Alpha-Toxin Contributes to Biofilm Formation among Staphylococcus aureus Wound Isolates

 ,
, 
Abstract
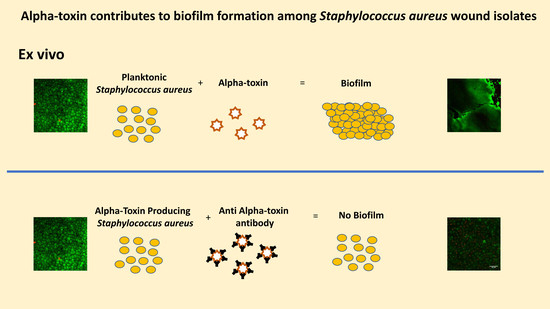
:
1. Introduction
2. Results
2.1. Strain Genotyping
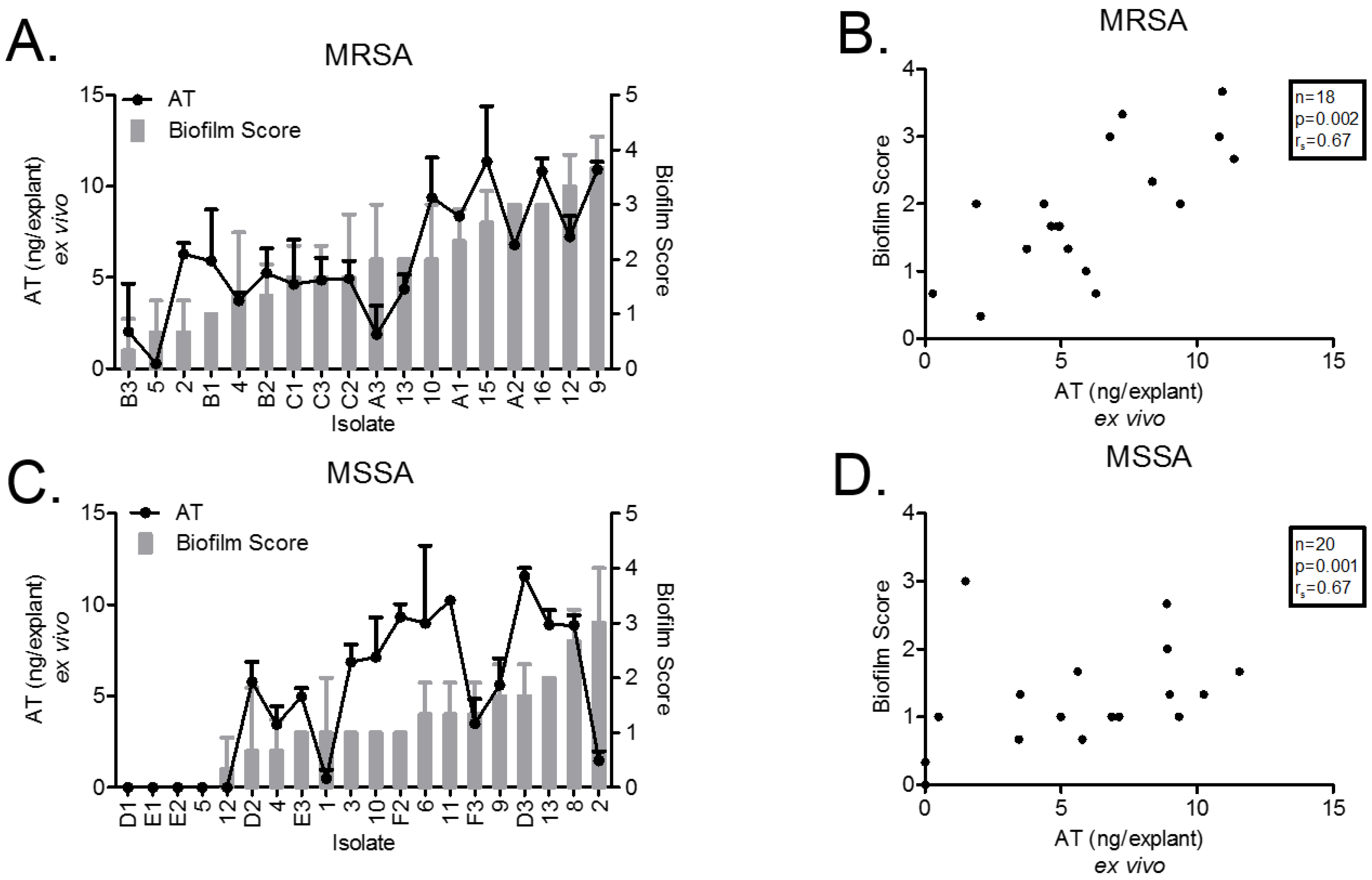
2.2. Ex Vivo AT Production on PVM Explants
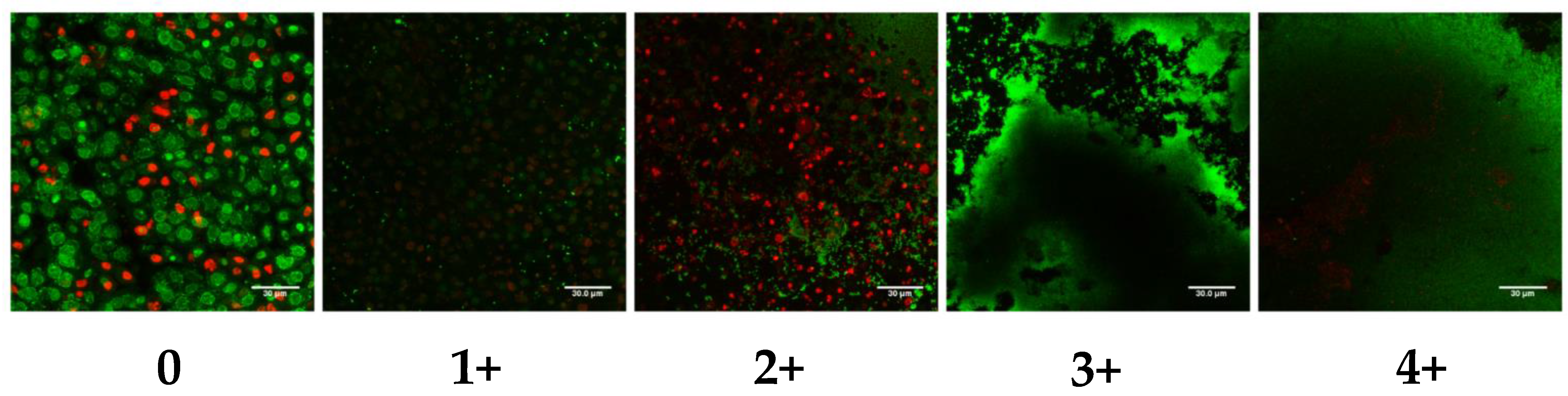
2.3. Ex Vivo Biofilm Production on PVM Explants
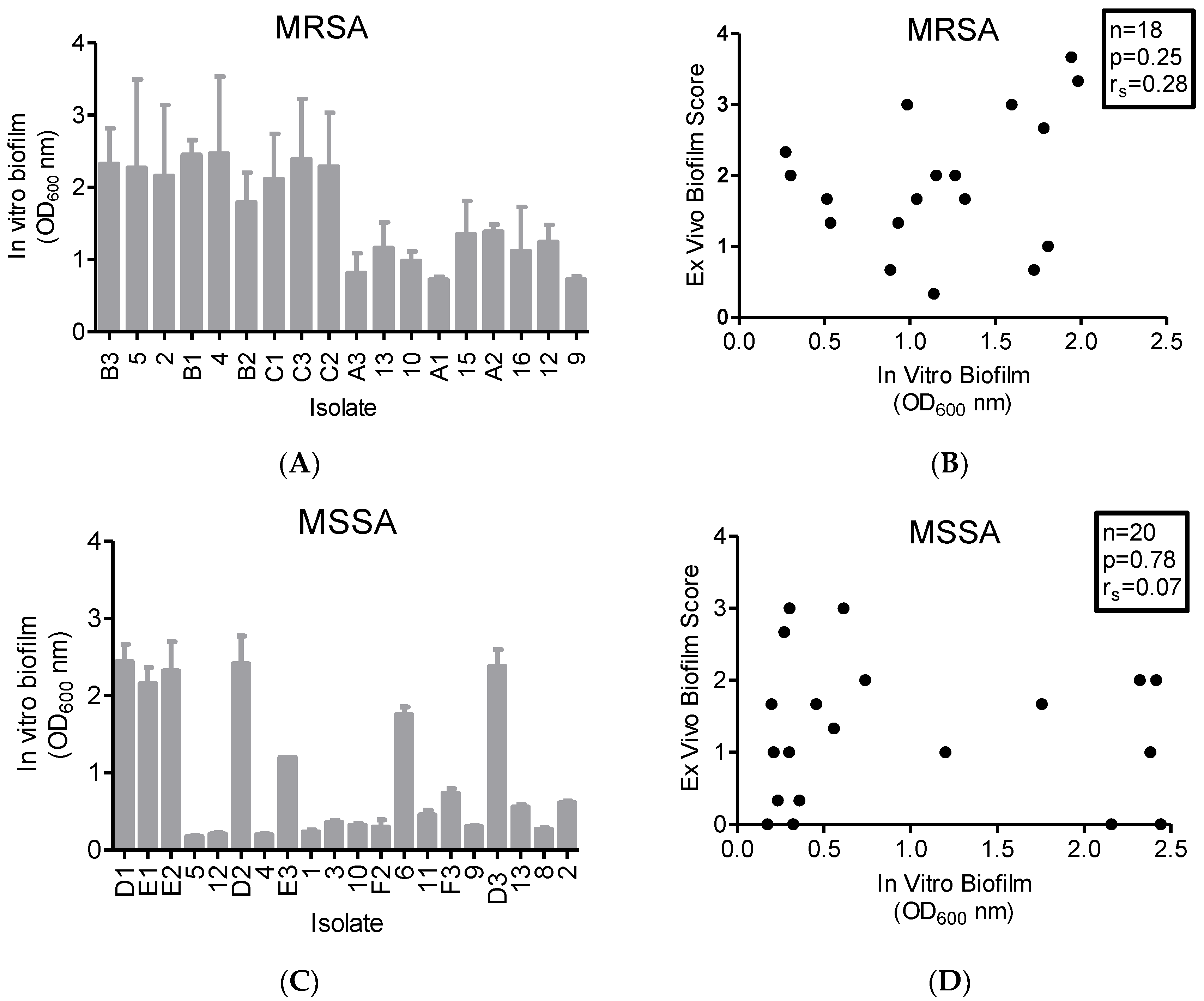
2.4. In Vitro Biofilm Production
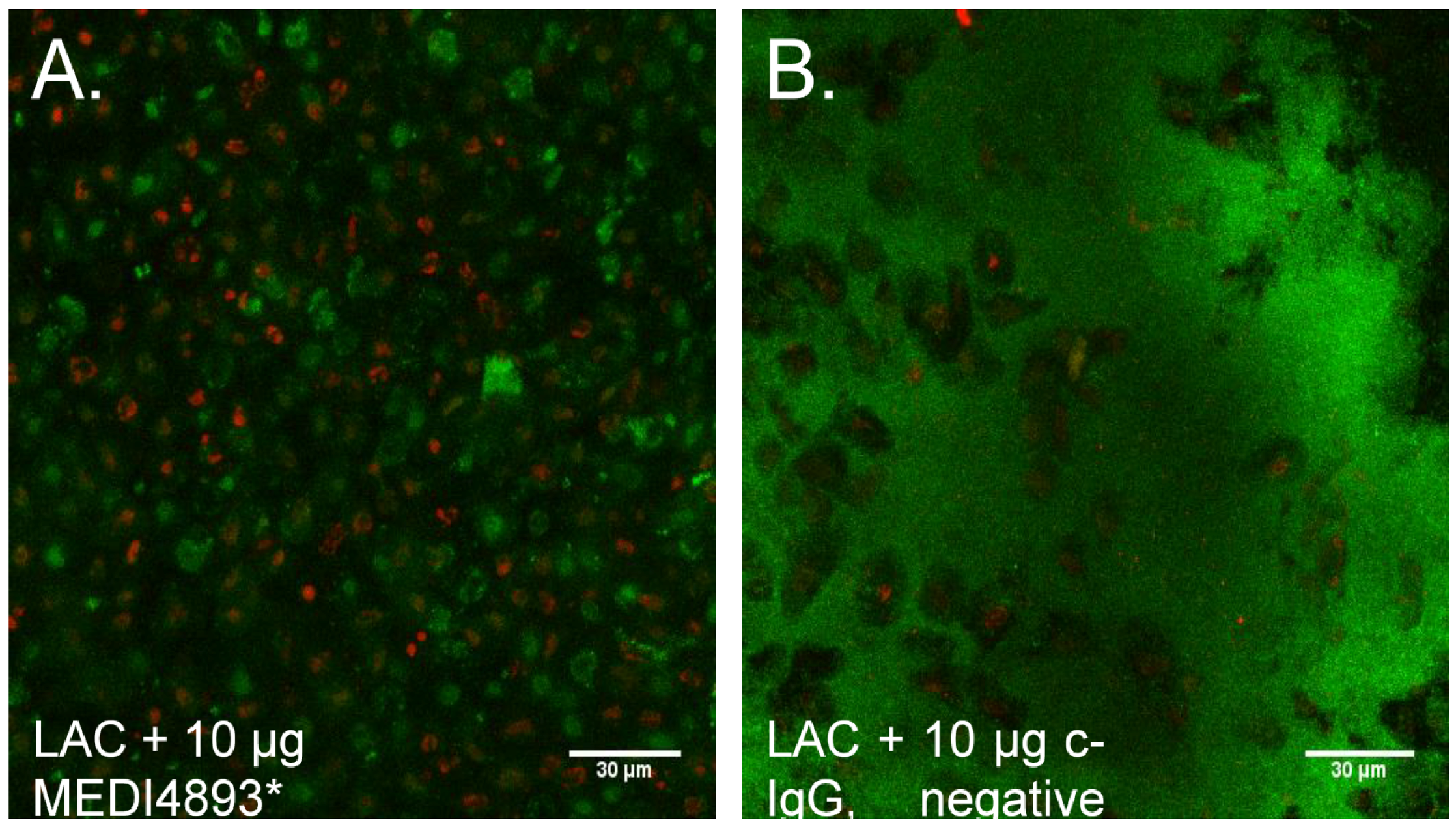
2.5. Neutralization of AT Prevents MRSA Biofilm Formation on Mucosal Tissue
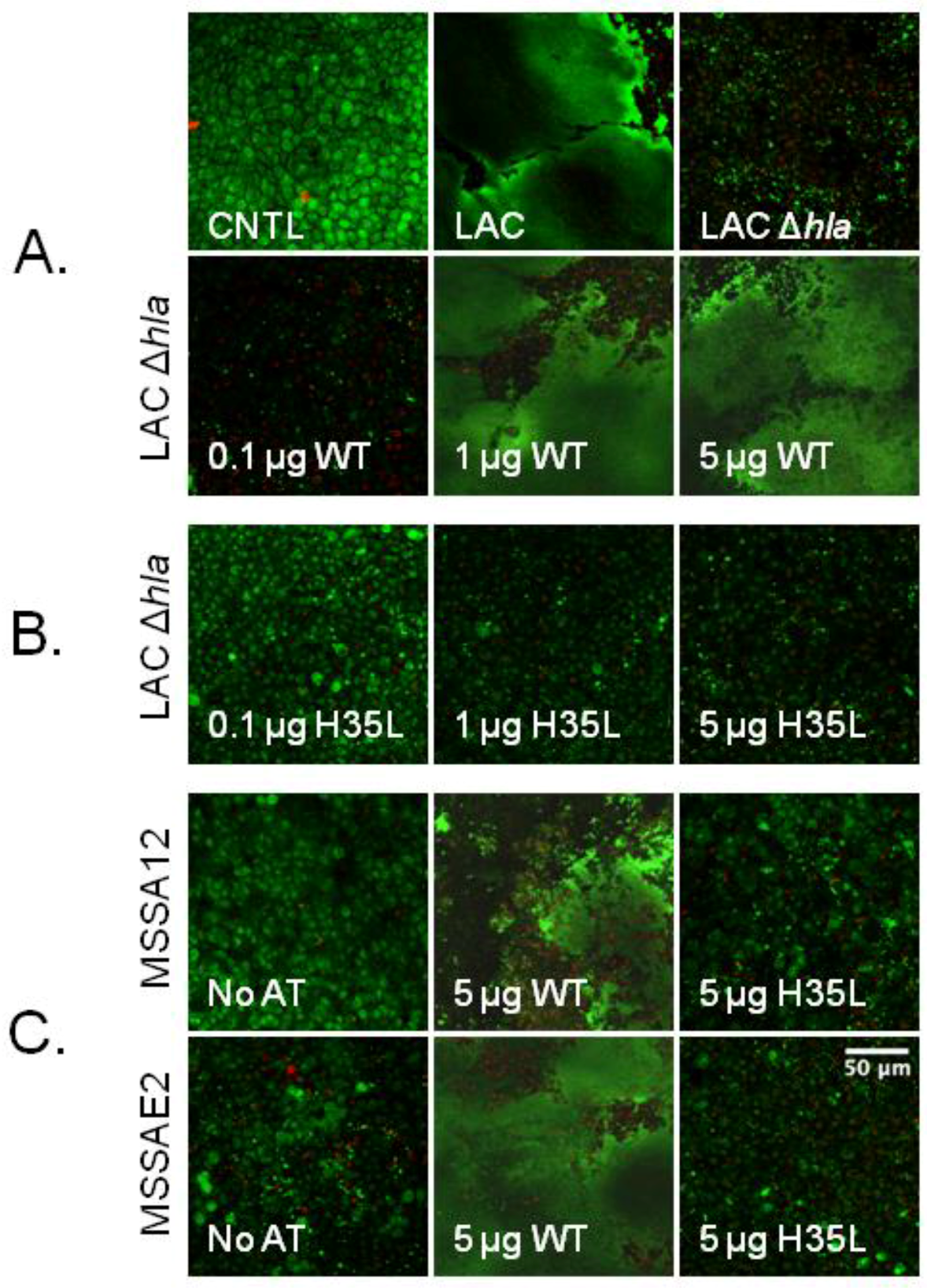
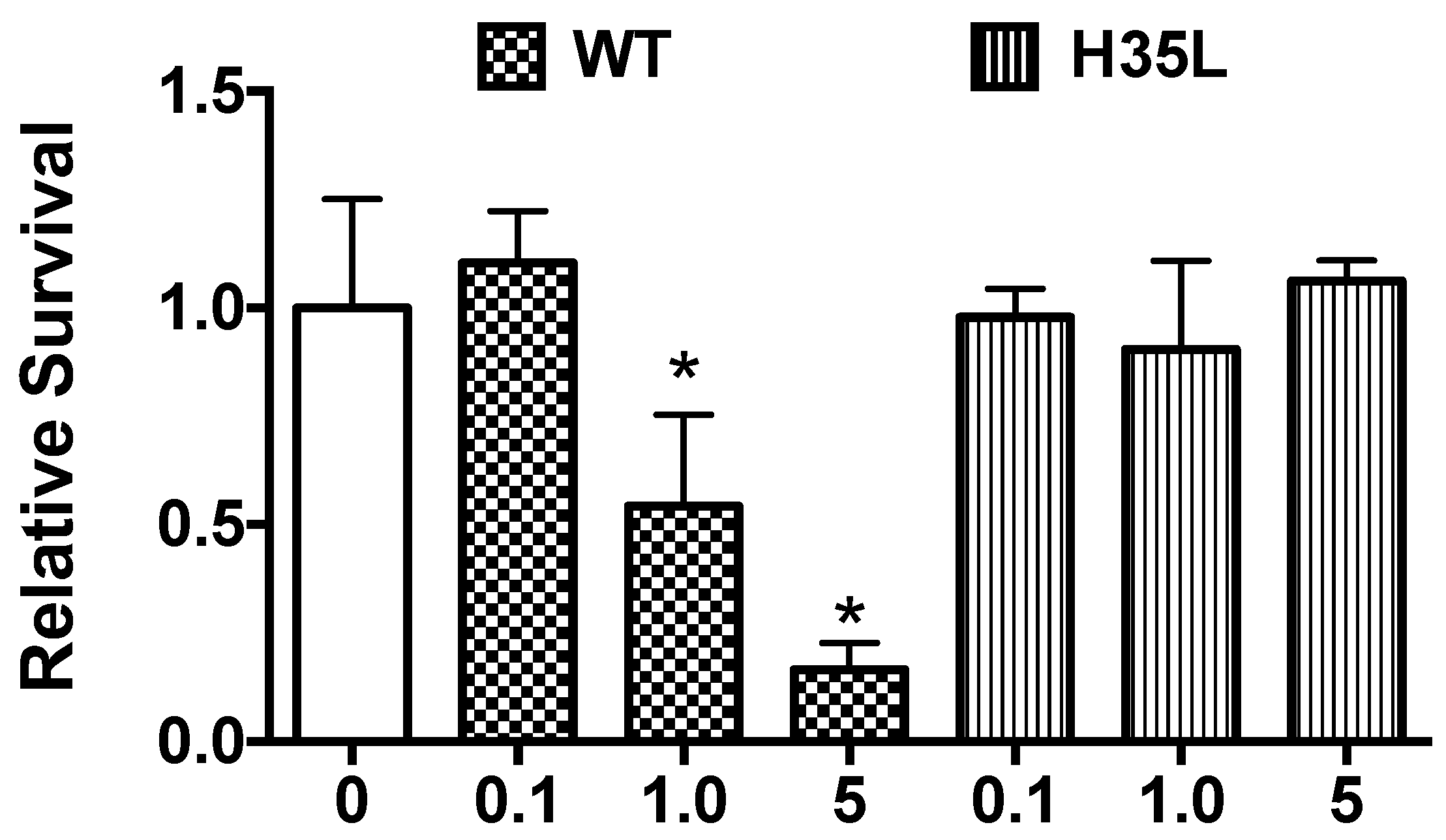
2.6. Rescue of Biofilm Defect with Exogenous AT
3. Discussion
4. Materials and Methods
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Elgharably, H.; Mann, E.; Awad, H.; Ganesh, K.; Ghatak, P.D.; Gordillo, G.; Sai-Sudhakar, C.B.; Roy, S.; Wozniak, D.J.; Sen, C.K. First evidence of sternal wound biofilm following cardiac surgery. PLoS ONE 2013, 8, e70360. [Google Scholar] [CrossRef] [PubMed]
- James, G.A.; Swogger, E.; Wolcott, R.; Pulcini, E.; Secor, P.; Sestrich, J.; Costerton, J.W.; Stewart, P.S. Biofilms in chronic wounds. Wound Repair Regener. 2008, 16, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Percival, S.L.; Hill, K.E.; Williams, D.W.; Hooper, S.J.; Thomas, D.W.; Costerton, J.W. A review of the scientific evidence for biofilms in wounds. Wound Repair Regener. 2012, 20, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.L.; Neumann, C.; Cook, C.; Chukwuma, U.; Ellis, M.W.; Hospenthal, D.R.; Murray, C.K. Epidemiology of Staphylococcus aureus blood and skin and soft tissue infections in the US military health system, 2005–2010. JAMA 2012, 308, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Tracy, L.A.; Furuno, J.P.; Harris, A.D.; Singer, M.; Langenberg, P.; Roghmann, M.C. Staphylococcus aureus infections in US veterans, Maryland, USA, 1999–2008. Emerg. Infect. Dis. 2011, 17, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.E.; Lowy, F.D.; Wright, L.N.; Larson, E.L. Meticillin-resistant Staphylococcus aureus among US prisoners and military personnel: Review and recommendations for future studies. Lancet Infect. Dis. 2006, 6, 335–341. [Google Scholar] [CrossRef]
- Diekema, D.J.; Richter, S.S.; Heilmann, K.P.; Dohrn, C.L.; Riahi, F.; Tendolkar, S.; McDanel, J.S.; Doern, G.V. Continued emergence of USA300 methicillin-resistant Staphylococcus aureus in the United States: Results from a nationwide surveillance study. Infect. Control Hosp. Epidemiol. 2014, 35, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, C.P.; Boyle-Vavra, S.; Adem, P.V.; Lee, J.C.; Husain, A.N.; Clasen, J.; Daum, R.S. Comparison of virulence in community-associated methicillin-resistant Staphylococcus aureus pulsotypes usa300 and usa400 in a rat model of pneumonia. J. Infect. Dis. 2008, 198, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Berube, B.J.; Bubeck Wardenberg, J. Staphylococcus aureus alpha-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [PubMed]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Wardenburg, J.B. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, T.K.; Pallister, K.B.; DuMont, A.L.; DeWald, M.; Watkins, R.L.; Pallister, E.Q.; Malone, C.; Griffith, S.; Horswill, A.R.; Torres, V.J.; et al. Alpha-toxin induces programmed cell death of human T cells, B cells, and monocytes during USA300 infection. PLoS ONE 2012, 7, e36532. [Google Scholar] [CrossRef] [PubMed]
- Sengers, R.C. Hemolytic action of staphylococcal alpha-hemolysin on human erythrocytes in a Na plus-and K plus-containing suspending fluid. Antonie Van Leeuwenhoek 1970, 36, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Tabor, D.E.; Yu, L.; Mok, H.; Tkaczyk, C.; Sellman, B.R.; Wu, Y.; Oganesyan, V.; Slidel, T.; Jafri, H.; McCarthy, M.; et al. Staphylococcus aureus alpha-toxin is conserved among diverse hospital respiratory isolates collected from a gobal surveillance study and is neutralized by monoclonal antibody MEDI4893. Antimicrob. Agents Chemother. 2016, 60, 5312–5321. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J.; Lin, Y.-C.; Gillman, A.N.; Parks, P.J.; Schlievert, P.M.; Peterson, M. Alpha-Toxin Promotes Mucosal Biofilm Formation by Staphylococcus aureus. Front. Cell. Infect. Microbiol. 2012, 2, 64. [Google Scholar] [CrossRef] [PubMed]
- Den Reijer, P.M.; Haisma, E.M.; Toom, N.A.L.; Willemse, J.; Koning, R.A.; Demmers, J.A.; Dekkers, D.H.; Rijkers, E.; El Ghalbzouri, A.; Nibbering, P.H.; et al. Detection of Alpha-Toxin and Other Virulence Factors in Biofilms of Staphylococcus aureus on Polystyrene and a Human Epidermal Model. PLoS ONE 2016, 11, e0145722. [Google Scholar]
- O’Reilly, M.; Kreiswirth, B.; Foster, T.J. Cryptic alpha-toxin gene in toxic shock syndrome and septicaemia strains of Staphylococcus aureus. Mol. Microbiol. 1990, 4, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Tenover, F.C.; McAllister, S.; Fosheim, G.; McDougal, L.K.; Carey, R.B.; Limbago, B.; Lonsway, D.; Patel, J.B.; Kuehnert, M.J.; Gorwitz, R. Characterization of Staphylococcus aureus isolates from nasal cultures collected from individuals in the United States in 2001 to 2004. J. Clin. Microbiol. 2008, 46, 2837–2841. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J.; Parks, P.J.; Peterson, M.L. A mucosal model to study microbial biofilm development and anti-biofilm therapeutics. J. Microbiol. Methods 2013, 92, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Ando, E.; Monden, K.; Mitsuhata, R.; Kariyama, R.; Kumon, H. Biofilm formation among methicillin-resistant Staphylococcus aureus isolates from patients with urinary tract infection. Acta Med. Okayama 2004, 58, 207–214. [Google Scholar] [PubMed]
- Enright, M.C.; Day, N.P.; Davies, C.E.; Peacock, S.J.; Spratt, B.G. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 2000, 38, 1008–1015. [Google Scholar] [PubMed]
- Kennedy, A.D.; Otto, M.; Braughton, K.R.; Whitney, A.R.; Chen, L.; Mathema, B.; Mediavilla, J.R.; Byrne, K.A.; Parkins, L.D.; Tenover, F.C.; et al. Epidemic community-associated methicillin-resistant Staphylococcus aureus: Recent clonal expansion and diversification. Proc. Natl. Acad. Sci. USA 2008, 105, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Tkaczyk, C.; Hamilton, M.M.; Datta, V.; Yang, X.P.; Hilliard, J.J.; Stephens, G.L.; Sadowska, A.; Hua, L.; O’Day, T.; Suzich, J.; et al. Staphylococcus aureus alpha toxin suppresses effective innate and adaptive immune responses in a murine dermonecrosis model. PLoS ONE 2013, 8, e75103. [Google Scholar] [CrossRef] [PubMed]
- David, M.Z.; Taylor, A.; Lynfield, R.; Boxrud, D.J.; Short, G.; Zychowski, D.; Boyle-Vavra, S.; Daum, R.S. Comparing pulsed-field gel electrophoresis with multilocus sequence typing, spa typing, staphylococcal cassette chromosome mec (SCCmec) typing, and PCR for panton-valentine leukocidin, arcA, and opp3 in methicillin-resistant Staphylococcus aureus isolates at a U.S. Medical Center. J. Clin. Microbiol. 2013, 51, 814–819. [Google Scholar] [PubMed]
- Fisher, A.; Webber, B.J.; Pawlak, M.T.; Johnston, L.; Tchandja, J.B.; Yun, H. Epidemiology, microbiology, and antibiotic susceptibility patterns of skin and soft tissue infections, Joint Base San Antonio-Lackland, Texas, 2012–2014. MSMR 2015, 22, 2–6. [Google Scholar] [PubMed]
- Pardos de la Gandara, M.; Garay, J.A.R.; Mwangi, M.; Tobin, J.N.; Tsang, A.; Khalida, C.; D’Orazio, B.; Kost, R.G.; Leinberger-Jabari, A.; Coffran, C.; et al. Molecular Types of Methicillin-Resistant Staphylococcus aureus and Methicillin-Sensitive S. aureus Strains Causing Skin and Soft Tissue Infections and Nasal Colonization, Identified in Community Health Centers in New York City. J. Clin. Microbiol. 2015, 53, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Stulik, L.; Malafa, S.; Hudcova, J.; Rouha, H.; Henics, B.Z.; Craven, D.E.; Sonnevend, A.M.; Nagy, E. alpha-Hemolysin activity of methicillin-susceptible Staphylococcus aureus predicts ventilator-associated pneumonia. Am. J. Respir. Crit. Care. Med. 2014, 190, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.J.; Shivshankar, P.; Stol, K.; Trakhtenbroit, S.; Sullam, P.M.; Sauer, K.; Hermans, P.W.; Orihuela, C.J. The pneumococcal serine-rich repeat protein is an intra-species bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. PLoS Pathog. 2010, 6, e1001044. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, V.; Peng, L.; Damschroder, M.M.; Cheng, L.; Sadowska, A.; Tkaczyk, C.; Sellman, B.R.; Wu, H.; Dall’Acqua, W.F. Mechanisms of neutralization of a human anti-alpha-toxin antibody. J. Biol. Chem. 2014, 289, 29874–29880. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.; Hilliard, J.J.; Shi, Y.; Tkaczyk, C.; Cheng, L.I.; Yu, X.; Datta, V.; Ren, S.; Feng, H.; Zinsou, R.; et al. Assessment of an anti-alpha-toxin monoclonal antibody for prevention and treatment of Staphylococcus aureus-induced pneumonia. Antimicrob. Agents Chemother. 2014, 58, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Tkaczyk, C.; Hua, L.; Varkey, R.; Shi, Y.; Dettinger, L.; Woods, R.; Barnes, A.; MacGill, R.S.; Wilson, S.; Chowdhury, P.; et al. Identification of anti-alpha toxin monoclonal antibodies that reduce the severity of Staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin. Vaccine Immunol. 2012, 19, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.A.; Gill, S.R.; Chang, R.F.; Phan, T.H.; Chen, J.H.; Davidson, M.G.; Lin, F.; Lin, J.; Carleton, H.A.; Mongodin, E.F.; et al. Complete genome sequence of USA300, an epidemic clone of community-acquired methicillin-resistant Staphylococcus aureus. Lancet 2006, 367, 731–739. [Google Scholar] [CrossRef]
- Spaan, A.N.; van Strijp, J.A.G.; Torres, V.J. Leukocidins: Staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol. 2017, 15, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Tawk, M.Y.; Zimmermann-Meisse, G.; Bossu, J.L.; Potrich, C.; Bourcier, T.; Serra, M.D.; Poulain, B.; Prevost, G.; Jover, E. Internalization of staphylococcal leukotoxins that bind and divert the C5a receptor is required for intracellular Ca(2+) mobilization by human neutrophils. Cell Microbiol. 2015, 17, 1241–1257. [Google Scholar] [CrossRef] [PubMed]
- Ortines, R.V.; Liu, H.; Cheng, L.I.; Cohen, T.S.; Lawlor, H.; Gami, A.; Wan, Y.; Dillen, C.A.; Archer, N.K.; Miller, R.J.; et al. Neutralizing alpha-toxin accelerates healing of Staphylococcus aureus-infected wounds in nondiabetic and diabetic mice. Antimicrob. Agents Chemother. 2018, 62, e02288-17. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, J.J.; Datta, V.; Tkaczyk, C.; Hamilton, M.; Sadowska, A.; Jones-Nelson, O.; O’Day, T.; Weiss, W.J.; Szarka, S.; Nguyen, V.; et al. Anti-alpha-toxin monoclonal antibody and antibiotic combination therapy improves disease outcome and accelerates healing in a Staphylococcus aureus dermonecrosis model. Antimicrob. Agents Chemother. 2015, 59, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Wardenburg, J.B.; Gardner, D.J.; Long, D.; Whitney, A.R.; Braughton, K.R.; Schneewind, O.; DeLeo, F.R. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 2010, 202, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- McDougal, L.K.; Steward, C.D.; Killgore, G.E.; Chaitram, J.M.; McAllister, S.K.; Tenover, F.C. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: Establishing a national database. J. Clin. Microbiol. 2003, 41, 5113–5120. [Google Scholar] [CrossRef] [PubMed]
- Christensen, G.D.; Simpson, W.A.; Younger, J.J.; Baddour, L.M.; Barrett, F.F.; Melton, D.M.; Beachey, E.H. Adherence of coagulase-negative staphylococci to plastic tissue culture plates: A quantitative model for the adherence of staphylococci to medical devices. J. Clin. Microbiol. 1985, 22, 996–1006. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methicillin Phenotype | Isolate | PFGE USA Type a | MLST b | Spa c | Q113Stop d |
|---|---|---|---|---|---|
| MRSA | 4 | 100 | ST231 | t002 | ND |
| 5 | 100 | ST5 | t088 | ND | |
| 12 | 100 | ST5 | t002 | ND | |
| 13 | 100 | ST5 | t002 | ND | |
| A2 | 100 | ST5 | t002 | ND | |
| A3 | 100 | ST5 | t002 | ND | |
| B1 | 100 | ST5 | t002 | ND | |
| C1 | 100 | ST5 | t105 | ND | |
| C2 | 100 | ST5 | t002 | ND | |
| C3 | 100 | ST5 | t002 | ND | |
| 9 | 300 | ST8 | t008 | ND | |
| 10 | 300 | ST8 | t008 | ND | |
| 15 | 300 | ST8 | t008 | ND | |
| 16 | 300 | ST8 | t008 | ND | |
| A1 | 300 | ST8 | t008 | ND | |
| B2 | 300 | ST8 | t008 | ND | |
| 2 | Non-typeable | ST5 | t242 | ND | |
| B3 | Non-typeable | ST88 | t11140 | ND | |
| MSSA | D3 | 100 | ST231 | t548 | no |
| D1 | 200 | ST30 | t012 | yes | |
| 1 | 200 | ST30 | t012 | yes | |
| 5 | 200 | ST30 | t1577 | yes | |
| E2 | 200 | ST30 | t012 | yes | |
| 13 | 200 | ST45 | t015 | no | |
| 11 | 300 | ST8 | unknown | no | |
| 8 | 400 | ST1 | t127 | no | |
| 9 | 400 | ST1 | t127 | no | |
| 2 | 600 | ST45 | t073 | no | |
| 3 | 900 | ST15 | t084 | no | |
| E1 | 900 | ST15 | t084 | no | |
| 12 | Non-typeable | ST30 | t1577 | yes | |
| E3 | Non-typeable | ST30 | t012 | yes | |
| 4 | Non-typeable | ST39 | t2271 | no | |
| 10 | Non-typeable | ST5 | t010 | no | |
| F2 | Non-typeable | ST5 | t002 | no | |
| 6 | Non-typeable | ST8 | unknown | no | |
| F3 | Non-typeable | ST97 | t521 | no | |
| D2 | non-typeable | ST97 | t267 | no |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, M.J.; Schaaf, E.; Breshears, L.M.; Wallis, H.W.; Johnson, J.R.; Tkaczyk, C.; Sellman, B.R.; Sun, J.; Peterson, M.L. Alpha-Toxin Contributes to Biofilm Formation among Staphylococcus aureus Wound Isolates. Toxins 2018, 10, 157. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10040157
Anderson MJ, Schaaf E, Breshears LM, Wallis HW, Johnson JR, Tkaczyk C, Sellman BR, Sun J, Peterson ML. Alpha-Toxin Contributes to Biofilm Formation among Staphylococcus aureus Wound Isolates. Toxins. 2018; 10(4):157. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10040157
Chicago/Turabian StyleAnderson, Michele J., Emily Schaaf, Laura M. Breshears, Heidi W. Wallis, James R. Johnson, Christine Tkaczyk, Bret R. Sellman, Jisun Sun, and Marnie L. Peterson. 2018. "Alpha-Toxin Contributes to Biofilm Formation among Staphylococcus aureus Wound Isolates" Toxins 10, no. 4: 157. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10040157