Evaluation of Aflatoxin M1 Effects on the Metabolomic and Cytokinomic Profiling of a Hepatoblastoma Cell Line


 and
and
Abstract
:1. Introduction
2. Results and Discussion
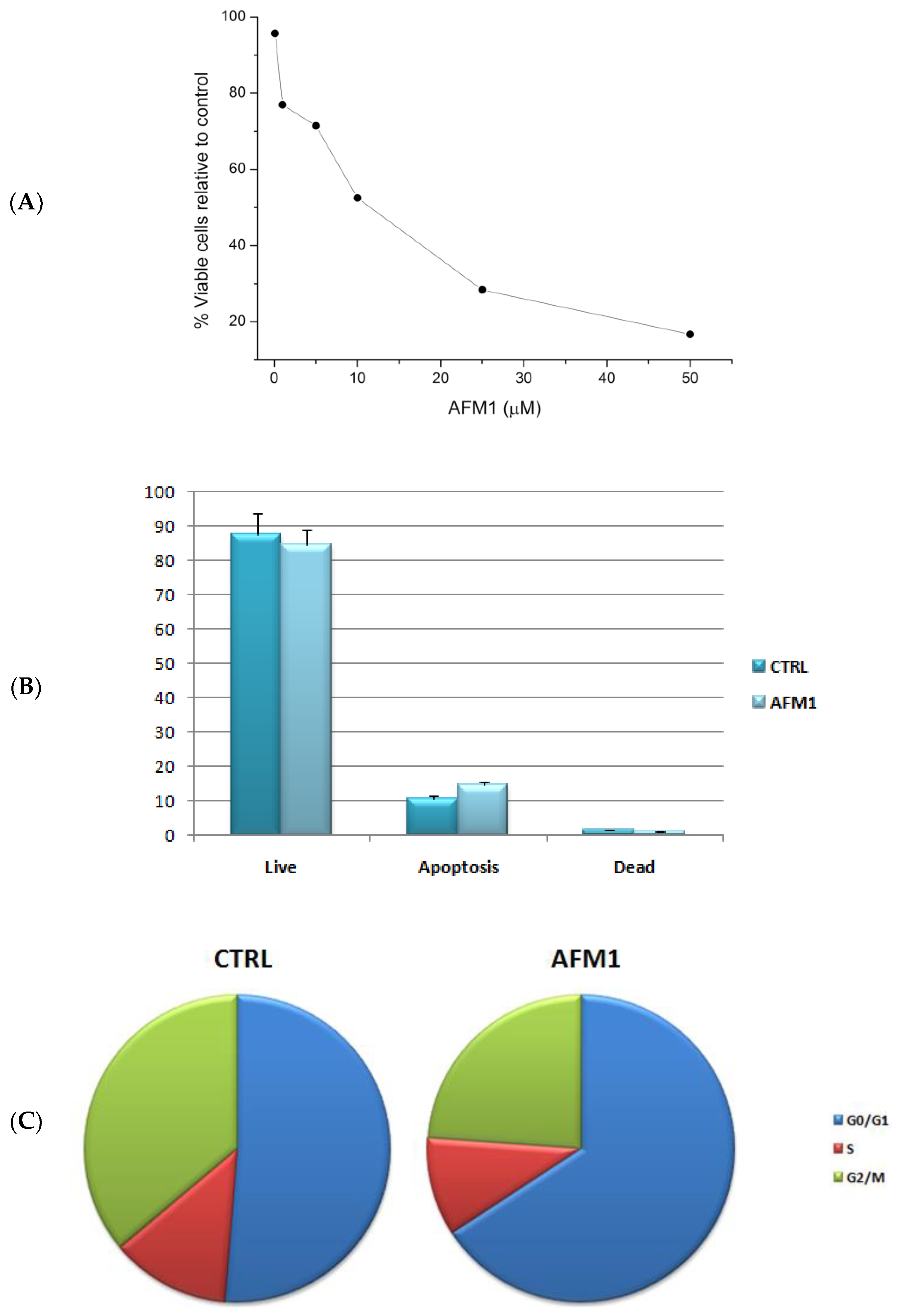
2.1. AFM1 Reduces Cell Proliferation and Induces a Block of the Cell Cycle in HepG2 Cells
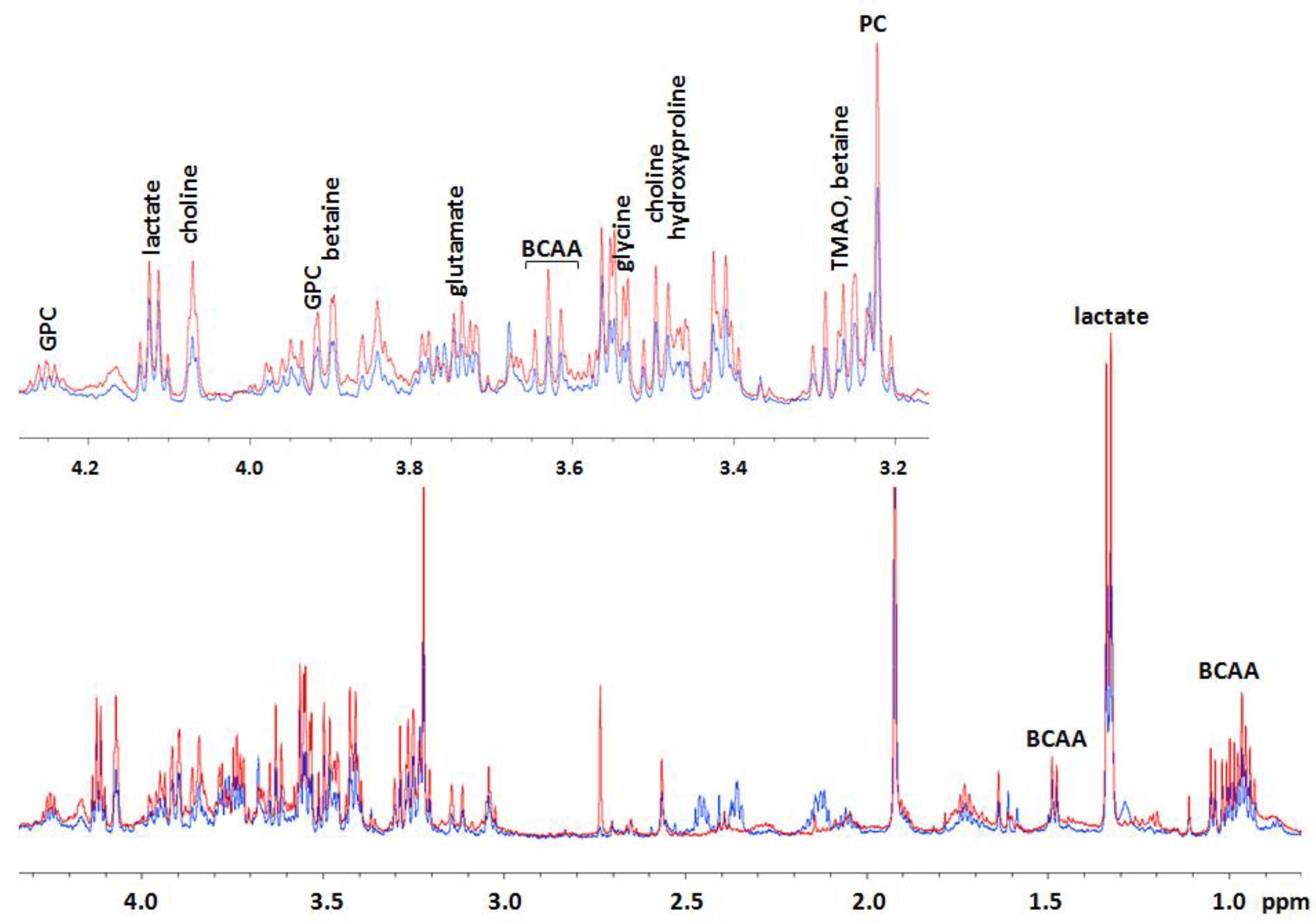
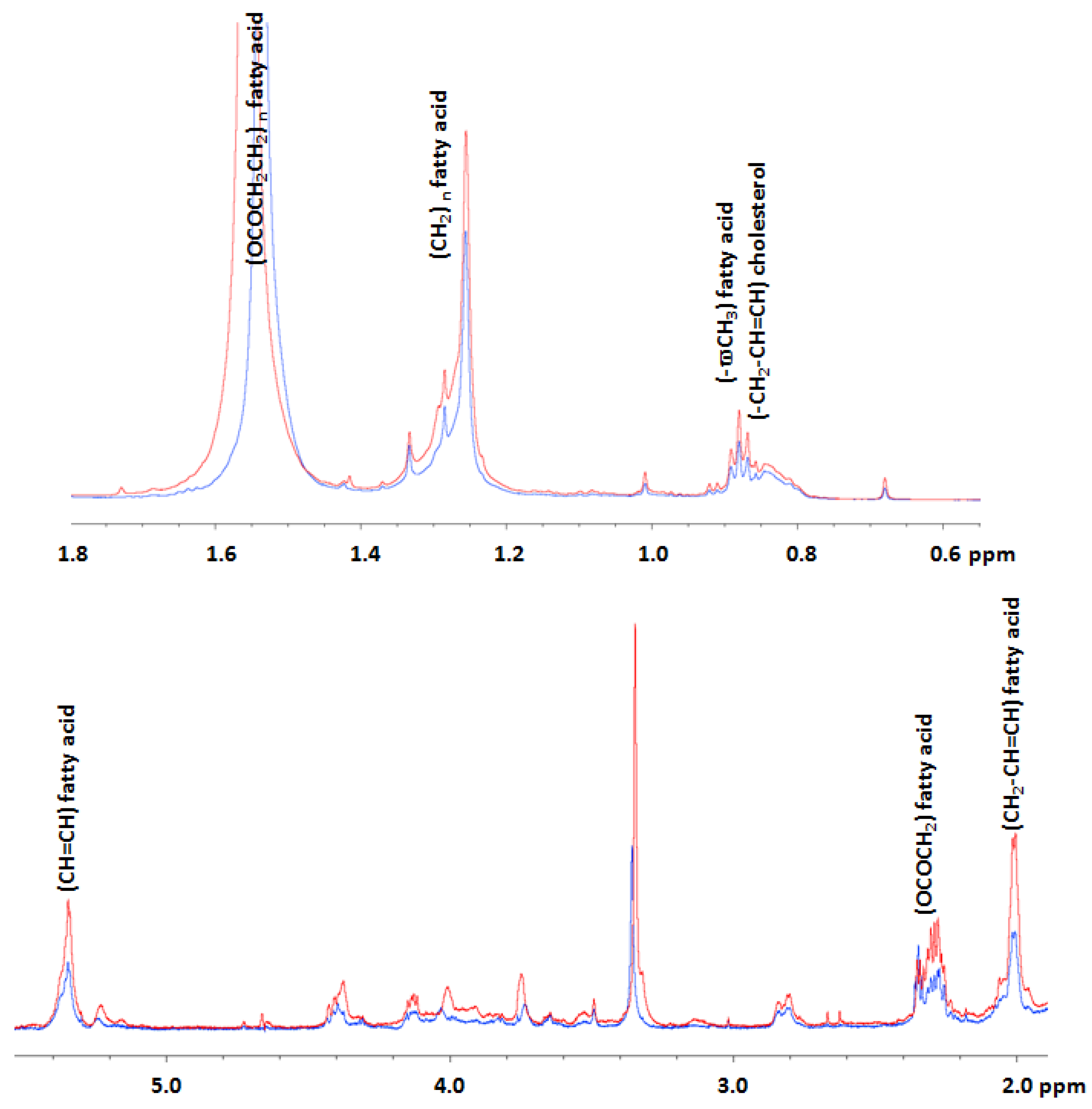
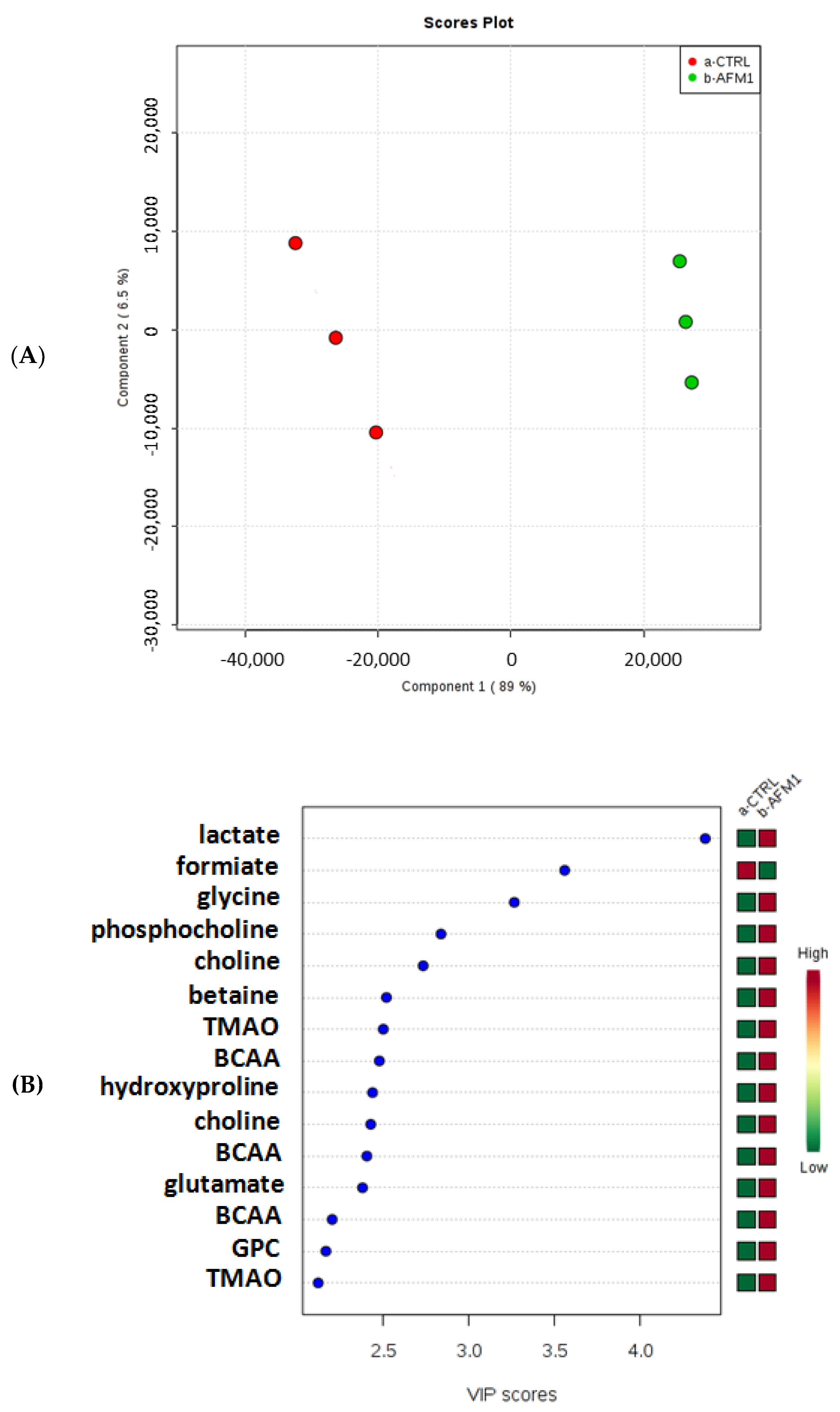
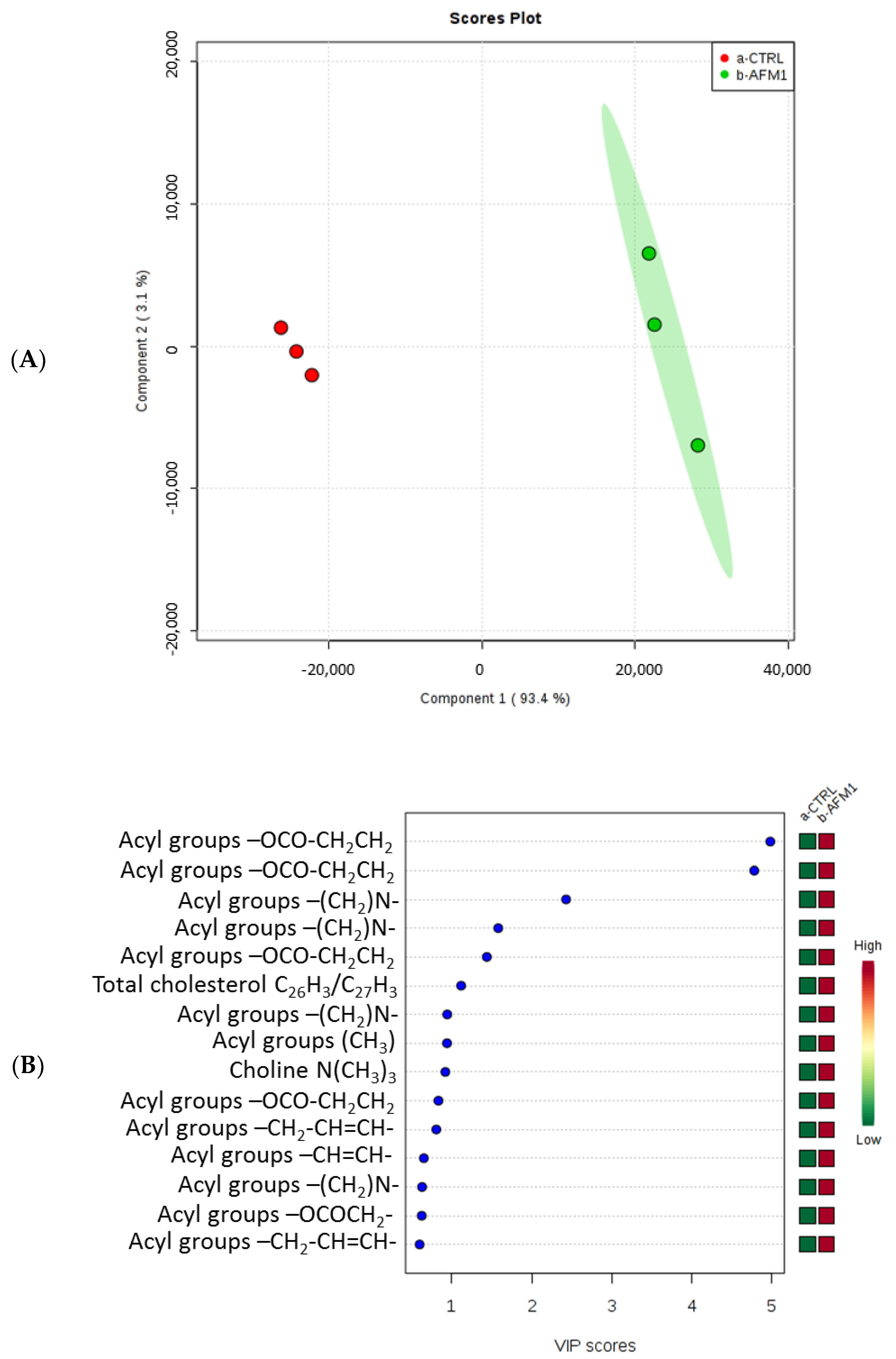
2.2. Effects of AFM1 on the Metabolome of HepG2 Cells
2.3. Evaluation of the Cytokine Levels of HepG2 Cells after AFM1 Treatment
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture
5.2. Cell Treatment and Cell Proliferation Assay
5.3. Apoptosis Evaluation at IC50 Concentration after 48 h of Treatment
5.4. Cell Cycle Assay at IC50 Concentration after 48 h of Treatment
5.5. Extraction of the Polar and Lipidic Fractions in Untreated and Treated HepG2 Cells
5.6. 1H-NMR Metabolomic Analysis of the Cellular Fractions
5.7. Statistical and Pathway Analysis
5.8. Bio-Plex Assay
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, D.; Subbarao, G.; Saxena, R. Hepatoblastoma. Semin. Diagn. Pathol. 2017, 34, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Adesina, A.M.; Lòpez-Terrada, D.; Wong, K.K.; Gunaratne, P.; Nquyen, Y.; Pulliam, J.; Margolin, J.; Finegold, M.J. Gene expression profiling reveals signatures characterizing histologic subtypes of hepatoblastoma and global deregulation in cell growth and survival pathways. Hum. Pathol. 2009, 40, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czauderna, P.; Garnier, H. Hepatoblastoma: Current understanding, recent advances, and controversies. F1000Research 2018, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Apte, U.; Micsenyi, A.; Kotsagrelos, E.; Luo, J.H.; Ranganathan, S.; Monga, D.K.; Bell, A.; Michalopoulos, G.K.; Monga, S.P. Epidermal growth factor receptor: A novel target of the Wnt/beta-catenin pathway in liver. Gastroenterology 2005, 129, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Litten, J.B.; Tomlinson, G.E. Liver tumors in children. Oncologist 2008, 13, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Pateva, I.B.; Egler, R.A.; Stearns, D.S. Hepatoblastoma in an 11-year-old: Case report and a review of the literature. Medicine 2017, 96, e5858. [Google Scholar] [CrossRef] [PubMed]
- Wogan, G.N. Aflatoxins as Risk Factors for Hepatocellular Carcinoma in Humans. Cancer Res. 1992, 52, 2114s–2118s. [Google Scholar] [PubMed]
- Neal, G.E.; Eaton, D.L.; Judah, D.J.; Verma, A. Metabolism and toxicity of aflatoxins M1 and B1 in human-derived in vitro systems. Toxicol. Appl. Pharmacol. 1998, 151, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, A.; Khotimah, K.; Rietjens, I.M.C.M. Risk assessment of aflatoxin B1 exposure from maize and peanut consumption in Indonesia using the margin of exposure and liver cancer risk estimation approaches. Food Chem. Toxicol. 2018, 113, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Jafari, T.; Fallah, A.A.; Kheiri, S.; Fadaei, A.; Amini, S.A. Aflatoxin M1 in human breast milk in Shahrekord, Iran and association with dietary factors. Food Addit. Contam. Part B 2017, 10, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, S.; Talpur, F.N.; Nizamani, S.M.; Afridi, H.I. Contamination profile of aflatoxin M1 residues in milk supply chain of Sindh, Pakistan. Toxicol. Rep. 2015, 2, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Ostry, V.; Malir, F.; Toman, J.; Grosse, Y. Mycotoxins as human carcinogens-the IARC Monographs classification. Mycotoxin Res. 2017, 33, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Zhang, H.; Li, S.; Wang, J.; Liu, J.; Ren, H.; Gao, Y. Lactoferrin inhibits aflatoxin B1- and aflatoxin M1-induced cytotoxicity and DNA damage in Caco-2, HEK, Hep-G2, and SK-N-SH cells. Toxicon 2018, 150, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Gao, J.; Huang, K.; Luo, Y.; Zhang, B.; Xu, W. miR-34a screened by miRNA profiling negatively regulates Wnt/β-catenin signaling pathway in Aflatoxin B1 induced hepatotoxicity. Sci. Rep. 2015, 5, 16732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, H.; Shim, Y.; Arumugam, P.; Song, J.M. Crosstalk-eliminated quantitative determination of aflatoxin B1-induced hepatocellular cancer stem cells based on concurrent monitoring of CD133, CD44, and aldehyde dehydrogenase1. Toxicol. Lett. 2016, 243, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Taee, A.F.M.; Hassan, A.A. Effect of aflatoxin on malondialdehyde, glutathione levels, and stress index in Toxoplasma gondii infected mice. Iraqi J. Vet. Sci. 2012, 26, 5–9. [Google Scholar]
- Costantini, S.; Castello, G.; Colonna, G. Human Cytokinome: A new challenge for systems biology. Bioinformation 2010, 5, 166–167. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.H.; Xie, X.; Xu, F.; Shi, X.; Wang, Y.; Deng, L. Distinctive pharmacological differences between liver cancer cell lines HepG2 and Hep3B. Cytotechnology 2015, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- López-Terrada, D.; Cheung, S.W.; Finegold, M.J.; Knowles, B.B. HepG2 is a hepatoblastoma-derived cell line. Hum. Pathol. 2009, 40, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Eichenmüller, M.; Trippel, F.; Kreuder, M.; Beck, A.; Schwarzmayr, T.; Häberle, B.; Cairo, S.; Leuschner, I.; von Schweinitz, D.; Strom, T.M.; et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J. Hepatol. 2014, 61, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Polychronaki, N.; West, R.M.; Turner, P.C.; Amra, H.; Abdel-Wahhab, M.; Mykkänen, H.; El-Nezami, H. A longitudinal assessment of aflatoxin M1 excretion in breast milk of selected Egyptian mothers. Food Chem. Toxicol. 2007, 45, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Magoha, H.; Kimanya, M.; De Meulenaer, B.; Roberfroid, D.; Lachat, C.; Kolsteren, P. Association between aflatoxin M1 exposure through breast milk and growth impairment in infants from Northern Tanzania. World Mycotoxin J. 2014, 7, 277–284. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, N.; Liu, J.; Li, F.D.; Li, S.L.; Wang, J.Q. Aflatoxin B1 and aflatoxin M1 induced cytotoxicity and DNA damage in differentiated and undifferentiated Caco-2 cells. Food Chem. Toxicol. 2015, 83, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.N.; Wang, J.Q.; Li, S.L.; Zhang, Y.D.; Zheng, N. Aflatoxin M1 cytotoxicity against human intestinal Caco-2 cells is enhanced in the presence of other mycotoxins. Food Chem. Toxicol. 2016, 96, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Luongo, D.; Russo, R.; Balestrieri, A.; Marzocco, S.; Bergamo, P.; Severino, L. In vitro study of AFB1 and AFM1 effects on human lymphoblastoid Jurkat T-cell model. J. Immunotoxicol. 2014, 11, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate Influx through the Endothelial Cell Monocarboxylate Transporter MCT1 Supports an NF-κB/IL-8 Pathway that Drives Tumor Angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.; Nie, X.; Feng, X.; Chen, W.; Yue, Y.; Tang, H.; Deng, F. Metabonomic Studies of Human Hepatocellular Carcinoma Using High-Resolution Magic-Angle Spinning 1H NMR Spectroscopy in Conjunction with Multivariate Data Analysis. J. Proteome Res. 2007, 6, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Salem, N.; Corn, D.J.; Erowku, B.; Tian, H.; Wang, F.; Lee, Z. Transport and Metabolism of Radiolabeled Choline in Hepatocellular Carcinoma. Mol. Pharm. 2010, 7, 2077–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, N.; Wildes, F.; Takagi, T.; Glunde, K.; Bhujwalla, Z.M. The Tumor Microenvironment Modulates Choline and Lipid Metabolism. Front. Oncol. 2016, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ye, Y.; An, Y.; Tian, Y.; Wang, Y.; Tang, H. Systems Responses of Rats to Aflatoxin B1 Exposure Revealed with Metabonomic Changes in Multiple Biological Matrices. J. Proteome Res. 2011, 10, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zeng, J.; Geng, P.; Fang, C.; Wang, Y.; Sun, M.; Wang, C.; Wang, J.; Yin, P.; Hu, C.; et al. Global Metabolic Profiling Identifies a Pivotal Role of Proline and Hydroxyproline Metabolism in Supporting Hypoxic Response in Hepatocellular Carcinoma. Clin. Cancer Res. 2018, 24, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terakura, D.; Shimizu, M.; Iwasa, J.; Baba, A.; Kochi, T.; Ohno, T.; Kubota, M.; Shirakami, Y.; Shiraki, M.; Takai, K.; et al. Preventive effects of branched-chain amino acid supplementation on the spontaneous development of hepatic preneoplastic lesions in C57BL/KsJ-db/db obese mice. Carcinogenesis 2012, 33, 2499–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarru, L.P.; Settivari, R.S.; Antoniou, E.; Ledoux, D.R.; Rottinghaus, G.E. Toxicological and gene expression analysis of the impact of aflatoxin B1 on hepatic function of male broiler chicks. Poult. Sci. 2009, 88, 360–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinger-Ziegelbauer, H.; Stuart, B.; Wahle, B.; Bomann, W.; Ahr, H.J. Characteristic expression profiles induced by genotoxic carcinogens in rat liver. Toxicol. Sci. 2004, 77, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Rotimi, O.A.; Rotimi, S.O.; Duru, C.U.; Ebebeinwe, O.J.; Abiodun, A.O.; Oyeniyi, B.O.; Faduyile, F.A. Acute aflatoxin B1-Induced hepatotoxicity alters gene expression and disrupts lipid and lipoprotein metabolism in rats. Toxicol. Rep. 2017, 4, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Li, T.; Zhang, X.; Gao, P.; Qiao, P.; Li, S.; Geng, Z. Expression and roles of fatty acid synthase in hepatocellular carcinoma. Oncol. Rep. 2014, 32, 2471–2476. [Google Scholar] [CrossRef] [PubMed]
- Hinton, D.M.; Myers, M.J.; Raybourne, R.A.; Francke-Carroll, S.; Sotomayor, R.E.; Shaddock, J.; Warbritton, A.; Chou, M.W. Immunotoxicity of aflatoxin B1 in rats: Effects on lymphocytes and the inflammatory response in a chronic intermittent dosing study. Toxicol. Sci. 2003, 73, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.; Tang, L.; Guo, X.; Wang, F.; Massey, M.E.; Su, J.; Guo, T.L.; Williams, J.H.; Phillips, T.D.; Wang, J.S. Aflatoxin B1 modulates the expression of phenotypic markers and cytokines by splenic lymphocytes of male F344 rats. J. Appl. Toxicol. 2014, 34, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jolly, P.E.; Ellis, W.O.; Wang, J.S.; Phillips, T.D.; Williams, J.H. Aflatoxin B1 albumin adduct levels and cellular immune status in Ghanaians. Int. Immunol. 2005, 17, 807–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nittoli, A.C.; Costantini, S.; Sorice, A.; Capone, F.; Ciarcia, R.; Marzocco, S.; Budillon, A.; Severino, L. Effects of α-zearalenol on the metabolome of two breast cancer cell lines by 1H-NMR approach. Metabolomics 2018, 14, 33. [Google Scholar] [CrossRef]
- Ruocco, N.; Costantini, S.; Zupo, V.; Lauritano, C.; Caramiello, D.; Ianora, A.; Budillon, A.; Romano, G.; Nuzzo, G.; D’Ippolito, G.; et al. Toxigenic effects of two benthic diatoms upon grazing activity of the sea urchin: Morphological, metabolomic and de novo transcriptomic analysis. Sci. Rep. 2018, 8, 5622. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, 251–257. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | Chemical shift (ppm) |
|---|---|
| Acetate | 1.91 |
| Alanine | 1.48; 3.75 |
| Arginine | 1.68; 1.9; 3.26; 3.76 |
| Betaine | 3.25; 3.89 |
| Choline | 3.19; 3.51; 4.06 |
| Formiate | 8.44 |
| Glucose | 3.23; 3.39; 3.46; 3.52; 3.73; 3.82; 3.88; 4.63; 5.22 |
| Glutamate | 2.04; 2.12; 2.34; 3.75 |
| Glutamine | 2.13; 2.45; 3.77 |
| Glycerophosphocholine (GPC) | 3.20; 3.36;3.90; 4.28 |
| Glycine | 3.54 |
| Histidine | 3.16; 3.23; 7.09; 7.9 |
| Hydroxyproline | 2.14; 2.42; 3.36; 3.46; 4.33 |
| Isoleucine | 0.93; 0.99; 1.24; 1.46; 1.97; 3.66 |
| Lactate | 1.33; 4.11 |
| Leucine | 0.96; 1.72; 3.72 |
| Lysine | 1.46; 1.71; 1.89; 3.02; 3.74 |
| Malate | 2.36; 2.66; 4.29 |
| Phenylalanine | 3.19; 7.32–7.42 |
| Phosphocholine (PC) | 3.21; 3.58; 4.17 |
| Proline | 1.99; 2.06; 2.34; 3.33; 3.41; 4.12 |
| Pyruvate | 2.46 |
| Threonine | 1.32; 3.58; 4.24 |
| Trimethylamine N-oxide (TMAO) | 3.25 |
| Tyrosine | 3.02; 3.17; 3.92; 6.9; 7.2 |
| Valine | 0.97; 1.04; 2.28; 3.60 |
| Cholesterol C18H3 | 0.67 |
| C26H3,C27H3 | 0.88 |
| C19H3 | 1.01–1.03 |
| C3H | 3.5 |
| C6H | 5.37 |
| Fatty acid residues ω-CH3 | 0.89 |
| (CH2)n | 1.3 |
| -COCH2-CH2 | 1.6 |
| -CH2-CH= | 2.04 |
| -CO-CH2 | 2.3 |
| -CH=CH-CH2-CH=CH | 2.76 |
| -CH=CH | 5.36 |
| Phosphatidylcholine (POCH2) | 4.33–4.43 |
| Phospholipids (-CH2-NH2) | 3.11–3.14 |
| (-CH2-N-(CH3)3) | 3.33 |
| Triglycerides C1H | 4.15 |
| C3H | 4.29 |
| C2H | 5.25 |
| Cytokines | Ratio (HepG2-AFM1 vs CTRL) |
|---|---|
| PDGF-ββ | 0.94 |
| IL-1β | 1.04 |
| IL-1ra | 1.24 |
| IL-2 | 0.92 |
| IL-4 | 0.68 |
| IL-5 | 0.98 |
| IL-6 | 1.30 |
| IL-7 | 0.92 |
| IL-8 | 1.73 |
| IL-9 | 0.96 |
| IL-10 | 0.95 |
| IL-12 | 0.90 |
| IL-13 | 1.00 |
| IL-15 | 0.90 |
| IL-17 | 0.88 |
| Eotaxin | 0.96 |
| FGF basic | 0.96 |
| G-CSF | 1.00 |
| GM-CSF | 0.86 |
| IFN-γ | 0.89 |
| IP-10 | 0.95 |
| MCP-1 | 0.92 |
| MIP-1α | 0.96 |
| MIP-1β | 0.86 |
| RANTES | 0.90 |
| TNF-a | 1.32 |
| VEGF | 1.00 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchese, S.; Sorice, A.; Ariano, A.; Florio, S.; Budillon, A.; Costantini, S.; Severino, L. Evaluation of Aflatoxin M1 Effects on the Metabolomic and Cytokinomic Profiling of a Hepatoblastoma Cell Line. Toxins 2018, 10, 436. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10110436
Marchese S, Sorice A, Ariano A, Florio S, Budillon A, Costantini S, Severino L. Evaluation of Aflatoxin M1 Effects on the Metabolomic and Cytokinomic Profiling of a Hepatoblastoma Cell Line. Toxins. 2018; 10(11):436. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10110436
Chicago/Turabian StyleMarchese, Silvia, Angela Sorice, Andrea Ariano, Salvatore Florio, Alfredo Budillon, Susan Costantini, and Lorella Severino. 2018. "Evaluation of Aflatoxin M1 Effects on the Metabolomic and Cytokinomic Profiling of a Hepatoblastoma Cell Line" Toxins 10, no. 11: 436. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10110436