Impacts VE-Cadherin and ZO-1 Expression in Human Endothelial Cell-to-Cell Junctions. Toxins 2018, 10, 404. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10100404
Maciel RAP, Cunha RS, Busato V, Franco CRC, Gregório PC, Dolenga CJR, Nakao LS, Massy ZA, Boullier A, Pecoits-Filho R, et al. Uremia Impacts VE-Cadherin and ZO-1 Expression in Human Endothelial Cell-to-Cell Junctions. Toxins. 2018; 10(10):404. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10100404
Chicago/Turabian StyleMaciel, Rayana A. P., Regiane S. Cunha, Valentina Busato, Célia R. C. Franco, Paulo C. Gregório, Carla J. R. Dolenga, Lia S. Nakao, Ziad A. Massy, Agnès Boullier, Roberto Pecoits-Filho, and et al. 2018. "Uremia Impacts VE-Cadherin and ZO-1 Expression in Human Endothelial Cell-to-Cell Junctions" Toxins 10, no. 10: 404. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10100404