SS1P Immunotoxin Induces Markers of Immunogenic Cell Death and Enhances the Effect of the CTLA-4 Blockade in AE17M Mouse Mesothelioma Tumors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
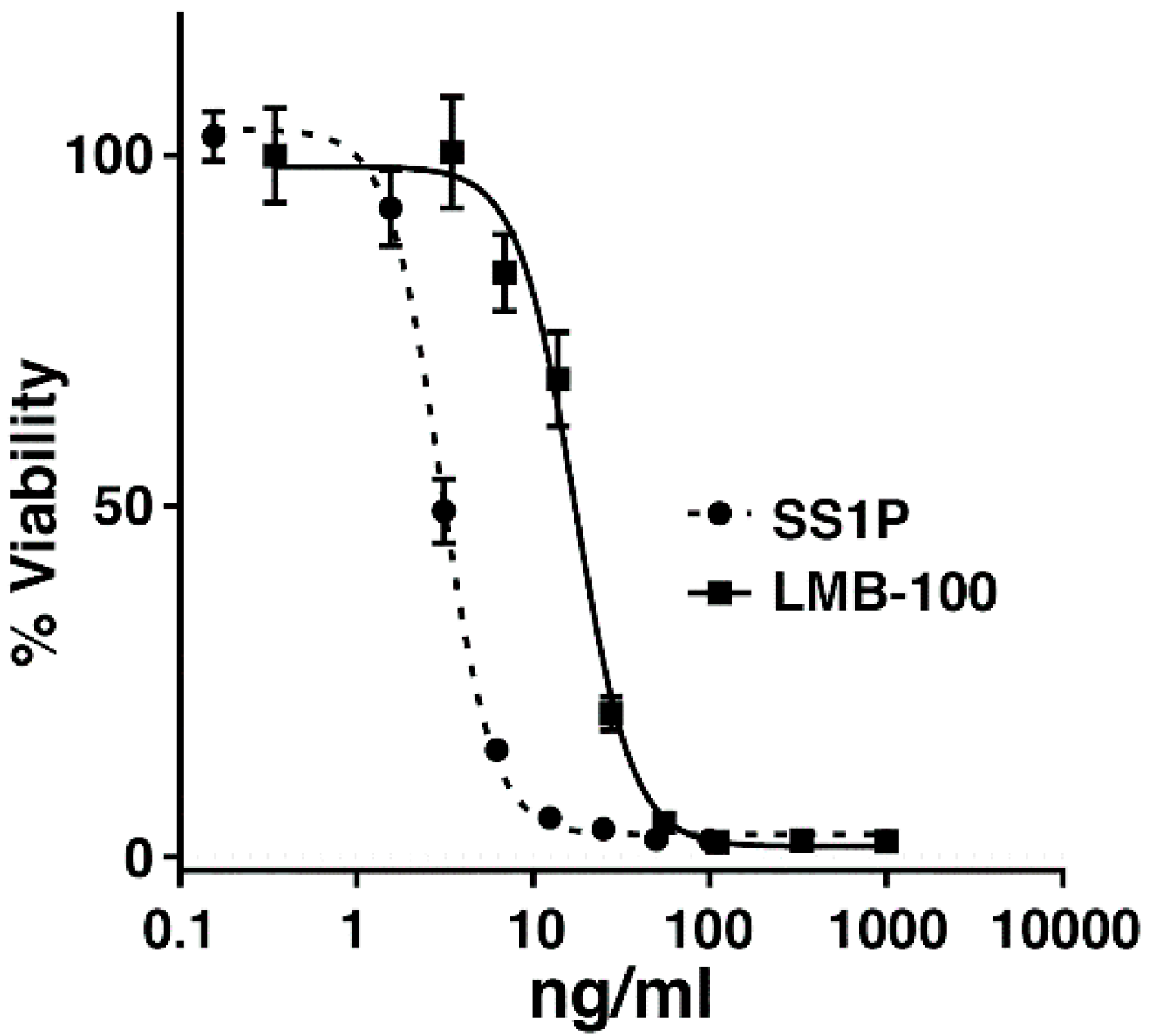
2.1. AE17 Cells Are Sensitive to Mesothelin Targeting Immunotoxins
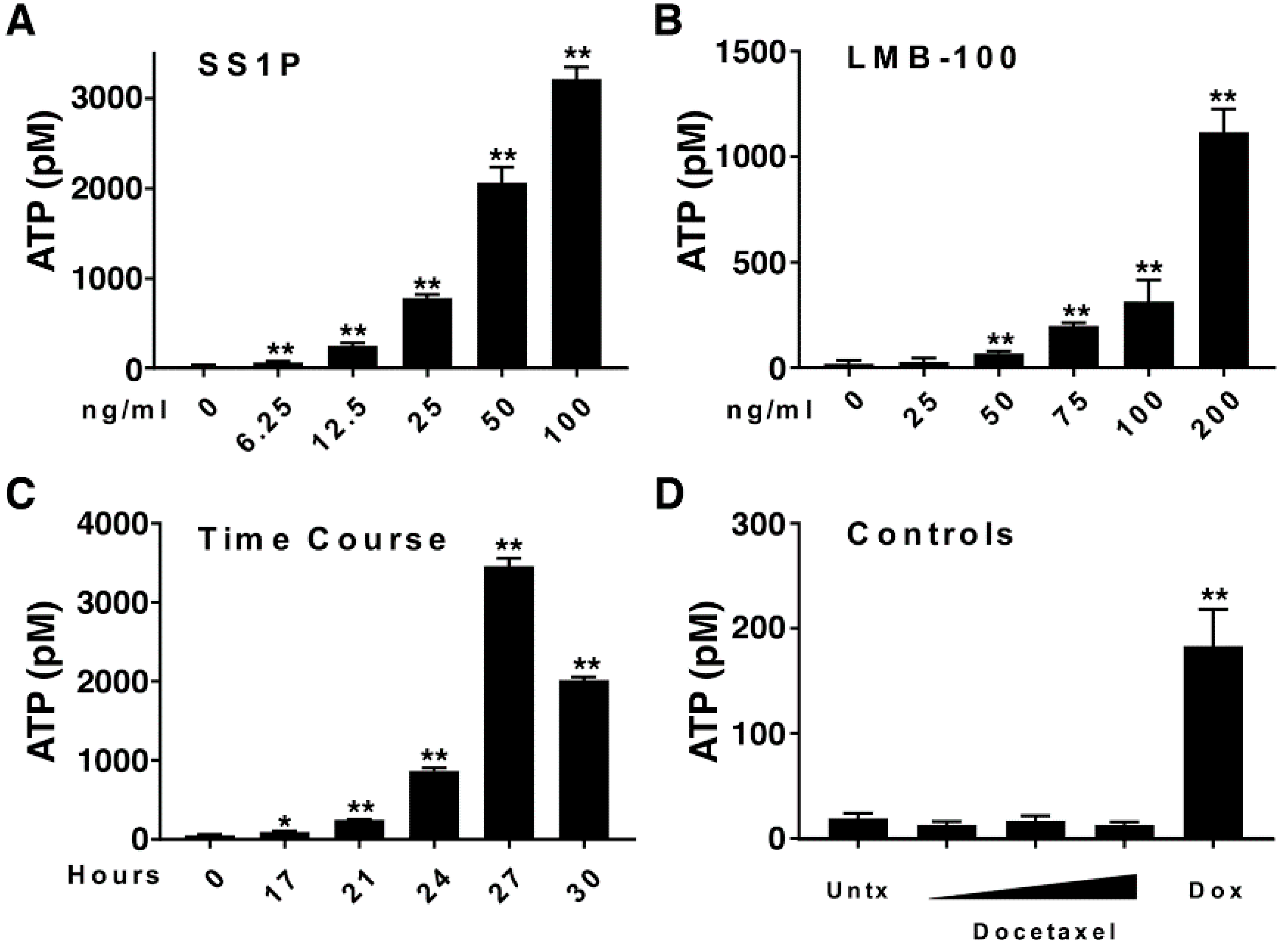
2.2. Anti-Mesothelin Immunotoxins Induce Extracellular Secretion of ATP
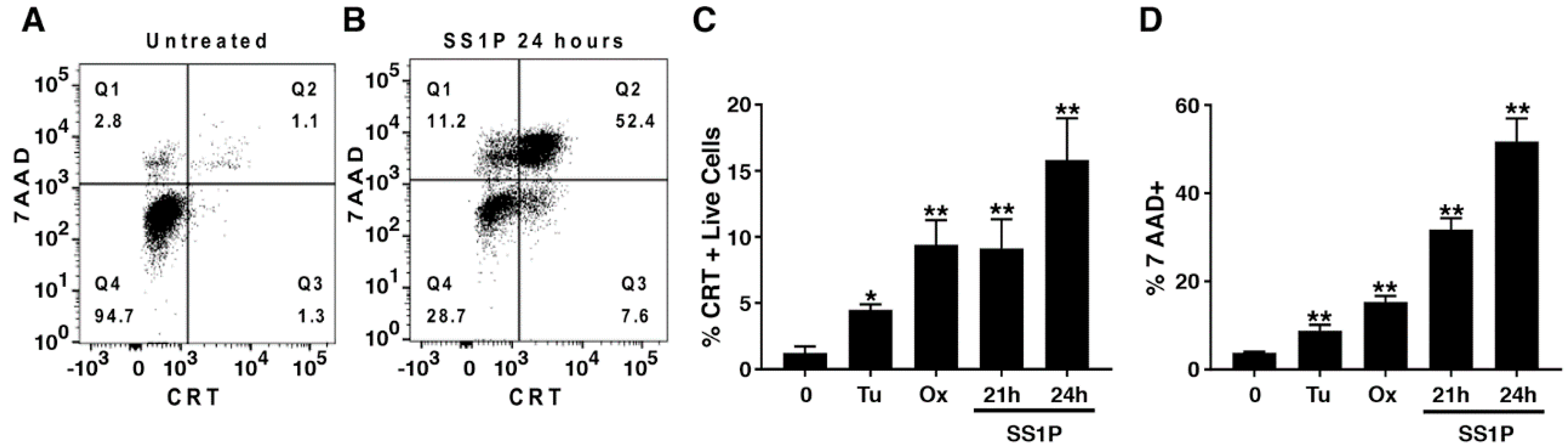
2.3. Anti-Mesothelin Immunotoxins Induce Surface Calreticulin
2.4. Direct Intra-Tumoral Injection of SS1P Inhibits Tumor Growth
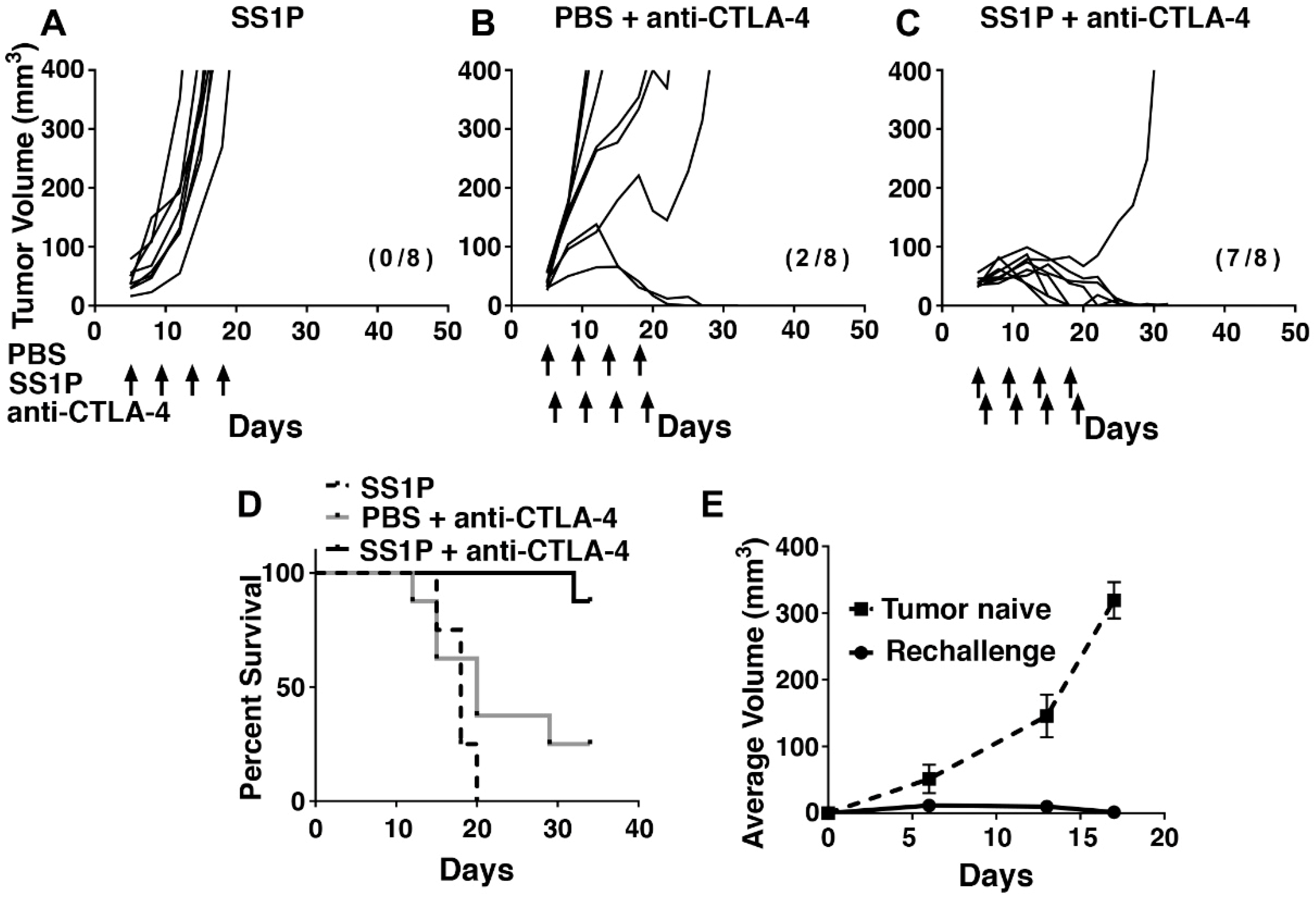
2.5. Local SS1P Renders AE17M Tumors More Sensitive to Anti-CTLA-4 Effect
2.6. Long-Term Anti-Tumor Immunity After SS1P and Anti-CTLA-4 Combination
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cytotoxicity Assays
4.3. Mouse Experiments
4.4. Flow Cytometry
4.5. ATP Assay
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the pd-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Shen, S.; Yamazaki, T.; Sukkurwala, A.Q.; Michaud, M.; Mignot, G.; et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, S.K.; Schroth, M.N.; Cho, J.J.; Kominos, S.K.; Vitanza-jack, V.B. Agricultural plants and soil as a reservoir for Pseudomonas aeruginosa. Appl. Microbiol. 1974, 28, 987–991. [Google Scholar] [PubMed]
- Wolf, P.; Elsasser-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Bera, T.K.; Pastan, I. Mesothelin is not required for normal mouse development or reproduction. Mol. Cell. Biol. 2000, 20, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Pastan, I.; Hassan, R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 2014, 74, 2907–2912. [Google Scholar] [CrossRef] [PubMed]
- Morello, A.; Sadelain, M.; Adusumilli, P.S. Mesothelin-targeted cars: Driving t cells to solid tumors. Cancer Discov. 2016, 6, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin immunotherapy for cancer: Ready for prime time? J. Clin. Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of ss1p, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. Infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Miller, A.C.; Sharon, E.; Thomas, A.; Reynolds, J.C.; Ling, A.; Kreitman, R.J.; Miettinen, M.M.; Steinberg, S.M.; Fowler, D.H.; et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Hollevoet, K.; Mason-Osann, E.; Liu, X.F.; Imhof-Jung, S.; Niederfellner, G.; Pastan, I. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin rg7787 in pancreatic cancer. Mol. Cancer Ther. 2014, 13, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Leshem, Y.; O’Brien, J.; Liu, X.; Bera, T.K.; Terabe, M.; Berzofsky, J.A.; Bossenmaier, B.; Niederfellner, G.; Tai, C.H.; Reiter, Y.; et al. Combining local immunotoxins targeting mesothelin with ctla-4 blockade synergistically eradicates murine cancer by promoting anticancer immunity. Cancer Immunol. Res. 2017, 5, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Poussin, M.; Hagemann, I.S.; Coukos, G.; Sandaltzopoulos, R.; Scholler, N.; Powell, D.J., Jr. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol. Ther. 2012, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Jackaman, C.; Bundell, C.S.; Kinnear, B.F.; Smith, A.M.; Filion, P.; van Hagen, D.; Robinson, B.W.; Nelson, D.J. Il-2 intratumoral immunotherapy enhances cd8+ t cells that mediate destruction of tumor cells and tumor-associated vasculature: A novel mechanism for il-2. J. Immunol. 2003, 171, 5051–5063. [Google Scholar] [CrossRef] [PubMed]
- Aymeric, L.; Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Martins, I.; Kroemer, G.; Smyth, M.J.; Zitvogel, L. Tumor cell death and atp release prime dendritic cells and efficient anticancer immunity. Cancer Res. 2010, 70, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Garnett, C.T.; Farsaci, B.; Palena, C.; Tsang, K.Y.; Ferrone, S.; Gameiro, S.R. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic t lymphocytes and is distinct from immunogenic cell death. Int. J. Cancer 2013, 133, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of lrp on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Senovilla, L.; Menger, L.; Chaput, N.; Kroemer, G. Immunogenic tumor cell death for optimal anticancer therapy: The calreticulin exposure pathway. Clin. Cancer Res. 2010, 16, 3100–3104. [Google Scholar] [CrossRef] [PubMed]
- Risberg, K.; Fodstad, O.; Andersson, Y. Synergistic anticancer effects of the 9.2.27pe immunotoxin and abt-737 in melanoma. PLoS ONE 2011, 6, e24012. [Google Scholar] [CrossRef] [PubMed]
- Antignani, A.; Sarnovsky, R.; FitzGerald, D.J. Abt-737 promotes the dislocation of er luminal proteins to the cytosol, including pseudomonas exotoxin. Mol. Cancer Ther. 2014, 13, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Lipp, H.P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.H.; Carter, S.K. Adriamycin. A new anticancer drug with significant clinical activity. Ann. Intern. Med. 1974, 80, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Hassan, R.; Fitzgerald, D.J.; Pastan, I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin ss1p. Clin. Cancer Res. 2009, 15, 5274–5279. [Google Scholar] [CrossRef] [PubMed]
- Luther, N.; Cheung, N.K.; Souliopoulos, E.P.; Karampelas, I.; Bassiri, D.; Edgar, M.A.; Guo, H.F.; Pastan, I.; Gutin, P.H.; Souweidane, M.M. Interstitial infusion of glioma-targeted recombinant immunotoxin 8h9scfv-pe38. Mol. Cancer Ther. 2010, 9, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, H.; Archer, G.E.; Herndon, J.E., II; Kuan, C.T.; Mitchell, D.A.; Bigner, D.D.; Pastan, I.H.; Sampson, J.H. Egfrviii-targeted immunotoxin induces antitumor immunity that is inhibited in the absence of cd4+ and cd8+ t cells. Cancer Immunol. Immunother. 2008, 57, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Terabe, M.; Kioi, M.; Berzofsky, J.A.; Puri, R.K. Intratumoral therapy with il13-pe38 results in effective ctl-mediated suppression of Il-13ralpha2-expressing contralateral tumors. Clin. Cancer Res. 2006, 12, 4678–4686. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Wang, Z.; Lasota, J.; Onda, M.; Czapiewski, P.; Langfort, R.; Rys, J.; Szpor, J.; Waloszczyk, P.; Okon, K.; et al. Comprehensive immunohistochemical study of mesothelin (msln) using different monoclonal antibodies 5b2 and mn-1 in 1562 tumors with evaluation of its prognostic value in malignant pleural mesothelioma. Oncotarget 2017, 8, 26744–26754. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Reardon, D.A.; Friedman, A.H.; Friedman, H.S.; Coleman, R.E.; McLendon, R.E.; Pastan, I.; Bigner, D.D. Sustained radiographic and clinical response in patient with bifrontal recurrent glioblastoma multiforme with intracerebral infusion of the recombinant targeted toxin tp-38: Case study. Neuro-Oncology 2005, 7, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Berger, M.S.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; Greer, K.; Herndon, J.E., 2nd; Kunwar, S.; et al. Intracerebral infusion of an egfr-targeted toxin in recurrent malignant brain tumors. Neuro-Oncology 2008, 10, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Asher, A.; Bucholz, R.; Berger, M.; Prados, M.; Chang, S.; Bruce, J.; Hall, W.; Rainov, N.G.; Westphal, M.; et al. Safety, tolerability, and tumor response of il4-pseudomonas exotoxin (nbi-3001) in patients with recurrent malignant glioma. J. Neuro-Oncol. 2003, 64, 125–137. [Google Scholar] [CrossRef]
- Rainov, N.G.; Heidecke, V. Long term survival in a patient with recurrent malignant glioma treated with intratumoral infusion of an il4-targeted toxin (nbi-3001). J. Neuro-Oncol. 2004, 66, 197–201. [Google Scholar] [CrossRef]
- MacDonald, G.C.; Rasamoelisolo, M.; Entwistle, J.; Cizeau, J.; Bosc, D.; Cuthbert, W.; Kowalski, M.; Spearman, M.; Glover, N. A phase I clinical study of vb4-845: Weekly intratumoral administration of an anti-epcam recombinant fusion protein in patients with squamous cell carcinoma of the head and neck. Drug Des. Dev. Ther. 2009, 2, 105–114. [Google Scholar]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leshem, Y.; King, E.M.; Mazor, R.; Reiter, Y.; Pastan, I. SS1P Immunotoxin Induces Markers of Immunogenic Cell Death and Enhances the Effect of the CTLA-4 Blockade in AE17M Mouse Mesothelioma Tumors. Toxins 2018, 10, 470. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10110470
Leshem Y, King EM, Mazor R, Reiter Y, Pastan I. SS1P Immunotoxin Induces Markers of Immunogenic Cell Death and Enhances the Effect of the CTLA-4 Blockade in AE17M Mouse Mesothelioma Tumors. Toxins. 2018; 10(11):470. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10110470
Chicago/Turabian StyleLeshem, Yasmin, Emily M. King, Ronit Mazor, Yoram Reiter, and Ira Pastan. 2018. "SS1P Immunotoxin Induces Markers of Immunogenic Cell Death and Enhances the Effect of the CTLA-4 Blockade in AE17M Mouse Mesothelioma Tumors" Toxins 10, no. 11: 470. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10110470