Purification and Characterization of JZTx-14, a Potent Antagonist of Mammalian and Prokaryotic Voltage-Gated Sodium Channels

 and
and 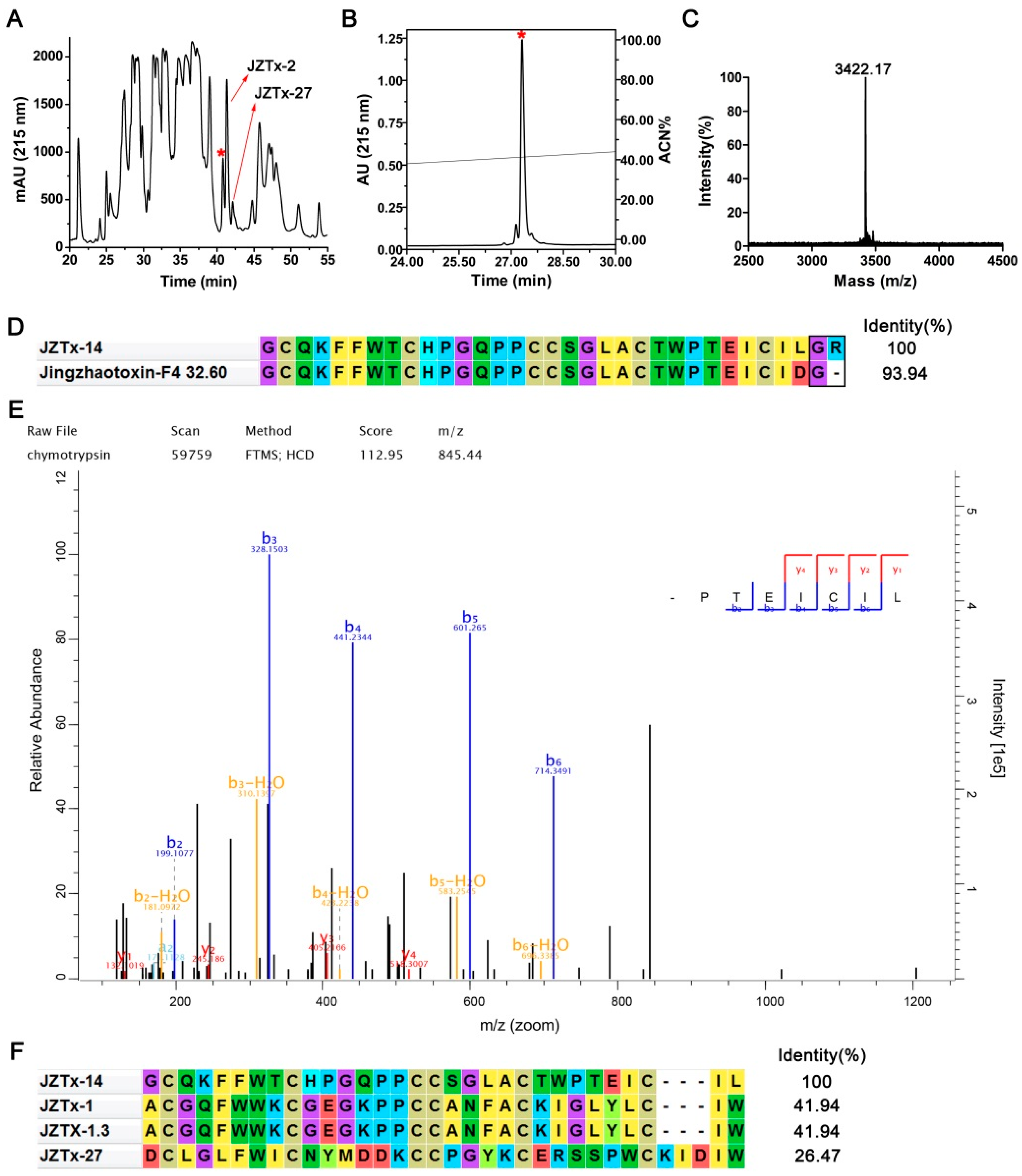{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Purification and Characterization of JZTx-14
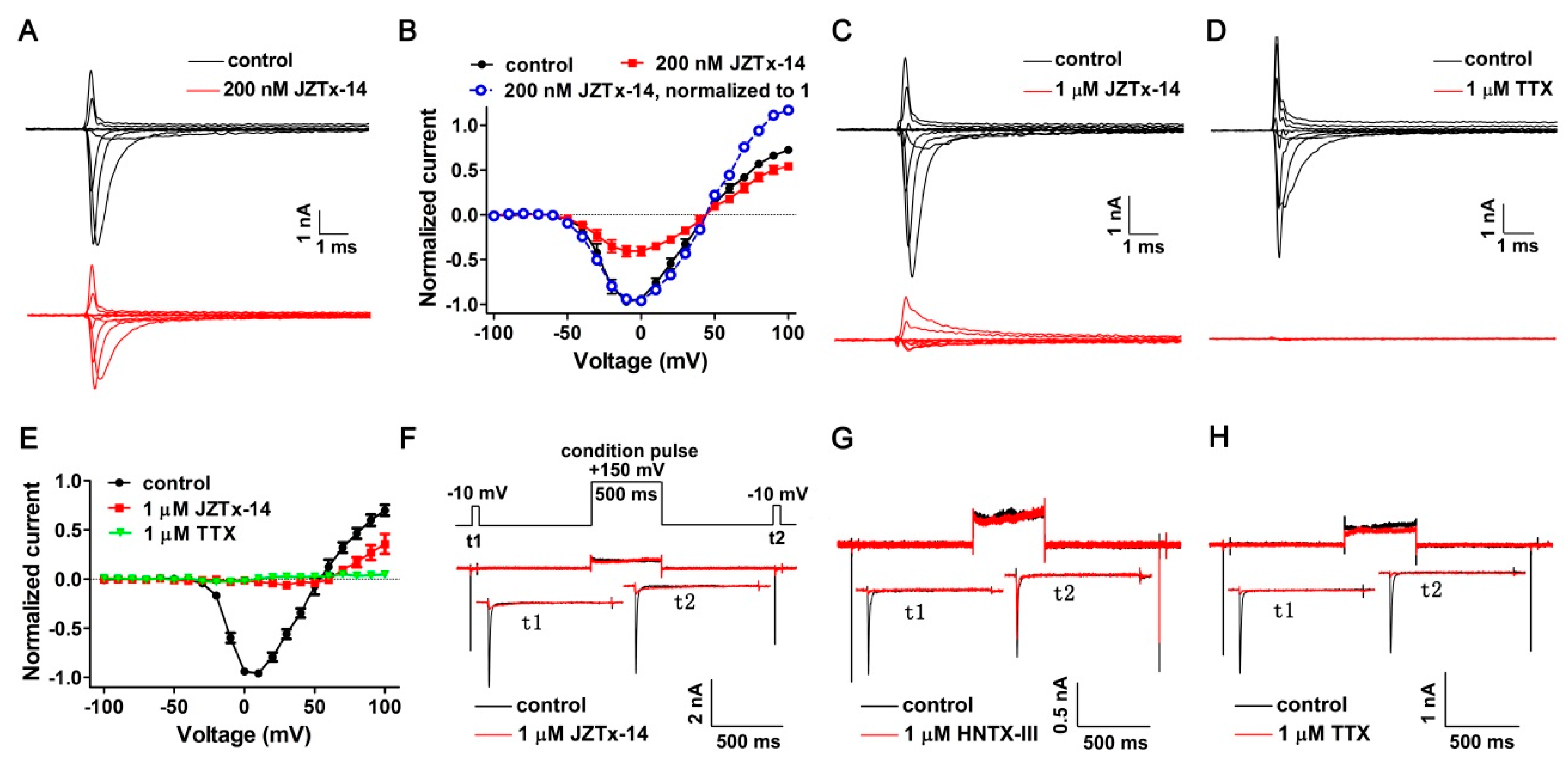
2.2. JZTx-14 Is a Potent but Non-Selective Mammalian NaVs Toxin
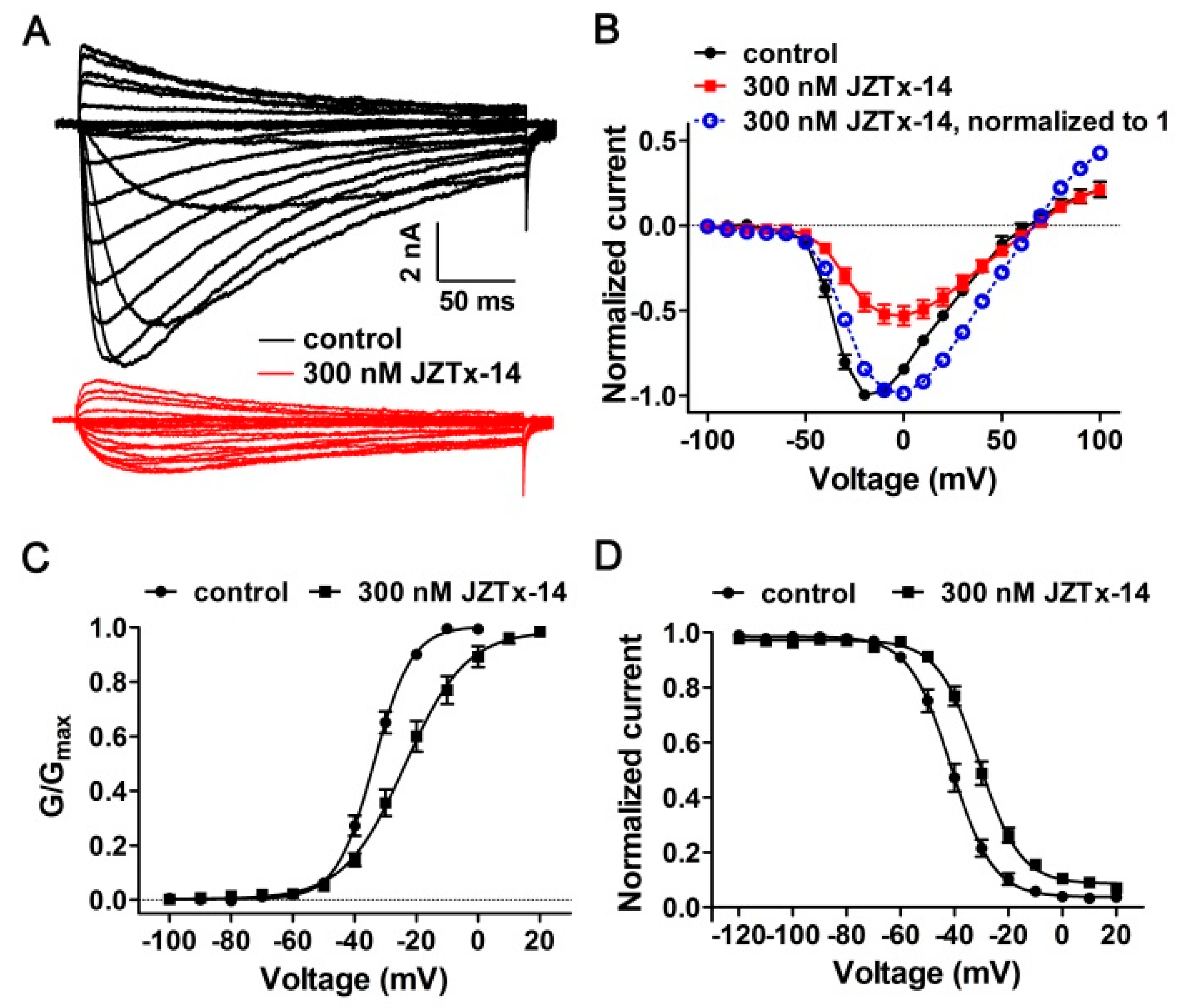
2.3. JZTx-14 Acts on Mammalian NaVs as a Gating Modifier
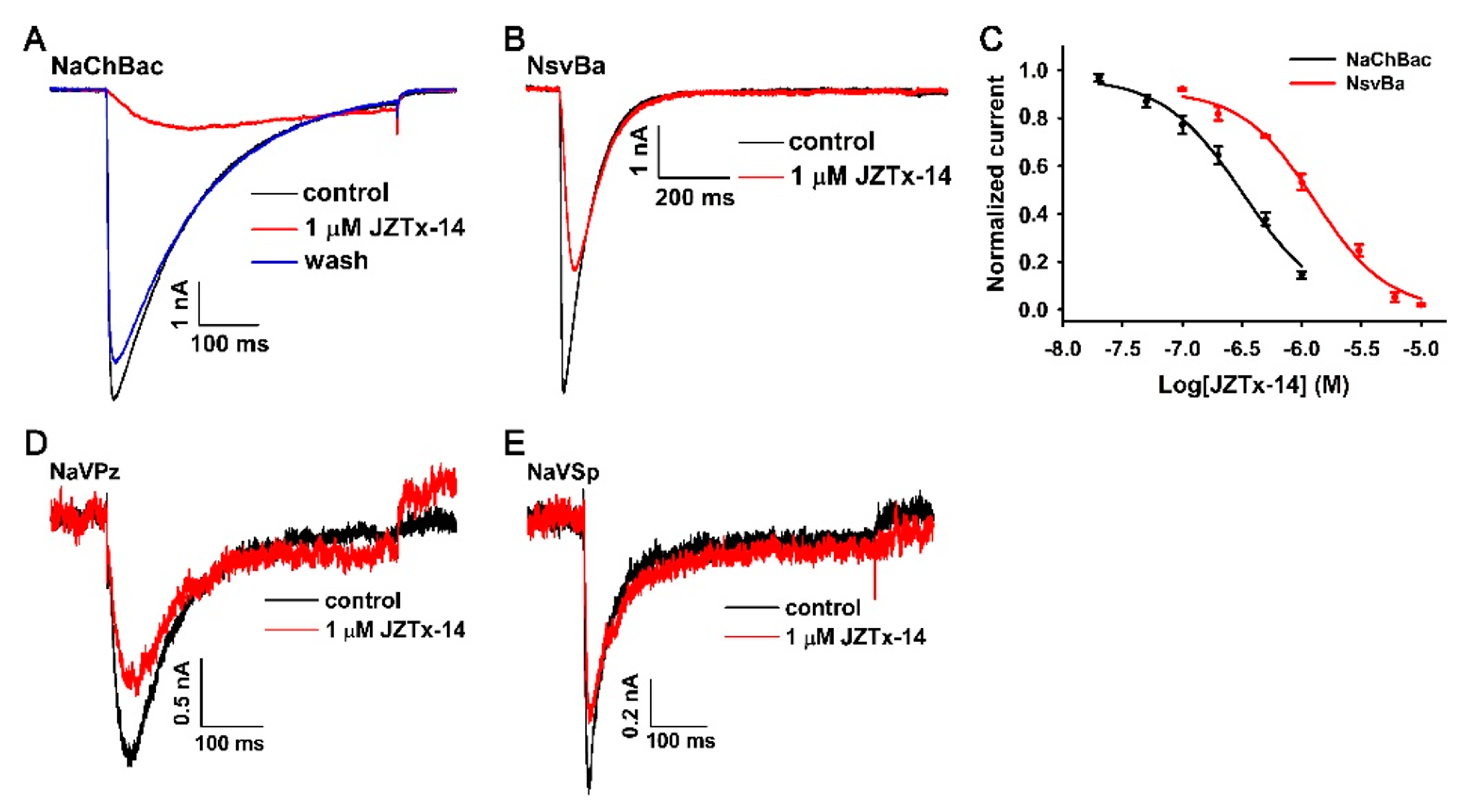
2.4. Effects of JZTx-14 on Bacterial NaVs
2.5. The Molecular Mechanism of JZTx-14 Interacting with NaChBac
3. Discussion
4. Materials and Methods
4.1. Toxin Purification and N-Terminal Sequence Determination
4.2. Mass Spectrometric Analysis and Toxin C-Terminal Sequence Determination
4.3. Constructs and Transfection
4.4. Whole-Cell Patch Clamp Recordings
4.5. Data Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Goldin, A.L. Diversity of mammalian voltage-gated sodium channels. Ann. N. Y. Acad. Sci. 1999, 868, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International union of pharmacology. XlVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Andavan, G.S.; Lemmens-Gruber, R. Voltage-gated sodium channels: Mutations, channelopathies and targets. Curr. Med. Chem. 2011, 18, 377–397. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Waxman, S.G. Noncanonical roles of voltage-gated sodium channels. Neuron 2013, 80, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.Z.; Zhou, Q.; Pan, X.J.; Li, Z.Q.; Wu, J.P.; Yan, N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zhou, Q.; Wang, L.; Wu, J.; Zhao, Y.; Huang, G.; Peng, W.; Shen, H.; Lei, J.; Yan, N. Structure of the Nav1.4-beta1 complex from electric eel. Cell 2017, 170, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science 2018, 2018. [Google Scholar] [CrossRef]
- Gilchrist, J.; Olivera, B.M.; Bosmans, F. Animal toxins influence voltage-gated sodium channel function. Handb. Exp. Pharmacol. 2014, 221, 203–229. [Google Scholar] [PubMed]
- Tang, C.; Zhou, X.; Zhang, Y.; Xiao, Z.; Hu, Z.; Zhang, C.; Huang, Y.; Chen, B.; Liu, Z.; Liang, S. Synergetic action of domain II and IV underlies persistent current generation in Nav1.3 as revealed by a tarantula toxin. Sci. Rep. 2015, 5, 9241. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Cestele, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- Moczydlowski, E.G. The molecular mystique of tetrodotoxin. Toxicon 2013, 63, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Ruben, P.C. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels 2008, 2, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, D.; Navarro, B.; Xu, H.; Yue, L.; Shi, Q.; Clapham, D.E. A prokaryotic voltage-gated sodium channel. Science 2001, 294, 2372–2375. [Google Scholar] [CrossRef] [PubMed]
- Koishi, R.; Xu, H.; Ren, D.; Navarro, B.; Spiller, B.W.; Shi, Q.; Clapham, D.E. A superfamily of voltage-gated sodium channels in bacteria. J. Biol. Chem. 2004, 279, 9532–9538. [Google Scholar] [CrossRef] [PubMed]
- Scheuer, T. Bacterial sodium channels: Models for eukaryotic sodium and calcium channels. Handb. Exp. Pharmacol. 2014, 221, 269–291. [Google Scholar] [PubMed]
- Payandeh, J.; Gamal El-Din, T.M.; Scheuer, T.; Zheng, N.; Catterall, W.A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 2012, 486, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagneris, C.; DeCaen, P.G.; Naylor, C.E.; Pryde, D.C.; Nobeli, I.; Clapham, D.E.; Wallace, B.A. Prokaryotic Navms channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc. Natl. Acad. Sci. USA 2014, 111, 8428–8433. [Google Scholar] [CrossRef] [PubMed]
- McCusker, E.C.; Bagneris, C.; Naylor, C.E.; Cole, A.R.; D’Avanzo, N.; Nichols, C.G.; Wallace, B.A. Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nat. Commun. 2012, 3, 1102. [Google Scholar] [CrossRef] [PubMed]
- Sula, A.; Booker, J.; Ng, L.C.; Naylor, C.E.; DeCaen, P.G.; Wallace, B.A. The complete structure of an activated open sodium channel. Nat. Commun. 2017, 8, 14205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Ren, W.; DeCaen, P.; Yan, C.; Tao, X.; Tang, L.; Wang, J.; Hasegawa, K.; Kumasaka, T.; He, J.; et al. Crystal structure of an orthologue of the nachbac voltage-gated sodium channel. Nature 2012, 486, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Shaya, D.; Findeisen, F.; Abderemane-Ali, F.; Arrigoni, C.; Wong, S.; Nurva, S.R.; Loussouarn, G.; Minor, D.L., Jr. Structure of a prokaryotic sodium channel pore reveals essential gating elements and an outer ion binding site common to eukaryotic channels. J. Mol. Biol. 2014, 426, 467–483. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Gamal El-Din, T.M.; Ing, C.; Lu, P.; Pomes, R.; Zheng, N.; Catterall, W.A. Structural basis for gating pore current in periodic paralysis. Nature 2018, 557, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Lenaeus, M.J.; Gamal El-Din, T.M.; Ing, C.; Ramanadane, K.; Pomes, R.; Zheng, N.; Catterall, W.A. Structures of closed and open states of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. USA 2017, 114, E3051–E3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boiteux, C.; Flood, E.; Allen, T.W. Comparison of permeation mechanisms in sodium-selective ion channels. Neurosci. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Xia, M.; Gong, H. Lysine and the Na+/K+ selectivity in mammalian voltage-gated sodium channels. PLoS ONE 2016, 11, e0162413. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Jih, T.Y.; Zhang, T.T.; Correa, A.M.; Hemmings, H.C., Jr. Isoflurane inhibits nachbac, a prokaryotic voltage-gated sodium channel. J. Pharmacol. Exp. Ther. 2007, 322, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Zhou, X.; Nguyen, P.T.; Zhang, Y.; Hu, Z.; Zhang, C.; Yarov-Yarovoy, V.; DeCaen, P.G.; Liang, S.; Liu, Z. A novel tarantula toxin stabilizes the deactivated voltage sensor of bacterial sodium channel. FASEB J. 2017, 31, 3167–3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Zhang, Y.; Tang, D.; Liang, S.; Chen, P.; Tang, C.; Liu, Z. A chimeric Nav1.8 channel expression system based on hek293t cell line. Front. Pharmacol. 2018, 9, 337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xiao, Z.; Xu, Y.; Zhang, Y.; Tang, D.; Wu, X.; Tang, C.; Chen, M.; Shi, X.; Chen, P.; et al. Electrophysiological and pharmacological analyses of Nav1.9 voltage-gated sodium channel by establishing a heterologous expression system. Front. Pharmacol. 2017, 8, 852. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cai, T.; Zhu, Q.; Deng, M.; Li, J.; Zhou, X.; Zhang, F.; Li, D.; Li, J.; Liu, Y.; et al. Structure and function of hainantoxin-III, a selective antagonist of neuronal tetrodotoxin-sensitive voltage-gated sodium channels isolated from the chinese bird spider ornithoctonus hainana. J. Biol. Chem. 2013, 288, 20392–20403. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Bingham, J.P.; Zhu, W.; Moczydlowski, E.; Liang, S.; Cummins, T.R. Tarantula huwentoxin-iv inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain ii voltage sensor in the closed configuration. J. Biol. Chem. 2008, 283, 27300–27313. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, E.; Cassulini, R.R.; Silva, D.F.; Clement, H.; Schiavon, E.; Zamudio, F.Z.; Odell, G.; Arcangeli, A.; Clare, J.J.; Alagon, A.; et al. Target promiscuity and heterogeneous effects of tarantula venom peptides affecting Na+ and K+ ion channels. J. Biol. Chem. 2010, 285, 4130–4142. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Martin-Eauclaire, M.F.; Swartz, K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 2008, 456, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Blumenthal, K.; Jackson, J.O., 2nd; Liang, S.; Cummins, T.R. The tarantula toxins protx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol. Pharmacol. 2010, 78, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Green, B.R.; Bulaj, G.; Norton, R.S. Structure and function of mu-conotoxins, peptide-based sodium channel blockers with analgesic activity. Future Med. Chem. 2014, 6, 1677–1698. [Google Scholar] [CrossRef] [PubMed]
- Fozzard, H.A.; Lipkind, G.M. The tetrodotoxin binding site is within the outer vestibule of the sodium channel. Mar. Drugs 2010, 8, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Green, B.R.; Catlin, P.; Fiedler, B.; Azam, L.; Chadwick, A.; Terlau, H.; McArthur, J.R.; French, R.J.; Gulyas, J.; et al. Structure/function characterization of micro-conotoxin kiiia, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J. Biol. Chem. 2007, 282, 30699–30706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Yarov-Yarovoy, V.; Scheuer, T.; Karbat, I.; Cohen, L.; Gordon, D.; Gurevitz, M.; Catterall, W.A. Structure-function map of the receptor site for beta-scorpion toxins in domain ii of voltage-gated sodium channels. J. Biol. Chem. 2011, 286, 33641–33651. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Cestele, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlov, E.; Bladen, C.; Winkfein, R.; Diao, C.; Dhaliwal, P.; French, R.J. The pore, not cytoplasmic domains, underlies inactivation in a prokaryotic sodium channel. Biophys. J. 2005, 89, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Irie, K.; Kitagawa, K.; Nagura, H.; Imai, T.; Shimomura, T.; Fujiyoshi, Y. Comparative study of the gating motif and c-type inactivation in prokaryotic voltage-gated sodium channels. J. Biol. Chem. 2010, 285, 3685–3694. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, K.; Wallace, B.A. Nachbac: The long lost sodium channel ancestor. Biochemistry 2011, 50, 6742–6752. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Zheng, N. Deciphering voltage-gated Na+ and Ca2+ channels by studying prokaryotic ancestors. Trends Biochem. Sci. 2015, 40, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Goodchild, S.J.; Ahern, C.A. Local anesthetic inhibition of a bacterial sodium channel. J. Gen. Physiol. 2012, 139, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.T.; Zhou, X.; Chen, J.; Tang, C.; Xiao, Z.; Ying, D.Z.; Liu, Z.H.; Liang, S.P. The venom of the spider selenocosmia jiafu contains various neurotoxins acting on voltage-gated ion channels in rat dorsal root ganglion neurons. Toxins 2014, 6, 988–1001. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Tang, D.; Liu, S.; Hu, H.; Liang, S.; Tang, C.; Liu, Z. Purification and Characterization of JZTx-14, a Potent Antagonist of Mammalian and Prokaryotic Voltage-Gated Sodium Channels. Toxins 2018, 10, 408. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10100408
Zhang J, Tang D, Liu S, Hu H, Liang S, Tang C, Liu Z. Purification and Characterization of JZTx-14, a Potent Antagonist of Mammalian and Prokaryotic Voltage-Gated Sodium Channels. Toxins. 2018; 10(10):408. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10100408
Chicago/Turabian StyleZhang, Jie, Dongfang Tang, Shuangyu Liu, Haoliang Hu, Songping Liang, Cheng Tang, and Zhonghua Liu. 2018. "Purification and Characterization of JZTx-14, a Potent Antagonist of Mammalian and Prokaryotic Voltage-Gated Sodium Channels" Toxins 10, no. 10: 408. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10100408