Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins



1
California Animal Health and Food Safety Laboratory, San Bernardino Branch, School of Veterinary Medicine, University of California-Davis, San Bernardino, CA 92408, USA
2
Department of Microbiology and Molecular Genetics, University of Pittsburgh School of Medicine, Room 420, Bridgeside Point II Building, 450 Technology Drive, Pittsburgh, PA 15219, USA
*
Author to whom correspondence should be addressed.
Toxins 2018, 10(5), 212; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10050212
Submission received: 27 April 2018
/
Revised: 18 May 2018
/
Accepted: 19 May 2018
/
Published: 22 May 2018
(This article belongs to the Collection Clostridium difficile/Clostridium sordellii and Clostridium perfringens Toxins)
Abstract
:Clostridium perfringens uses its large arsenal of protein toxins to produce histotoxic, neurologic and intestinal infections in humans and animals. The major toxins involved in diseases are alpha (CPA), beta (CPB), epsilon (ETX), iota (ITX), enterotoxin (CPE), and necrotic B-like (NetB) toxins. CPA is the main virulence factor involved in gas gangrene in humans, whereas its role in animal diseases is limited and controversial. CPB is responsible for necrotizing enteritis and enterotoxemia, mostly in neonatal individuals of many animal species, including humans. ETX is the main toxin involved in enterotoxemia of sheep and goats. ITX has been implicated in cases of enteritis in rabbits and other animal species; however, its specific role in causing disease has not been proved. CPE is responsible for human food-poisoning and non-foodborne C. perfringens-mediated diarrhea. NetB is the cause of necrotic enteritis in chickens. In most cases, host–toxin interaction starts on the plasma membrane of target cells via specific receptors, resulting in the activation of intracellular pathways with a variety of effects, commonly including cell death. In general, the molecular mechanisms of cell death associated with C. perfringens toxins involve features of apoptosis, necrosis and/or necroptosis.
Key Contribution: This review summarizes current knowledge of Clostridium perfringens toxins action on target tissues and cells. There is a special emphasis on cell death pathways activated by these toxins.
1. Introduction
The ability of Clostridium perfringens to produce a large repertoire of toxins makes it a serious pathogen of humans and animals, being able to produce histotoxic, enteric and/or enterotoxemic diseases [1,2]. Toxin production varies significantly among C. perfringens strains, which is the basis for a classification system that was traditionally based upon the presence of encoding genes for alpha (CPA), beta (CPB), epsilon (ETX) and iota (ITX) toxins [1]. This typing system, however, was recently revised to include two additional types, i.e., C. perfringens type F strains producing enterotoxin (CPE) but not CPB, ETX or ITX, and C. perfringens type G strains producing necrotic enteritis B-like toxin (NetB) [3] (Table 1).
The cellular action of many C. perfringens toxins involves initial binding to a receptor located on the plasma membrane of target cells, followed by activation of intracellular pathways and a variety of cytopathic effects that eventually lead to cell death [1,4]. Recent studies of pathogen-induced host cell death have revealed a variety of complex mechanisms involved in this outcome, once believed to be simple and merely incidental [5,6,7]. Beyond the classic concepts of necrosis (an accidental and uncontrolled type of cell death) and apoptosis (a programmed mechanism of cell death), it is now well known that many pathogens are capable of triggering several other pathways which have been extensively reviewed [8,9,10,11]. These include necroptosis (or “programed necrosis”), autophagy, pyroptosis, anoikis, and several others. Understanding the mechanisms of action and factors needed for a pathogen to kill host cells is critical to deciphering bacterial pathogenesis, and it represents a key piece of knowledge to explore novel therapeutic approaches. Here we review the current knowledge on the mechanisms of action of C. perfringens toxins, with special emphasis on their effects on cell death.
2. C. perfringens Alpha Toxin (CPA)
2.1. Toxin Genetics, Structure and Role in Disease
The gene encoding CPA is situated in a stable region within the bacterial chromosome and it is present in all C. perfringens isolates [1,12]. CPA is a zinc metalloenzyme composed of 370 amino acids, which binds to host cell membranes in the presence of calcium ions [13]. It is divided into two main domains: the catalytic N-domain and the membrane binding C-domain [13,14]. Only the latter is immunoprotective [1]. CPA also has a central loop domain containing a ganglioside (GM1a) binding site [14].
The lethal, hemolytic, and dermonecrotic CPA is the most important virulence factor involved in human gas gangrene (clostridial myonecrosis) [15] which is characterized by necrosis of skeletal muscle, subcutaneous edema and emphysema, shock, multiple organ failure and death [16,17]. In addition, a member of the cholesterol-dependent cytolysin family of pore-forming toxins, named Perfringolysin O (PFO) [18], has been shown to act synergistically with CPA to produce the pathological effects observed in gas gangrene [19]. The role of PFO by itself in the disease, however, seems to be minor [1]. Although C. perfringens type A has been associated with cases of gas gangrene in animals, the role of CPA in these species has not been established [4].
2.2. General Mechanism of Action
The activity of CPA is highly complex, and it varies among cell types due to factors such as the ratio of phosphatidylcholine (PC) to sphingomyelin (SM) in the plasma membrane, and it is also influenced by local toxin concentrations. For these reasons, many different pathways are affected during CPA action (Figure 1). CPA hydrolyzes PC and SM in the plasma membrane, producing diacylglycerol (DAG) and ceramide (CER), respectively [14]. In addition to these activities, CPA can indirectly activate endogenous host enzymes which have similar phospholipase and sphingomyelinase properties through the interaction with Gi type GTP-binding proteins (Gi-GTP-BPs) (Figure 1) [20]. However, CPA action is not limited only to membrane disruption. The ganglioside-binding site of the toxin in the central loop domain promotes the interaction and tethering of tropomyosin receptor kinase A (TrkA) on cell membranes [21], leading to activation of the MEK/ERK pathway [14]. Interestingly, ganglioside-deficient DonQ or GM95 cells are highly sensitive to CPA action [22,23,24]. Gangliosides are glycosphingolipids that may reduce the fluidity of the plasma membrane, resulting in a tighter packing of phospholipids, and are also associated with electrostatic changes affecting the availability of substrates [22,25]. These effects could interfere with the membrane-disrupting activity of CPA, thus protecting cells from cell damage. Sialic acids are important in the structure of gangliosides [26], and their removal by C. perfringens sialidases increased cell sensitivity to CPA in vitro and in vivo [22], implying a potential synergism between CPA and sialidases. This synergistic effect of sialidases has also been proved to increase binding/activity of other C. perfringens toxins (particularly NanI sialidase) including CPB, CPE and ETX [27,28].
As mentioned before, CPA can bind to and act on membrane PC and SM, in addition to activating Gi-GTP-BPs, triggering different pathways depending on the cell type involved (Figure 1). For instance, horse erythrocytes are lysed by intrinsic CPA phospholipase activity on PC, and probably also through activation of endogenous phosphatidylinositol-specific phospholipase C (PI-PLC) [29], whereas sheep erythrocytes which contain only traces of PC are affected by CPA mainly by activation of the SM metabolism via GTP-BPs [20,30]. In rabbit neutrophils and DonQ cells, CPA-induced superoxide anion generation occurs through the activation of endogenous phospholipase C (PLC) and phosphorylation of PI3K via TrkA receptor, and further phosphorylation of PDK1, PKCθ, the MEK1/2, ERK1/2 system and NF-κB [31,32]. This pathway is also important in the generation of IL-8 by A549 cells, in addition to the activation of p38 MAPK, which is believed to stabilize IL-8 mRNA [21]. In this regard, the scarcity of neutrophils within gas gangrene-affected tissues and their accumulation on the vascular endothelium could be due, at least in part, to the effect of elevated IL-8 promoting firm adhesion of neutrophils to extracellular matrix proteins [21]. Recent studies have also reported impaired differentiation and replenishment of neutrophils in peripheral blood induced by CPA [33], effects that have been attributed to alterations in GM1a-containing lipid rafts in those cells [34]. Furthermore, the persistence of an anaerobic microenvironment during gas gangrene is believed to be the result of CPA-induced activation of arachidonic acid (AA) metabolism through phospholipase A2 (PLA2), generating prostaglandins, tromboxanes and leukotriens, which are associated with inflammation, muscle contraction, platelet aggregation and vasoconstriction, resulting in decreased perfusion [12].
2.3. Mechanisms of Cell Death
Lytic concentrations of CPA can result in extensive degradation of plasma membrane and lactate dehydrogenase (LDH) release [22,35], which is characteristic of necrosis [36]. Sub-lytic concentrations of CPA are associated with activation of the MEK/ERK pathway and generation of reactive oxygen species (ROS) [23], which in certain amounts can lead to oxidative stress in cells and activate intrinsic mechanisms of apoptosis [37]. A recent study [24] suggests that the SM metabolism induced by a low CPA dose is associated with the generation of pro-apoptotic mediators, such as CER, N-acylethanolamine and saturated fatty acids, and the release of mitochondrial cytochrome C, activation of caspase-3 and increased exposure of phosphatidylserine in GM95 cells. The role of CER as a second messenger involved in apoptosis has been previously described [38,39]. CER is processed to sphingosine (SPH) which can accumulate inside of and induce rupture of lysosomes, releasing lysosomal proteases involved in apoptosis [40]. ROS production can also lead to lysosomal damage [41] when CPA is endocytosed through cholesterol-containing caveolae and, within endosomal compartments, it activates signaling pathways along its trafficking routes [32]. High levels of intracytoplasmic Ca2+ (iCa2+) commonly play a role in pre-lethal events of both apoptosis and necrosis [42,43]. CPA leads to the formation of sphingosine-1-phosphate (S1P) from sphingosine (SPH) [30], and inositol trisphosphate (IP3) from PIP2 [12] (Figure 1); both S1P and IP3 are associated with mobilization and increase of iCa2+ [44]. To our knowledge, however, the role of iCa2+ as a trigger for specific cell death pathways associated with CPA has not been investigated.
3. C. perfringens Beta Toxin (CPB)
3.1. Toxin Genetics, Structure and Role in Disease
The cpb gene is carried on large virulence plasmids which can contain additional toxin genes such as cpe or the toxin perfringens Large (tpeL) [45,46]. CPB is encoded as a prototoxin of 336 amino acids, including a 27-amino acid signal sequence which is removed during secretion, resulting in a mature toxin of ~35 kDa [47,48]. The deduced amino acid sequence of CPB shares significant homology with delta toxin and beta-pore forming toxins (β-PFTs) of Staphylococcus aureus [47]. CPB is classified as a clostridial β-PFT of the α-hemolysin family [49], and it is characterized for being extremely sensitive to trypsin and other proteases [50]. Due to this trypsin sensitivity, CPB is only active in the presence of trypsin inhibitors, both in vitro and in vivo [4]. CPB variants exist that may possess different trypsin sensitivity and in vitro cytotoxic activity [51].
CPB is produced by C. perfringens types B and C and it is responsible for diseases in several animal species and humans [1]. Type B isolates have been associated with fatal hemorrhagic dysentery in sheep, whereas type C isolates cause necrotic enteritis and/or enterotoxemias in several livestock species, and enteritis necroticans (Pigbel) in humans [1,4,52]. Studies using isogenic-null mutants demonstrated that CPB is necessary and sufficient to reproduce the intestinal pathology of type C isolates in rabbit intestinal loops [53,54], and in goats, which are natural hosts for C. perfringens type C disease [55]. CPB was also shown to be responsible for lethality in a mouse enterotoxemia model [56].
3.2. General Mechanism of Action
The pathology of the spontaneous disease associated with CPB is characterized by hemorrhage and necrosis of the epithelium of the small and sometimes large intestine. CPB-associated damage begins in the intestinal mucosa but may progress to all layers of the intestine. Fibrin thrombi occluding the superficial microvasculature present in the lamina propria are characteristic of CPB-associated intestinal disease [4]. Whether the mucosal epithelium is the primary target for the toxin to induce damage or if this is secondary to vascular thrombosis has been a frequent matter of debate. In the rabbit intestinal loop model, inoculation of the wild-type C. perfringens type C isolate CN3685 produced necrosis of the villous tip before thrombosis was apparent, suggesting early intestinal epithelial injury [53]. On the other hand, CPB was demonstrated by immunohistochemistry only in endothelial cells within the lamina propria in naturally-occurring cases of necrotic enteritis in piglets [57] and in a human patient [58], as well as in an experimental infection model in pigs associated with early vascular lesions [59]. In all those studies, however, tissue necrosis was already present and CPB binding to epithelial cells was not detected. Further CPB immunohistochemical studies on porcine jejunal explants showed no binding of the toxin to epithelial cells, and the presence of an intact epithelial layer inhibited CPB detection into deeper intestinal layers of the intestine [60]. The same study showed no cytopathic effect in cultured intestinal porcine epithelial cells (IPEC-J2) and primary jejunal epithelial cells when co-incubated with recombinant CPB (rCPB). An initial epithelial damage was proposed for CPB to reach the microvasculature in the lamina propria in the porcine intestinal model. However, since the supernatants of two C. perfringens type C strains induced IPEC-J2 cell damage that was not prevented with anti-CPB antibodies, it was concluded that CPB was not responsible for the cytopathic effect observed [60]. In addition, an indirect vascular effect of CPB in mouse skin was observed, in which the toxin induced the release of substance P (SP), an NK1 agonist from sensory neurons, leading to the release of TNF-α and plasma extravasation [61,62].
As an oligomerizing, pore-forming toxin, CPB is capable of forming functional pores in plasma membranes of susceptible cells [63,64]. These pores allow the efflux of K+ and the entry of Ca2+, Na+ and Cl− into the cells, followed by cell swelling [64]. Potassium efflux induced by CPB promotes phosphorylation of p38 MAP and JNK kinases, which activate pathways associated with host cell survival and adaptation [65]. Susceptibility of cells to CPB action has been tested in different human hematopoietic tumor cell lines, with THP-1 and U937 cells showing the highest CPB-induced cytotoxicity compared to HL-60, BALL-1 and MOLT-4 [65]. Using primary porcine aortic endothelial cells, CPB was demonstrated to induce rapid disruption of the actin cytoskeleton, cell border retraction and cell shrinkage [66]. Differences in cell susceptibility clearly suggest that the toxin binds to specific receptors. It has been recently shown that CPB putatively binds to the ATP-gated P2X7 receptor, after which ATP is released from the cell through an ATP channel called Pannexin 1 (Panx1) [67,68]. How this ATP release related to CPB binding occurs is not known; however, it is not associated with cell lysis [68]. Perhaps, an initial influx of Ca2+ through the CPB pores could induce mitochondrial ATP production and later release from the affected cell [69]. It is presumed that this rapid peak of ATP release from cells through Panx1 could stimulate further CPB oligomer formation and consequent cytotoxicity (Figure 2) [68]. Functional and physical interactions of Panx1 with P2X7 receptor bound to CPB would influence Panx1 channels opening promoting ATP release [68]. Both enterocytes and endothelial cells express the P2X7 receptor and Panx1 [70,71], but their role in CPB-associated disease in vivo needs further investigation.
3.3. Mechanisms of Cell Death
Studies using porcine endothelial cells in vitro have shown that rCPB rapidly induces cellular events consistent with necrosis, including LDH release and propidium iodide (PI) intake [72]. Both events were inhibited by calpain inhibitors and necrostatin-1, a RIP-1 inhibitor, which suggested that CPB-induced necrotic cell death was not a passive event but occurred through a programmed biochemical pathway. The authors concluded their necrostatin effects indicated CPB induces necroptosis. However, it is worth mentioning that RIP-1 (the necrostain target) can also be involved in apoptosis [73]. On the other hand, the referred study detected low levels of caspase-3 activation, but not appreciable DNA fragmentation, suggesting that apoptosis was not an important cell death pathway at the toxin concentrations and times of incubation tested. How caspase-3 activation occurred in those cells was not explored. It may be hypothesized that the rise of iCa2+ could play a key role since it has been associated with both apoptosis and necroptosis through the activation of calpains [8,43].
4. C. perfringens Epsilon Toxin (ETX)
4.1. Toxin Genetics, Structure and Role in Disease
The etx gene is localized on plasmids, where it can be associated with other toxin genes such as cpb2 and cpe [74]. ETX is secreted as a poorly active protototoxin of ~33 kDa, which is activated by intestinal proteases produced by the host such as trypsin, α-chymotrypsin, carboxypeptidases, and/or sometimes by λ-protease, which is produced by some strains of C. perfringens [75,76,77]. Removal of the 13 N-terminal and 29 C-terminal residues by trypsin or α-chymotrypsin results in a mature toxin that is 1000 times more toxic than the prototoxin. C. perfringens λ-protease removes only 10 of the N-terminal residues [75,78]. In an ex vivo study using caprine intestinal contents, it was demonstrated that ETX prototoxin is processed in a step-wise fashion into a stable, active ~27 kDa band on SDS-PAGE [79]. The ~27 kDa band was shown to contain three ETX species with varying C-terminal residues. Each of those ETX species was cytotoxic. This additional processing of ETX C-terminal sequences is apparently caused by intestinal carboxypeptidases, based upon inhibitor studies [79]. Those findings suggest that ETX activation in vivo is more complex than previously reported when using only purified proteases [77,79]. This toxin is classified as a heptameric β-PFTs of the aerolysin family given its structural similarity to aerolysin produced by Aeromonas sp., although no significant amino acid sequence identity is shared between them [49].
ETX, the third most potent clostridial toxin known after C. botulinum and C. tetani toxins, is synthesized by strains of C. perfringens types B and D. ETX-producing C. perfringens type D strains are the most common cause of clostridial enterotoxemia in sheep, goats and less frequently cattle [4,80,81]. Studies using isogenic C. perfringens etx -null mutants in sheep, goats and mice models have confirmed that ETX is the main virulence factor responsible for all lesions and symptoms due to C. perfringens type D enterotoxemia [82,83]. Because enterotoxemia results from toxin absorption from the gut into the circulation, toxoid vaccines are used to protect livestock against type D infections [84].
4.2. General Mechanism of Action
ETX is produced in the intestine, and even though fibrinonecrotic colitis may be present in cases of caprine enterotoxemia, the toxin mainly targets distant organs such as the central nervous system, lungs and heart [4]. It is believed that ETX increases intestinal permeability, hence facilitating its own absorption [4,84]. This effect is not fully understood, but it involves the opening of the mucosa tight junctions and degenerative changes in the lamina propria of the gut [85]. Microscopically, lesions in the brain tend to be multifocal and characterized by perivascular and intramural vascular edema, hemorrhage, and in more chronic cases, necrosis of the white matter and swelling of astrocytes and axons [4]. The latter is commonly symmetrical and bilateral [86]. The origin of these lesions is believed to be the result of an initial binding of the toxin to endothelial cells of the brain–blood barrier (BBB) since intravenous inoculation of ETX labeled with green-fluorescent protein (GFP) in mice revealed binding of the toxin to those cells [87]. After ETX binding, endothelial cells become swollen, vacuolated and necrotic [88]. By disrupting the BBB, leakage of fluids and proteins occurs, leading to an edema that causes mechanical damage and hypoxia of the neural parenchyma [83]. In response to this edema, astrocytes of rats and sheep overexpress aquaporin-4, which is believed to be the host’s attempt to reduce the excess of fluid from perivascular spaces [83,89]. In addition to this indirect effect on the brain, ETX can directly affect certain types of neurons and oligodendrocytes, but not astrocytes [90,91,92]. Mice injected with ETX fused to GFP also revealed severe kidney alterations, including degeneration of distal tubules and hemorrhages in the medulla [93]. However, renal lesions are not usually observed in natural disease and the so-called “pulpy kidney” is believed to be a postmortem change rather than a true lesion [84].
Binding and cytotoxic activities of ETX have been extensively studied using cell lines from renal origin of different species in which spontaneous cases of type D enterotoxemia have not been reported, including dogs (Madin–Darby canine kidney cells, MDCK), mice and humans. However, kidney cell lines from species naturally affected by ETX such as sheep and cattle are resistant [94].
The initial steps of ETX action involve binding to an unidentified cell surface receptor and heptamerization into a prepore on the membrane surface, followed by insertion into the plasma membrane to form an active pore [95,96,97]. Recently, it was reported that ETX activates neutral sphingomyelinase (nSMase) which in turn facilitates oligomer formation by producing ceramide in the plasma membrane [98]. In fact, knockdown of nSMase blocked oligomer formation and ETX-induced cell death in ACHN cells (derived from human renal adenocarcinoma) (Figure 3) [98]. Since that particular study used ETX isolated from C. perfringens culture, it is possible that impurities containing CPA, which also induces CER formation [24], could have been in part responsible for this effect, which might indicate a synergistic effect between these two toxins. This possibility, however, has not been explored. In vitro, ETX binds to the hepatitis A virus cellular receptor (HAVCR1) in MDCK and ACHN cells, facilitating cytotoxicity [99]. Nevertheless, induced expression of HAVCR1 in cells resistant to ETX did not result in sensitivity to the toxin [99]. The myelin and lymphocyte (MAL) protein has been found to be required for ETX cytotoxicity in several cell types [100]. MAL is expressed in many cells known to be targeted by ETX, including endothelial cells, renal cells and oligodendrocytes. Neurons, which have been suggested but not definitively proved to be sensitive to ETX, do not express MAL [101]. It has been hypothesized that MAL may act as a specific receptor for ETX, as a protein involved in assembly of a multi-protein complex required for ETX interaction with the plasma membrane, or even allowing a mechanism independent of pore formation [100,102].
4.3. Mechanisms of Cell Death
Mechanisms of cell death and intracellular pathways associated with ETX action are not fully characterized. However, the toxin induces cellular changes that are compatible with necrosis, including initial and marked cell swelling, followed by disappearance of mitochondria, blebbing, membrane disruption, ATP depletion, reduction of nucleus size and increased PI uptake; no DNA fragmentation occurs [81,103,104]. Pore formation in affected cells leads to a rapid loss of intracellular K+, entry of Cl− and Na+, followed later by an increase of iCa2+ [103]. Whether this imbalance of intracellular electrolytes is involved in activating specific downstream signaling events leading to cell death has not been determined, although it seems likely. ETX-affected cells lose important coenzymes critical for energy production, particularly nicotinamide adenine dinucleotide (NAD+ and NADH) and coenzyme A, contributing to the dissipation of the mitochondrial membrane potential and opening of the mitochondrial permeability transition pore [104,105]. Consistent with those findings, AMP-activated protein kinase, an intracellular sensor of ATP, is stimulated in ETX-affected cells [104]. In addition, apoptosis-inducing factor (AIF), a potent caspase-independent cell death factor, is translocated from mitochondria to the nucleus (Figure 3) [104]. Paracellular permeability to macromolecules is not significantly increased in renal cells treated with ETX in vitro, and actin cytoskeleton and organization of tight and adherens junctions are not affected [104,106]. In the brain, ETX can bind to certain types of neurons and cause a decrease in electrical membrane resistance [104], which is attributed to pore formation. This induces membrane depolarization associated with an increase of iCa2+, followed by the release of the excitatory neurotransmitter, glutamate [92,107]. Interestingly, ETX does not form pores in the plasma membrane of oligodendrocytes [92]; thus, the associated glutamate release, Ca2+ fluctuations and demyelination occurs by a different mechanism, involving intracellular pathways occurring as a consequence of ETX–MAL interaction [92,102].
5. C. perfringens Iota Toxin (ITX)
5.1. Toxin Genetics, Structure and Role in Disease
ITX is a clostridial binary toxin composed of an enzyme component (Ia) and a binding component (Ib) (Figure 4) [108]. Both components are encoded by two separate genes, iap and iab, which are located on large, potentially conjugative plasmids [109]. Ib is synthetized as an inactive form (100 kDa) which becomes active after proteolytic removal of a 20 kDa N-terminal fragment by trypsin or chymotrypsin [110].
ITX is produced by C. perfringens type E strains. Enteric disease associated with this toxinotype has been suggested in many animal species. However, the diagnosis in most of those cases was based only on isolation of C. perfringens type E from intestinal content of animals with hemorrhagic enteritis, which is not considered to be a diagnostic criterion for C. perfringens type E infection. In addition, Koch’s molecular postulates have not been fulfilled for type E disease. The specific role of ITX in disease remains, therefore, unclear [111]. In the past, cases of enterotoxemia in rabbits were attributed to C. perfringens type E, based on the detection of ITX in the intestine of affected animals. Nevertheless, it has been hypothesized that those cases were probably caused by an iota-like toxin produced by Clostridium spiroforme, a toxin that cross-reacts with ITX by currently available toxin detection methods. To date, there are no reliable diagnostic criteria or gold standards for the diagnosis of C. perfringens type E-associated disease in animals [4,111].
5.2. General Mechanism of Action
The lipolysis-stimulated lipoprotein receptor (LSR) has been reported as a cellular receptor for Ib, which also mediates the toxin entering into host cells [112]. It has also been shown that entrance of ITX into host cells can involve cell-surface antigen CD44-associated endocytosis [113]. Once bound to its receptor, Ib assembles into heptamers which insert into the host plasma membrane forming functional channels, allowing the movement of ions, and the translocation and endocytosis of Ia (Figure 4) [114,115,116,117]. The enzymatic Ia component is also secreted as an inactive form that needs proteolytic removal of 9 to 11 N-terminal residues [110]. The Ia C-domain is responsible for the ADP-ribosylating activity of the toxin, which involves the covalent attachment of ADP-ribose onto an Arg at residue 177 of actin [118]. This leads to the depolymerization of actin filaments and increase of G-actin monomers [118,119]. Depolymerization of the actin cytoskeleton results in changes in cell morphology and disorganization of intercellular tight and basolateral junctions, leading to an increase permeability of cultured monolayers of intestinal cells [114]. Both ITX components internalize in targeted cells via a Rho-dependent, clathrin-independent pathway and reach endocytic vesicles [120]. A small proportion of Ib is recycled back to the plasma membrane, enhancing further uptake of Ia [121]. After endocytosis, Ia translocates from late endosomes into the cytoplasm where it exerts its ADP-ribosylating activity (Figure 4). This translocation requires the acidic environment present in late endosomes [122].
5.3. Mechanisms of Cell Death
It has been demonstrated that Ib by itself can induce cytotoxic activity, particularly in two human cell lines, namely A431 (epithelial carcinoma) and A549 (lung adenocarcinoma) [123]. Those cytotoxic effects involved marked cell swelling, mitochondrial dysfunction, ATP depletion and increased IP intake, all features consistent with necrosis. Even though activation of the pro-apoptotic molecules Bax and Bak and cytochrome C release was also observed, no activation of caspase-3 was detected and incubation of cells with the pan-caspase inhibitor Z-VAD-FMK did not protect cells from Ib-induced loss of viability. In that study, internalization of Ib was indicated as required for cell survival, suggesting a role for endocytosis in protecting cells against pore formation associated with Ib. In another study, however, ITX treatment of Vero cells induced caspase-3 activation after 12 to 15 h of incubation [124]. This delayed induction of apoptotic cell death was attributed to the cytosolic stability of the ADP-ribosylating Ia targeting actin (Figure 4).
6. C. perfringens Enterotoxin (CPE)
6.1. Toxin Genetics, Structure and Role in Disease
The cpe gene can be positioned on either the chromosome or on plasmids, and the expression of the toxin only occurs during sporulation [125,126]. CPE consists of a single polypeptide containing 319 amino acids [127]. The C-terminal half of the toxin lacks cytotoxic activity, but it mediates receptor binding, which involves the presence of several tyrosine residues located on the last 30 amino acids of CPE [128,129,130]. On the other hand, the N-terminal half is particularly important for cytotoxicity given its key role in CPE oligomerization and pore formation [131].
CPE is a pore-forming toxin produced during sporulation mainly by strains of C. perfringens type F [3] (Table 1), formerly known as CPE+ type A strains. However, CPE-producing strains belonging to C and D toxinotypes are also common [132]. Type E strains can carry a silent cpe gene [133] or produce a variant CPE [134]. Experimental and epidemiological evidence indicates CPE as the main virulence factor involved in C. perfringens type F-associated food poisoning of humans [125,127]. Molecular Koch’s postulates have been fulfilled in rabbits for CPE-producing type F strains [135]. C. perfringens type F strains are also involved in about 5–10% of all cases of antibiotic-associated diarrhea [136]. Symptoms of CPE-associated food poisoning include diarrhea and abdominal cramps, and they usually resolve spontaneously after a day or two [125]. However, under certain predisposing conditions, including constipation or fecal impaction, CPE can be lethal. It is thought that by reducing the effect of diarrhea in those situations, the toxin would prolong its contact with the intestine, facilitating the absorption and action on extra-intestinal organs [137,138]. This enterotoxemic effect is supported by studies performed in mouse-ligated intestinal loops, in which the inoculated CPE gained access to the circulation and caused hyperpotassemia resulting in death [139].
6.2. General Mechanism of Action
The cellular action of CPE has been previously described in detail [127]. The cellular receptors for CPE are several members of the claudin family, essential components of tight junctions located at the apical contact region between epithelial and endothelial cells [127]. Specifically, claudins 3, 4, 6, 8, and 14 are proven CPE receptors [140,141,142]. Initial binding of CPE to claudin receptors results in the formation of a “small complex” of ~90 kDa [143]. The interaction of several (about 6) of these small complexes leads to CPE oligomerization and formation of a prepore on the plasma membrane surface [144]. The result is a “large complex” of ~450 kDa, named CH-1, which contains the CPE hexamer, receptor and non-receptor claudins [143]. The formation of a cation-permeating pore is initiated by the assembly of β-hairpin loops from CPE into a β-barrel that rapidly inserts into the plasma membrane of the target cell [145]. This CPE pore allows the influx of calcium which is essential for CPE to cause cell death (Figure 5 [146,147,148]).
6.3. Mechanisms of Cell Death
When enterocyte-like Caco-2 cells are exposed to low CPE doses, a low number of pores are formed and a modest influx of calcium follows, resulting in low calpain activation and apoptosis, characterized by cytochrome C release and caspase-3 activation [148,149]. However, with higher CPE doses, the formation of many pores results in massive influx of calcium and strong calpain activation, leading to minimal caspase-3 activation and morphologic cellular changes consistent with necrosis [149]. Extended incubation times in vitro intensify those morphological changes, resulting in the exposure of the basolateral cell surface; this allows additional binding of CPE and the formation of an even larger complex of ~600 kDa named CH-2 that contains claudin and occludin proteins [143,150]. In small intestinal-loops made in mice and rabbits, CPE induced dose-dependent histologic damage characterized by severe villous shortening, desquamation, and changes in epithelial cells consistent with necrosis (e.g., pyknosis, karyorrhexis) [139,151]. Histologic damage also occurs in the rabbit colon [152]. In contrast to the in vitro findings previously commented, treatment of mice intestinal loops with CPE induced a dose- and time- dependent caspase-3 activation [153]. This activation, however, was not essential for the development of intestinal lesions, because the use of the pan-caspase inhibitor Q-VD-OPh did not protect from intestinal damage or enterotoxemic death [153]. The role of calpain activation and a potential involvement of necroptosis in CPE-associated disease have not been explored.
7. C. perfringens Necrotic Enteritis B-Like Toxin (NetB)
7.1. Toxin Genetics, Structure and Role in Disease
The netB gene is located on a 42 kb pathogenicity locus named NELoc1, present on large, conjugative plasmids [154,155,156,157]. Like CPB, NetB shares partial sequence similarity with a β-PFT, named alpha hemolysin, from S. aureus [158].
According to the updated toxinotyping scheme, NetB is produced by C. perfringens type G strains (Table 1) [3]. Molecular Koch’s postulates for this toxin have been fulfilled in chicken disease models [159]. Those studies confirmed that NetB is the primary virulence factor involved in the development of avian necrotic enteritis, which is also supported by strong epidemiological evidence [159,160,161].
7.2. General Mechanism of Action
Death of enterocytes in cases of avian necrotic enteritis has been described as a consequence of an initial destruction of the lamina propria, the extracellular matrix and intercellular junctions [162]. NetB forms heptameric, hydrophilic pores with a central diameter of approximately 26 Å [163]. The specific receptor for NetB, however, has not been identified [161].
7.3. Mechanisms of Cell Death
As for many other pore-forming toxins, NetB pores allow an influx of ions such as Na+, Cl− and Ca2+ that may lead to osmotic cell lysis [160]. The toxin induces rounding and lysis of LMH cells, with subsequent LDH release [160]; although this is consistent with features of necrosis, specific pathways involved in cell death, to our knowledge, have not been explored for NetB action on the intestine.
8. Concluding Remarks
The body of knowledge about the molecular mechanisms of action of the main C. perfringens toxins has increased significantly over the past few years. It is interesting to observe, for example, how the hemolytic activity of CPA varies according to the affected host (horse vs sheep), and how ETX may induce intestinal lesions in goats in some cases of enterotoxemia, but rarely in other species. Host and cell susceptibility seem to be key elements for consideration when studying the pathogenesis of C. perfringens infections, and therefore the selection of either in vitro or animal models for C. perfringens-associated disease should address these variations. Since most C. perfringens toxins bind to and act on a receptor located on the plasma membrane of the host cell, this early step in the pathogenesis represents an evident therapeutic target for treating C. perfringens-associated diseases. In this regard, for instance, the therapeutic agent Mepacrine has been shown to protect enterocyte-like cells against CPE action in vitro by interfering with pore formation and insertion [164]. The potential effect of Mepacrine against CPE is currently being investigated in a mouse model. The final consequence in the action of most C. perfringens toxins is cell death. In the past few years significant progress has been made in understanding the complex intracellular pathways involved in this outcome by using different approaches. However, there are still many gaps in this knowledge, and dissecting the complex interaction between C. perfringens toxins and target cells may lead to the identification of additional pharmacological targets.
Author Contributions
M.A.N., B.A.M. and F.A.U. wrote the manuscript. M.A.N. prepared the figures. All authors revised, read, and approved the final manuscript.
Funding
Preparation of this review was supported, in part, by the National Institute of Allergy and Infectious Diseases grant number AI0198844-35 (B.A.M.). Mauricio A. Navarro is supported by Becas Chile, CONICYT, Gobierno de Chile.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Uzal, F.A.; Freedman, J.C.; Shrestha, A.; Theoret, J.R.; Garcia, J.; Awad, M.M.; Adams, V.; Moore, R.J.; Rood, J.I.; McClane, B.A. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014, 9, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Revitt-Mills, S.A.; Rood, J.I.; Adams, V. Clostridium Perfringens Extracellular Toxins and Enzymes: 20 and Counting. Microbiol. Aust. 2015, 36, 114–117. [Google Scholar] [CrossRef]
- Rood, J.I.; Adams, V.; Lacey, J.; Lyras, D.; McClane, B.A.; Melville, S.B.; Moore, R.J.; Popoff, M.R.; Sarker, M.R.; Songer, J.G.; et al. Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 2018. [Google Scholar] [CrossRef]
- Uzal, F.A.; Vidal, J.E.; McClane, B.A.; Gurjar, A.A. Clostridium Perfringens Toxins Involved in Mammalian Veterinary Diseases. Open Toxinol. J. 2010, 2, 24–42. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010, 8, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell death and infection: A double-edged sword for host and pathogen survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Los, M.J. Autophagy, apoptosis, mitoptosis and necrosis: Interdependence between those pathways and effects on cancer. Arch. Immunol. Ther. Exp. 2013, 61, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Ichim, G.; Green, D.R. Die another way—Non-apoptotic mechanisms of cell death. J. Cell Sci. 2014, 127, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A. Death in the fast lane: What’s next for necroptosis? FEBS J. 2016, 283, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Titball, R.W.; Naylor, C.E.; Basak, A.K. The Clostridium perfringens alpha-toxin. Anaerobe 1999, 5, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Jewell, S.A.; Titball, R.W.; Huyet, J.; Naylor, C.E.; Basak, A.K.; Gologan, P.; Winlove, C.P.; Petrov, P.G. Clostridium perfringens α-toxin interaction with red cells and model membranes. Soft Matter 2015, 11, 7748–7761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, M.; Terao, Y.; Sakurai, J.; Nagahama, M. Membrane-Binding Mechanism of Clostridium perfringens Alpha-Toxin. Toxins 2015, 7, 5268–5275. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Bryant, A.E.; Stevens, D.L.; Rood, J.I. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol. Microbiol. 1995, 15, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.E. Biology and pathogenesis of thrombosis and procoagulant activity in invasive infections caused by group A streptococci and Clostridium perfringens. Clin. Microbiol. Rev. 2003, 16, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.O.S.; Uzal, F.A.; Oliveira, C.A.; Lobato, F.C.F. Gas gangrene (malignant edema). In Clostridial Diseases of Animals; Uzal, F.A., Songer, J.G., Prescott, J.F., Popoff, M.R., Eds.; Wiley Blackwell: Ames, IA, USA, 2016; pp. 243–254. [Google Scholar]
- Heuck, A.P.; Moe, P.C.; Johnson, B.B. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell. Biochem. 2010, 51, 551–577. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Ellemor, D.M.; Boyd, R.L.; Emmins, J.J.; Rood, J.I. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 2001, 69, 7904–7910. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Matsuno, T.; Shiihara, R.; Ochi, S.; Yamauchi, R.; Saito, Y.; Imagawa, H.; Nagahama, M.; Nishizawa, M.; Sakurai, J. The relationship between the metabolism of sphingomyelin species and the hemolysis of sheep erythrocytes induced by Clostridium perfringens alpha-toxin. J. Lipid Res. 2008, 49, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Kabura, M.; Takagishi, T.; Suzue, A.; Tominaga, K.; Urano, S.; Nagahama, M.; Kobayashi, K.; Furukawa, K.; Furukawa, K.; et al. Clostridium perfringens alpha-toxin recognizes the GM1a-TrkA complex. J. Biol. Chem. 2012, 287, 33070–33079. [Google Scholar] [CrossRef] [PubMed]
- Flores-Díaz, M.; Alape-Girón, A.; Clark, G.; Catimel, B.; Hirabayashi, Y.; Nice, E.; Gutiérrez, J.-M.; Titball, R.; Thelestam, M. A cellular deficiency of gangliosides causes hypersensitivity to Clostridium perfringens phospholipase C. J. Biol. Chem. 2005, 280, 26680–26689. [Google Scholar] [CrossRef] [PubMed]
- Monturiol-Gross, L.; Flores-Díaz, M.; Pineda-Padilla, M.J.; Castro-Castro, A.C.; Alape-Giron, A. Clostridium perfringens phospholipase C induced ROS production and cytotoxicity require PKC, MEK1 and NFκB activation. PLoS ONE 2014, 9, e86475. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.M.; Valero, J.G.; Pérez-Cormenzana, M.; Cano, A.; Alonso, C.; Goñi, F.M. Lipidomic profile of GM95 cell death induced by Clostridium perfringens alpha-toxin. Chem. Phys. Lipids 2017, 203, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Maggio, B. The surface behavior of glycosphingolipids in biomembranes: A new frontier of molecular ecology. Prog. Biophys. Mol. Biol. 1994, 62, 55–117. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.-T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides—An overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sayeed, S.; Robertson, S.; Chen, J.; McClane, B.A. Sialidases affect the host cell adherence and epsilon toxin-induced cytotoxicity of Clostridium perfringens type D strain CN3718. PLoS Pathog. 2011, 7, e1002429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theoret, J.R.; Li, J.; Navarro, M.A.; Garcia, J.P.; Uzal, F.A.; McClane, B.A. Native or Proteolytically Activated NanI Sialidase Enhances the Binding and Cytotoxic Activity of Clostridium perfringens Enterotoxin and Beta Toxin. Infect. Immun. 2018, 86, e00730-17. [Google Scholar] [CrossRef] [PubMed]
- Ochi, S.; Oda, M.; Nagahama, M.; Sakurai, J. Clostridium perfringens alpha-toxin-induced hemolysis of horse erythrocytes is dependent on Ca2+ uptake. Biochim. Biophys. Acta 2003, 1613, 79–86. [Google Scholar] [CrossRef]
- Ochi, S.; Oda, M.; Matsuda, H.; Ikari, S.; Sakurai, J. Clostridium perfringens alpha-toxin activates the sphingomyelin metabolism system in sheep erythrocytes. J. Biol. Chem. 2004, 279, 12181–12189. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Ikari, S.; Matsuno, T.; Morimune, Y.; Nagahama, M.; Sakurai, J. Signal transduction mechanism involved in Clostridium perfringens alpha-toxin-induced superoxide anion generation in rabbit neutrophils. Infect. Immun. 2006, 74, 2876–2886. [Google Scholar] [CrossRef] [PubMed]
- Monturiol-Gross, L.; Flores-Díaz, M.; Campos-Rodríguez, D.; Mora, R.; Rodríguez-Vega, M.; Marks, D.L.; Alape-Girón, A. Internalization of Clostridium perfringens α-toxin leads to ERK activation and is involved on its cytotoxic effect. Cell. Microbiol. 2014, 16, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Takehara, M.; Takagishi, T.; Seike, S.; Ohtani, K.; Kobayashi, K.; Miyamoto, K.; Shimizu, T.; Nagahama, M. Clostridium perfringens α-Toxin Impairs Innate Immunity via Inhibition of Neutrophil Differentiation. Sci. Rep. 2016, 6, 28192. [Google Scholar] [CrossRef] [PubMed]
- Takehara, M.; Takagishi, T.; Seike, S.; Oishi, K.; Fujihara, Y.; Miyamoto, K.; Kobayashi, K.; Nagahama, M. Clostridium perfringens α-Toxin Impairs Lipid Raft Integrity in Neutrophils. Biol. Pharm. Bull. 2016, 39, 1694–1700. [Google Scholar] [CrossRef] [PubMed]
- Flores-Díaz, M.; Thelestam, M.; Clark, G.C.; Titball, R.W.; Alape-Girón, A. Effects of Clostridium perfringens phospholipase C in mammalian cells. Anaerobe 2004, 10, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.-M.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.N.; Krönke, M. Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 1998, 60, 643–665. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Ceramide in apoptosis: An overview and current perspectives. Biochim. Biophys. Acta 2002, 1585, 114–125. [Google Scholar] [CrossRef]
- Kågedal, K.; Zhao, M.; Svensson, I.; Brunk, U.T. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem. J. 2001, 359, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.-C.; Appelqvist, H.; Nilsson, C.; Kågedal, K.; Roberg, K.; Ollinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis Int. J. Program. Cell Death 2010, 15, 527–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Blom, T.; Slotte, J.P.; Pitson, S.M.; Törnquist, K. Enhancement of intracellular sphingosine-1-phosphate production by inositol 1,4,5-trisphosphate-evoked calcium mobilisation in HEK-293 cells: Endogenous sphingosine-1-phosphate as a modulator of the calcium response. Cell Signal. 2005, 17, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Gurjar, A.; Li, J.; McClane, B.A. Characterization of toxin plasmids in Clostridium perfringens type C isolates. Infect. Immun. 2010, 78, 4860–4869. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, S.; Li, J.; McClane, B.A. Characterization of virulence plasmid diversity among Clostridium perfringens type B isolates. Infect. Immun. 2010, 78, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.E.; Brown, J.E.; Oyston, P.C.; Sakurai, J.; Titball, R.W. Molecular genetic analysis of beta-toxin of Clostridium perfringens reveals sequence homology with alpha-toxin, gamma-toxin, and leukocidin of Staphylococcus aureus. Infect. Immun. 1993, 61, 3958–3965. [Google Scholar] [PubMed]
- Sakurai, J.; Duncan, C.L. Some properties of beta-toxin produced by Clostridium perfringens type C. Infect. Immun. 1978, 21, 678–680. [Google Scholar] [PubMed]
- Popoff, M.R. Clostridial pore-forming toxins: Powerful virulence factors. Anaerobe 2014, 30, 220–238. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Fujii, Y. Purification and characterization of Clostridium perfringens beta toxin. Toxicon 1987, 25, 1301–1310. [Google Scholar] [CrossRef]
- Theoret, J.R.; Uzal, F.A.; McClane, B.A. Identification and characterization of Clostridium perfringens beta toxin variants with differing trypsin sensitivity and in vitro cytotoxicity activity. Infect. Immun. 2015, 83, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Anderson, M.; Blanchard, P.; Mete, A.; Uzal, F.A. The pathology of enterotoxemia by Clostridium perfringens type C in calves. J. Vet. Diagn. Investig. 2013, 25, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, S.; Uzal, F.A.; Fisher, D.J.; Saputo, J.; Vidal, J.E.; Chen, Y.; Gupta, P.; Rood, J.I.; McClane, B.A. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol. Microbiol. 2008, 67, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.E.; McClane, B.A.; Saputo, J.; Parker, J.; Uzal, F.A. Effects of Clostridium perfringens beta-toxin on the rabbit small intestine and colon. Infect. Immun. 2008, 76, 4396–4404. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Beingesser, J.; Fisher, D.J.; Sayeed, S.; McClane, B.A.; Posthaus, H.; Uzal, F.A. The effect of Clostridium perfringens type C strain CN3685 and its isogenic beta toxin null mutant in goats. Vet. Microbiol. 2012, 157, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Saputo, J.; Sayeed, S.; Vidal, J.E.; Fisher, D.J.; Poon, R.; Adams, V.; Fernandez-Miyakawa, M.E.; Rood, J.I.; McClane, B.A. Development and application of new mouse models to study the pathogenesis of Clostridium perfringens type C enterotoxemias. Infect. Immun. 2009, 77, 5291–5299. [Google Scholar] [CrossRef] [PubMed]
- Miclard, J.; Jäggi, M.; Sutter, E.; Wyder, M.; Grabscheid, B.; Posthaus, H. Clostridium perfringens beta-toxin targets endothelial cells in necrotizing enteritis in piglets. Vet. Microbiol. 2009, 137, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Miclard, J.; van Baarlen, J.; Wyder, M.; Grabscheid, B.; Posthaus, H. Clostridium perfringens beta-toxin binding to vascular endothelial cells in a human case of enteritis necroticans. J. Med. Microbiol. 2009, 58, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, V.L.; Martel, A.; Pasmans, F.; Van Immerseel, F.; Posthaus, H. Endothelial binding of beta toxin to small intestinal mucosal endothelial cells in early stages of experimentally induced Clostridium perfringens type C enteritis in pigs. Vet. Pathol. 2013, 50, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Roos, S.; Wyder, M.; Candi, A.; Regenscheit, N.; Nathues, C.; van Immerseel, F.; Posthaus, H. Binding studies on isolated porcine small intestinal mucosa and in vitro toxicity studies reveal lack of effect of C. perfringens beta-toxin on the porcine intestinal epithelium. Toxins 2015, 7, 1235–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahama, M.; Morimitsu, S.; Kihara, A.; Akita, M.; Setsu, K.; Sakurai, J. Involvement of tachykinin receptors in Clostridium perfringens beta-toxin-induced plasma extravasation. Br. J. Pharmacol. 2003, 138, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Kihara, A.; Kintoh, H.; Oda, M.; Sakurai, J. Involvement of tumour necrosis factor-alpha in Clostridium perfringens beta-toxin-induced plasma extravasation in mice. Br. J. Pharmacol. 2008, 153, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Hayashi, S.; Morimitsu, S.; Sakurai, J. Biological activities and pore formation of Clostridium perfringens beta toxin in HL 60 cells. J. Biol. Chem. 2003, 278, 36934–36941. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Ochi, S.; Oda, M.; Miyamoto, K.; Takehara, M.; Kobayashi, K. Recent insights into Clostridium perfringens beta-toxin. Toxins 2015, 7, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Shibutani, M.; Seike, S.; Yonezaki, M.; Takagishi, T.; Oda, M.; Kobayashi, K.; Sakurai, J. The p38 MAPK and JNK pathways protect host cells against Clostridium perfringens beta-toxin. Infect. Immun. 2013, 81, 3703–3708. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, C.; Popescu, F.; Wyder, M.; Sutter, E.; Zeeh, F.; Frey, J.; von Schubert, C.; Posthaus, H. Rapid cytopathic effects of Clostridium perfringens beta-toxin on porcine endothelial cells. Infect. Immun. 2010, 78, 2966–2973. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Seike, S.; Shirai, H.; Takagishi, T.; Kobayashi, K.; Takehara, M.; Sakurai, J. Role of P2X7 receptor in Clostridium perfringens beta-toxin-mediated cellular injury. Biochim. Biophys. Acta 2015, 1850, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Seike, S.; Takehara, M.; Kobayashi, K.; Nagahama, M. Role of pannexin 1 in Clostridium perfringens beta-toxin-caused cell death. Biochim. Biophys. Acta 2016, 1858, 3150–3156. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Ralevic, V. P2X receptors in the cardiovascular system. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 663–674. [Google Scholar] [CrossRef]
- Penuela, S.; Harland, L.; Simek, J.; Laird, D.W. Pannexin channels and their links to human disease. Biochem. J. 2014, 461, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Autheman, D.; Wyder, M.; Popoff, M.; D’Herde, K.; Christen, S.; Posthaus, H. Clostridium perfringens beta-toxin induces necrostatin-inhibitable, calpain-dependent necrosis in primary porcine endothelial cells. PLoS ONE 2013, 8, e64644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, F.; Yang, S.; Wang, B.; Moynagh, P.N. RIP kinases: Key decision makers in cell death and innate immunity. Cell Death Differ. 2015, 22, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, S.; Li, J.; McClane, B.A. Virulence plasmid diversity in Clostridium perfringens type D isolates. Infect. Immun. 2007, 75, 2391–2398. [Google Scholar] [CrossRef] [PubMed]
- Minami, J.; Katayama, S.; Matsushita, O.; Matsushita, C.; Okabe, A. Lambda-toxin of Clostridium perfringens activates the precursor of epsilon-toxin by releasing its N- and C-terminal peptides. Microbiol. Immunol. 1997, 41, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Harkness, J.M.; Li, J.; McClane, B.A. Identification of a lambda toxin-negative Clostridium perfringens strain that processes and activates epsilon prototoxin intracellularly. Anaerobe 2012, 18, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.C.; McClane, B.A.; Uzal, F.A. New insights into Clostridium perfringens epsilon toxin activation and action on the brain during enterotoxemia. Anaerobe 2016, 41, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Bokori-Brown, M.; Savva, C.G.; Fernandes da Costa, S.P.; Naylor, C.E.; Basak, A.K.; Titball, R.W. Molecular basis of toxicity of Clostridium perfringens epsilon toxin. FEBS J. 2011, 278, 4589–4601. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.C.; Li, J.; Uzal, F.A.; McClane, B.A. Proteolytic processing and activation of Clostridium perfringens epsilon toxin by caprine small intestinal contents. mBio 2014, 5, e01994-14. [Google Scholar] [CrossRef] [PubMed]
- Filho, E.J.F.; Carvalho, A.U.; Assis, R.A.; Lobato, F.F.; Rachid, M.A.; Carvalho, A.A.; Ferreira, P.M.; Nascimento, R.A.; Fernandes, A.A.; Vidal, J.E.; et al. Clinicopathologic features of experimental Clostridium perfringens type D enterotoxemia in cattle. Vet. Pathol. 2009, 46, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R. Epsilon toxin: A fascinating pore-forming toxin. FEBS J. 2011, 278, 4602–4615. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Adams, V.; Beingesser, J.; Hughes, M.L.; Poon, R.; Lyras, D.; Hill, A.; McClane, B.A.; Rood, J.I.; Uzal, F.A. Epsilon toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats, and mice. Infect. Immun. 2013, 81, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Giannitti, F.; Finnie, J.W.; Manavis, J.; Beingesser, J.; Adams, V.; Rood, J.I.; Uzal, F.A. Comparative neuropathology of ovine enterotoxemia produced by Clostridium perfringens type D wild-type strain CN1020 and its genetically modified derivatives. Vet. Pathol. 2015, 52, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Giannitti, F.; Finnie, J.W.; Garcia, J.P. Diseases produced by Clostridium perfringens type D. In Clostridial Diseases of Animals; Wiley Blackwell: Ames, IA, USA, 2016; pp. 157–172. [Google Scholar]
- Goldstein, J.; Morris, W.E.; Loidl, C.F.; Tironi-Farinati, C.; Tironi-Farinatti, C.; McClane, B.A.; Uzal, F.A.; Fernandez Miyakawa, M.E. Clostridium perfringens epsilon toxin increases the small intestinal permeability in mice and rats. PLoS ONE 2009, 4, e7065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzal, F.A.; Songer, J.G. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. J. Vet. Diagn. Investig. 2008, 20, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Soler-Jover, A.; Dorca, J.; Popoff, M.R.; Gibert, M.; Saura, J.; Tusell, J.M.; Serratosa, J.; Blasi, J.; Martín-Satué, M. Distribution of Clostridium perfringens epsilon toxin in the brains of acutely intoxicated mice and its effect upon glial cells. Toxicon 2007, 50, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. Anaerobe 2004, 10, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Finnie, J.W.; Manavis, J.; Blumbergs, P.C. Aquaporin-4 in acute cerebral edema produced by Clostridium perfringens type D epsilon toxin. Vet. Pathol. 2008, 45, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Dorca-Arévalo, J.; Soler-Jover, A.; Gibert, M.; Popoff, M.R.; Martín-Satué, M.; Blasi, J. Binding of epsilon-toxin from Clostridium perfringens in the nervous system. Vet. Microbiol. 2008, 131, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Wioland, L.; Dupont, J.-L.; Bossu, J.-L.; Popoff, M.R.; Poulain, B. Attack of the nervous system by Clostridium perfringens Epsilon toxin: From disease to mode of action on neural cells. Toxicon 2013, 75, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Wioland, L.; Dupont, J.-L.; Doussau, F.; Gaillard, S.; Heid, F.; Isope, P.; Pauillac, S.; Popoff, M.R.; Bossu, J.-L.; Poulain, B. Epsilon toxin from Clostridium perfringens acts on oligodendrocytes without forming pores, and causes demyelination. Cell. Microbiol. 2015, 17, 369–388. [Google Scholar] [CrossRef] [PubMed]
- Soler-Jover, A.; Blasi, J.; Gómez de Aranda, I.; Navarro, P.; Gibert, M.; Popoff, M.R.; Martín-Satué, M. Effect of epsilon toxin-GFP on MDCK cells and renal tubules in vivo. J. Histochem. Cytochem. 2004, 52, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.W.; Williamson, E.D.; Havard, H.; Modi, N.; Brown, J. Evaluation of a new cytotoxicity assay for Clostridium perfringens type D epsilon toxin. FEMS Microbiol. Lett. 1994, 116, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Gibert, M.; Gillet, D.; Laurent-Winter, C.; Boquet, P.; Popoff, M.R. Clostridium perfringens epsilon-toxin acts on MDCK cells by forming a large membrane complex. J. Bacteriol. 1997, 179, 6480–6487. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Matsushita, O.; Minami, J.; Katayama, S.; Shimamoto, S.; Okabe, A. Cleavage of a C-terminal peptide is essential for heptamerization of Clostridium perfringens epsilon-toxin in the synaptosomal membrane. J. Biol. Chem. 2001, 276, 13778–13783. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.L.; Li, J.; Uzal, F.A.; McClane, B.A. Evidence for a prepore stage in the action of Clostridium perfringens epsilon toxin. PLoS ONE 2011, 6, e22053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagishi, T.; Oda, M.; Takehara, M.; Kobayashi, K.; Nagahama, M. Oligomer formation of Clostridium perfringens epsilon-toxin is induced by activation of neutral sphingomyelinase. Biochim. Biophys. Acta 2016, 1858, 2681–2688. [Google Scholar] [CrossRef] [PubMed]
- Ivie, S.E.; Fennessey, C.M.; Sheng, J.; Rubin, D.H.; McClain, M.S. Gene-trap mutagenesis identifies mammalian genes contributing to intoxication by Clostridium perfringens ε-toxin. PLoS ONE 2011, 6, e17787. [Google Scholar] [CrossRef] [PubMed]
- Rumah, K.R.; Ma, Y.; Linden, J.R.; Oo, M.L.; Anrather, J.; Schaeren-Wiemers, N.; Alonso, M.A.; Fischetti, V.A.; McClain, M.S.; Vartanian, T. The Myelin and Lymphocyte Protein MAL Is Required for Binding and Activity of Clostridium perfringens ε-Toxin. PLoS Pathog. 2015, 11, e1004896. [Google Scholar] [CrossRef] [PubMed]
- Schaeren-Wiemers, N.; Valenzuela, D.M.; Frank, M.; Schwab, M.E. Characterization of a rat gene, rMAL, encoding a protein with four hydrophobic domains in central and peripheral myelin. J. Neurosci. 1995, 15, 5753–5764. [Google Scholar] [CrossRef] [PubMed]
- Khalili, S.; Jahangiri, A.; Hashemi, Z.S.; Khalesi, B.; Mard-Soltani, M.; Amani, J. Structural pierce into molecular mechanism underlying Clostridium perfringens Epsilon toxin function. Toxicon 2017, 127, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Maier, E.; Gibert, M.; Popoff, M.R.; Benz, R. Clostridium perfringens epsilon toxin induces a rapid change of cell membrane permeability to ions and forms channels in artificial lipid bilayers. J. Biol. Chem. 2001, 276, 15736–15740. [Google Scholar] [CrossRef] [PubMed]
- Chassin, C.; Bens, M.; de Barry, J.; Courjaret, R.; Bossu, J.L.; Cluzeaud, F.; Ben Mkaddem, S.; Gibert, M.; Poulain, B.; Popoff, M.R.; et al. Pore-forming epsilon toxin causes membrane permeabilization and rapid ATP depletion-mediated cell death in renal collecting duct cells. Am. J. Physiol. Renal Physiol. 2007, 293, F927–F937. [Google Scholar] [CrossRef] [PubMed]
- Fennessey, C.M.; Ivie, S.E.; McClain, M.S. Coenzyme depletion by members of the aerolysin family of pore-forming toxins leads to diminished ATP levels and cell death. Mol. Biosyst. 2012, 8, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Gibert, M.; Gourch, A.; Bens, M.; Vandewalle, A.; Popoff, M.R. Clostridium perfringens epsilon toxin rapidly decreases membrane barrier permeability of polarized MDCK cells. Cell. Microbiol. 2003, 5, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Lonchamp, E.; Dupont, J.-L.; Wioland, L.; Courjaret, R.; Mbebi-Liegeois, C.; Jover, E.; Doussau, F.; Popoff, M.R.; Bossu, J.-L.; de Barry, J.; et al. Clostridium perfringens epsilon toxin targets granule cells in the mouse cerebellum and stimulates glutamate release. PLoS ONE 2010, 5, e13046. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Nagahama, M.; Oda, M.; Tsuge, H.; Kobayashi, K. Clostridium perfringens iota-toxin: Structure and function. Toxins 2009, 1, 208–228. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Miyamoto, K.; McClane, B.A. Comparison of virulence plasmids among Clostridium perfringens type E isolates. Infect. Immun. 2007, 75, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Gibert, M.; Petit, L.; Raffestin, S.; Okabe, A.; Popoff, M.R. Clostridium perfringens iota-toxin requires activation of both binding and enzymatic components for cytopathic activity. Infect. Immun. 2000, 68, 3848–3853. [Google Scholar] [CrossRef] [PubMed]
- Songer, J.G. Infections by Clostridium perfringens type E. In Clostridial Diseases of Animals; Uzal, F.A., Songer, J.G., Prescott, J.F., Popoff, M.R., Eds.; Wiley Blackwell: Ames, IA, USA, 2016; pp. 173–176. [Google Scholar]
- Schmidt, G.; Papatheodorou, P.; Aktories, K. Novel receptors for bacterial protein toxins. Curr. Opin. Microbiol. 2015, 23, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Wigelsworth, D.J.; Ruthel, G.; Schnell, L.; Herrlich, P.; Blonder, J.; Veenstra, T.D.; Carman, R.J.; Wilkins, T.D.; Van Nhieu, G.T.; Pauillac, S.; et al. CD44 Promotes intoxication by the clostridial iota-family toxins. PLoS ONE 2012, 7, e51356. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.F.; Mainguy, G.; Gibert, M.; Marvaud, J.C.; Stiles, B.G.; Popoff, M.R. Transcytosis of iota-toxin across polarized CaCo-2 cells. Mol. Microbiol. 2002, 43, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Nagayasu, K.; Kobayashi, K.; Sakurai, J. Binding component of Clostridium perfringens iota-toxin induces endocytosis in Vero cells. Infect. Immun. 2002, 70, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Umezaki, M.; Tashiro, R.; Oda, M.; Kobayashi, K.; Shibutani, M.; Takagishi, T.; Ishidoh, K.; Fukuda, M.; Sakurai, J. Intracellular trafficking of Clostridium perfringens iota-toxin b. Infect. Immun. 2012, 80, 3410–3416. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; Benz, R.; Popoff, M.R. Pore-forming activity of clostridial binary toxins. Biochim. Biophys. Acta 2016, 1858, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, H.; Nagahama, M.; Nishimura, H.; Hisatsune, J.; Sakaguchi, Y.; Itogawa, Y.; Katunuma, N.; Sakurai, J. Crystal structure and site-directed mutagenesis of enzymatic components from Clostridium perfringens iota-toxin. J. Mol. Biol. 2003, 325, 471–483. [Google Scholar] [CrossRef]
- Tsuge, H.; Nagahama, M.; Oda, M.; Iwamoto, S.; Utsunomiya, H.; Marquez, V.E.; Katunuma, N.; Nishizawa, M.; Sakurai, J. Structural basis of actin recognition and arginine ADP-ribosylation by Clostridium perfringens-toxin. Proc. Natl. Acad. Sci. USA 2008, 105, 7399–7404. [Google Scholar] [CrossRef] [PubMed]
- Gibert, M.; Monier, M.-N.; Ruez, R.; Hale, M.L.; Stiles, B.G.; Benmerah, A.; Johannes, L.; Lamaze, C.; Popoff, M.R. Endocytosis and toxicity of clostridial binary toxins depend on a clathrin-independent pathway regulated by Rho-GDI. Cell. Microbiol. 2011, 13, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Takehara, M.; Takagishi, T.; Seike, S.; Oda, M.; Sakaguchi, Y.; Hisatsune, J.; Ochi, S.; Kobayashi, K.; Nagahama, M. Cellular Entry of Clostridium perfringens Iota-Toxin and Clostridium botulinum C2 Toxin. Toxins 2017, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Gibert, M.; Marvaud, J.C.; Pereira, Y.; Hale, M.L.; Stiles, B.G.; Boquet, P.; Lamaze, C.; Popoff, M.R. Differential requirement for the translocation of clostridial binary toxins: Iota toxin requires a membrane potential gradient. FEBS Lett. 2007, 581, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Umezaki, M.; Oda, M.; Kobayashi, K.; Tone, S.; Suda, T.; Ishidoh, K.; Sakurai, J. Clostridium perfringens iota-toxin b induces rapid cell necrosis. Infect. Immun. 2011, 79, 4353–4360. [Google Scholar] [CrossRef] [PubMed]
- Hilger, H.; Pust, S.; von Figura, G.; Kaiser, E.; Stiles, B.G.; Popoff, M.R.; Barth, H. The long-lived nature of Clostridium perfringens iota toxin in mammalian cells induces delayed apoptosis. Infect. Immun. 2009, 77, 5593–5601. [Google Scholar] [CrossRef] [PubMed]
- McClane, B.A.; Robertson, S.L.; Li, J. Clostridium perfringens. In Food Microbiology: Fundamentals and Frontiers; Doyle, M.P., Buchanan, R.L., Eds.; ASM Press: Washington, DC, USA, 2013; pp. 465–489. [Google Scholar]
- McClane, B.A.; Lyerly, D.M.; Wilkins, T.D. Enterotoxic clostridia: Clostridium perfringens type A and Clostridium difficile. In Gram-Positive Pathogens; Fischetti, V.A., Novick, R.P., Ferretti, R.P., Portnoy, D.A., Rood, J.I., Eds.; ASM Press: Washington, DC, USA, 2006; pp. 703–714. [Google Scholar]
- Freedman, J.C.; Shrestha, A.; McClane, B.A. Clostridium perfringens Enterotoxin: Action, Genetics, and Translational Applications. Toxins 2016, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Hanna, P.C.; Mietzner, T.A.; Schoolnik, G.K.; McClane, B.A. Localization of the receptor-binding region of Clostridium perfringens enterotoxin utilizing cloned toxin fragments and synthetic peptides. The 30 C-terminal amino acids define a functional binding region. J. Biol. Chem. 1991, 266, 11037–11043. [Google Scholar] [PubMed]
- Harada, M.; Kondoh, M.; Ebihara, C.; Takahashi, A.; Komiya, E.; Fujii, M.; Mizuguchi, H.; Tsunoda, S.-I.; Horiguchi, Y.; Yagi, K.; et al. Role of tyrosine residues in modulation of claudin-4 by the C-terminal fragment of Clostridium perfringens enterotoxin. Biochem. Pharmacol. 2007, 73, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Komiya, E.; Kakutani, H.; Yoshida, T.; Fujii, M.; Horiguchi, Y.; Mizuguchi, H.; Tsutsumi, Y.; Tsunoda, S.; Koizumi, N.; et al. Domain mapping of a claudin-4 modulator, the C-terminal region of C-terminal fragment of Clostridium perfringens enterotoxin, by site-directed mutagenesis. Biochem. Pharmacol. 2008, 75, 1639–1648. [Google Scholar] [CrossRef] [PubMed]
- Smedley, J.G.; McClane, B.A. Fine mapping of the N-terminal cytotoxicity region of Clostridium perfringens enterotoxin by site-directed mutagenesis. Infect. Immun. 2004, 72, 6914–6923. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Miyamoto, K.; Sayeed, S.; McClane, B.A. Organization of the cpe locus in CPE-positive Clostridium perfringens type C and D isolates. PLoS ONE 2010, 5, e10932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billington, S.J.; Wieckowski, E.U.; Sarker, M.R.; Bueschel, D.; Songer, J.G.; McClane, B.A. Clostridium perfringens type E animal enteritis isolates with highly conserved, silent enterotoxin gene sequences. Infect. Immun. 1998, 66, 4531–4536. [Google Scholar] [PubMed]
- Miyamoto, K.; Yumine, N.; Mimura, K.; Nagahama, M.; Li, J.; McClane, B.A.; Akimoto, S. Identification of novel Clostridium perfringens type E strains that carry an iota toxin plasmid with a functional enterotoxin gene. PLoS ONE 2011, 6, e20376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarker, M.R.; Carman, R.J.; McClane, B.A. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 1999, 33, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Carman, R.J. Clostridium perfringens in spontaneous and antibiotic associated diarrhoea of man and other animals. Rev. Med. Microbiol. 1997, 8, S43–S45. [Google Scholar] [CrossRef]
- Bos, J.; Smithee, L.; McClane, B.; Distefano, R.F.; Uzal, F.; Songer, J.G.; Mallonee, S.; Crutcher, J.M. Fatal necrotizing colitis following a foodborne outbreak of enterotoxigenic Clostridium perfringens type A infection. Clin. Infect. Dis. 2005, 40, e78–e83. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Fatal foodborne Clostridium perfringens illness at a state psychiatric hospital—Louisiana, 2010. MMWR Morb. Mortal. Wkly. Rep. 2012, 61, 605–608. [Google Scholar]
- Caserta, J.A.; Robertson, S.L.; Saputo, J.; Shrestha, A.; McClane, B.A.; Uzal, F.A. Development and application of a mouse intestinal loop model to study the in vivo action of Clostridium perfringens enterotoxin. Infect. Immun. 2011, 79, 3020–3027. [Google Scholar] [CrossRef] [PubMed]
- Katahira, J.; Inoue, N.; Horiguchi, Y.; Matsuda, M.; Sugimoto, N. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J. Cell Biol. 1997, 136, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Katahira, J.; Horiguchi, Y.; Sonoda, N.; Furuse, M.; Tsukita, S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett. 2000, 476, 258–261. [Google Scholar] [CrossRef]
- Shrestha, A.; McClane, B.A. Human claudin-8 and -14 are receptors capable of conveying the cytotoxic effects of Clostridium perfringens enterotoxin. mBio 2013, 4, e00594-12. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.L.; Smedley, J.G.; Singh, U.; Chakrabarti, G.; Van Itallie, C.M.; Anderson, J.M.; McClane, B.A. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell. Microbiol. 2007, 9, 2734–2755. [Google Scholar] [CrossRef] [PubMed]
- Smedley, J.G.; Uzal, F.A.; McClane, B.A. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect. Immun. 2007, 75, 2381–2390. [Google Scholar] [CrossRef] [PubMed]
- McClane, B.A.; Uzal, F.A.; Miyakawa, M.F.; Lyerly, D.; Wilkins, T.D. The enterotoxic clostridia. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenburg, E., Schleifer, H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 688–752. [Google Scholar]
- McClane, B.A.; Wnek, A.P.; Hulkower, K.I.; Hanna, P.C. Divalent cation involvement in the action of Clostridium perfringens type A enterotoxin. Early events in enterotoxin action are divalent cation-independent. J. Biol. Chem. 1988, 263, 2423–2435. [Google Scholar] [PubMed]
- McClane, B.A. Clostridium perfringens enterotoxin acts by producing small molecule permeability alterations in plasma membranes. Toxicology 1994, 87, 43–67. [Google Scholar] [CrossRef]
- Chakrabarti, G.; McClane, B.A. The importance of calcium influx, calpain and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell. Microbiol. 2005, 7, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, G.; Zhou, X.; McClane, B.A. Death pathways activated in CaCo-2 cells by Clostridium perfringens enterotoxin. Infect. Immun. 2003, 71, 4260–4270. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.; Van Itallie, C.M.; Mitic, L.L.; Anderson, J.M.; McClane, B.A. CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J. Biol. Chem. 2000, 275, 18407–18417. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.; Klein, E.; McClane, B.A. Clostridium perfringens type A enterotoxin induces concurrent development of tissue damage and fluid accumulation in the rabbit ileum. J. Diarrheal Dis. 1994, 12, 200–207. [Google Scholar]
- Garcia, J.P.; Li, J.; Shrestha, A.; Freedman, J.C.; Beingesser, J.; McClane, B.A.; Uzal, F.A. Clostridium perfringens type A enterotoxin damages the rabbit colon. Infect. Immun. 2014, 82, 2211–2218. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.C.; Navarro, M.A.; Morrell, E.; Beingesser, J.; Shrestha, A.; McClane, B.A.; Uzal, F.A. Evidence that Clostridium perfringens Enterotoxin-Induced Intestinal Damage and Enterotoxemic Death in Mice Can Occur Independently of Intestinal Caspase-3 Activation. Infect. Immun. 2018, IAI.00931-17. [Google Scholar] [CrossRef] [PubMed]
- Lepp, D.; Roxas, B.; Parreira, V.R.; Marri, P.R.; Rosey, E.L.; Gong, J.; Songer, J.G.; Vedantam, G.; Prescott, J.F. Identification of novel pathogenicity loci in Clostridium perfringens strains that cause avian necrotic enteritis. PLoS ONE 2010, 5, e10795. [Google Scholar] [CrossRef]
- Bannam, T.L.; Yan, X.-X.; Harrison, P.F.; Seemann, T.; Keyburn, A.L.; Stubenrauch, C.; Weeramantri, L.H.; Cheung, J.K.; McClane, B.A.; Boyce, J.D.; et al. Necrotic enteritis-derived Clostridium perfringens strain with three closely related independently conjugative toxin and antibiotic resistance plasmids. mBio 2011, 2, e00190-11. [Google Scholar] [CrossRef] [PubMed]
- Parreira, V.R.; Costa, M.; Eikmeyer, F.; Blom, J.; Prescott, J.F. Sequence of two plasmids from Clostridium perfringens chicken necrotic enteritis isolates and comparison with C. perfringens conjugative plasmids. PLoS ONE 2012, 7, e49753. [Google Scholar] [CrossRef] [PubMed]
- Lacey, J.A.; Keyburn, A.L.; Ford, M.E.; Portela, R.W.; Johanesen, P.A.; Lyras, D.; Moore, R.J. Conjugation-mediated horizontal gene transfer of Clostridium perfringens plasmids in the chicken gastrointestinal tract results in the formation of new virulent strains. Appl. Environ. Microbiol. 2017, 83, e01814-17. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-X.; Porter, C.J.; Hardy, S.P.; Steer, D.; Smith, A.I.; Quinsey, N.S.; Hughes, V.; Cheung, J.K.; Keyburn, A.L.; Kaldhusdal, M.; et al. Structural and functional analysis of the pore-forming toxin NetB from Clostridium perfringens. mBio 2013, 4, e00019-13. [Google Scholar] [CrossRef] [PubMed]
- Keyburn, A.L.; Yan, X.-X.; Bannam, T.L.; Van Immerseel, F.; Rood, J.I.; Moore, R.J. Association between avian necrotic enteritis and Clostridium perfringens strains expressing NetB toxin. Vet. Res. 2010, 41, 21. [Google Scholar] [CrossRef] [PubMed]
- Rood, J.I.; Keyburn, A.L.; Moore, R.J. NetB and necrotic enteritis: The hole movable story. Avian Pathol. J. WVPA 2016, 45, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.F.; Parreira, V.R.; Mehdizadeh Gohari, I.; Lepp, D.; Gong, J. The pathogenesis of necrotic enteritis in chickens: What we know and what we need to know: A review. Avian Pathol. 2016, 45, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Olkowski, A.A.; Wojnarowicz, C.; Chirino-Trejo, M.; Laarveld, B.; Sawicki, G. Sub-clinical necrotic enteritis in broiler chickens: Novel etiological consideration based on ultra-structural and molecular changes in the intestinal tissue. Res. Vet. Sci. 2008, 85, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Savva, C.G.; Fernandes da Costa, S.P.; Bokori-Brown, M.; Naylor, C.E.; Cole, A.R.; Moss, D.S.; Titball, R.W.; Basak, A.K. Molecular architecture and functional analysis of NetB, a pore-forming toxin from Clostridium perfringens. J. Biol. Chem. 2013, 288, 3512–3522. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.C.; Hendricks, M.R.; McClane, B.A. The Potential Therapeutic Agent Mepacrine Protects Caco-2 Cells against Clostridium perfringens Enterotoxin Action. mSphere 2017, 2, e00352-17. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Intracellular pathways involved in C. perfringens alpha-toxin (CPA) intracellular action. Note the binding C-domain (C), catalytic N-domain (N) and ganglioside-binding loop domain (L) of CPA (PDB ID: 1CA1). CPA directly hydrolyzes phosphatidylcholine (PC) and sphingomyelin (SM) present in the plasma membrane of target cells. CPA can also activate Gi-type GTP-binding protein (Gi-GTP-BP) present in the plasma membrane, which in turn will activate endogenous phospholipases (PI-PLC) and sphingomyelinases (SMase). Phospholipase activity results in the formation of diacylglycerol (DAG), and inositol trisphosphate (IP3); the latter mobilizes and increases intracytoplasmic calcium ions (iCa2+). Sphingomyelinase action results in ceramide (CER), sphingosine (SPH) and sphingosine-1-phosphate (S1P) formation. In addition, interaction of CPA with the TrkA receptor leads to PDK1 and PKCθ phosphorylation, resulting in activation of the MEK/ERK signaling cascade and NF-κB, which is involved in reactive oxygen species (ROS) and IL-8 formation. A dashed red arrow represents what is concluded and/or suggested by the current authors.
Figure 1.
Intracellular pathways involved in C. perfringens alpha-toxin (CPA) intracellular action. Note the binding C-domain (C), catalytic N-domain (N) and ganglioside-binding loop domain (L) of CPA (PDB ID: 1CA1). CPA directly hydrolyzes phosphatidylcholine (PC) and sphingomyelin (SM) present in the plasma membrane of target cells. CPA can also activate Gi-type GTP-binding protein (Gi-GTP-BP) present in the plasma membrane, which in turn will activate endogenous phospholipases (PI-PLC) and sphingomyelinases (SMase). Phospholipase activity results in the formation of diacylglycerol (DAG), and inositol trisphosphate (IP3); the latter mobilizes and increases intracytoplasmic calcium ions (iCa2+). Sphingomyelinase action results in ceramide (CER), sphingosine (SPH) and sphingosine-1-phosphate (S1P) formation. In addition, interaction of CPA with the TrkA receptor leads to PDK1 and PKCθ phosphorylation, resulting in activation of the MEK/ERK signaling cascade and NF-κB, which is involved in reactive oxygen species (ROS) and IL-8 formation. A dashed red arrow represents what is concluded and/or suggested by the current authors.

Figure 2.
Pathways involved in C. perfringens beta-toxin (CPB) intracellular action. CPB structure was predicted by the SwissModel online software (Genbank ID: L13198) using standard settings. Initial binding of CPB to its potential receptor, the ATP-gated P2X7 receptor, induces a rapid peak of ATP release from target cells. This ATP loss is not associated with cell lysis and it would occur through the ATP-release channel pannexin 1. The released ATP would stimulate further CPB binding and oligomer formation, facilitating the pore-forming activity of the toxin. Pore formation results in Ca2+ influx and loss of intracytoplasmic K+ (iK+). Increase of iCa2+ is associated with calpain activation and necroptosis, which is inhibited by Nec-1; only low levels of caspase-3 activation occurs, suggesting that apoptosis is not a significant mechanism of cell death. Decrease iK+ is associated with the activation of MAPK and JNK, which activate host cell survival and defense pathways. Dashed red arrows represent what is concluded and/or suggested by the current authors. A dashed black arrow shows direction of movement.
Figure 2.
Pathways involved in C. perfringens beta-toxin (CPB) intracellular action. CPB structure was predicted by the SwissModel online software (Genbank ID: L13198) using standard settings. Initial binding of CPB to its potential receptor, the ATP-gated P2X7 receptor, induces a rapid peak of ATP release from target cells. This ATP loss is not associated with cell lysis and it would occur through the ATP-release channel pannexin 1. The released ATP would stimulate further CPB binding and oligomer formation, facilitating the pore-forming activity of the toxin. Pore formation results in Ca2+ influx and loss of intracytoplasmic K+ (iK+). Increase of iCa2+ is associated with calpain activation and necroptosis, which is inhibited by Nec-1; only low levels of caspase-3 activation occurs, suggesting that apoptosis is not a significant mechanism of cell death. Decrease iK+ is associated with the activation of MAPK and JNK, which activate host cell survival and defense pathways. Dashed red arrows represent what is concluded and/or suggested by the current authors. A dashed black arrow shows direction of movement.

Figure 3.
Pathways involved in C. perfringens epsilon-toxin (ETX) intracellular action. ETX (PDB ID: 1UYJ) action involves binding to its receptor (ETX-r), such as the myelin and lymphocyte (MAL) protein. Neutral sphingomyelinase (nSMase) may be activated resulting in sphingomyelin (SM) hydrolysis and ceramide (CER) production, which would facilitate ETX oligomer formation. Oligomerization results in a heptameric pore that induces a rapid loss of iK+, entry of Cl− and Na+ (not shown), followed later by an increase of iCa2+. ETX-affected cells also lose important coenzymes required for energy production, including NAD+, NADH and CoA, contributing the formation of the mitochondrial permeability transition pore (MPTP). This would facilitate the translocation of the apoptosis-inducing factor (AIF), a caspase independent cell death factor, from mitochondria to the nucleus. Dashed red arrows represent what is concluded and/or suggested by the current authors. A dashed black arrow shows direction of movement.
Figure 3.
Pathways involved in C. perfringens epsilon-toxin (ETX) intracellular action. ETX (PDB ID: 1UYJ) action involves binding to its receptor (ETX-r), such as the myelin and lymphocyte (MAL) protein. Neutral sphingomyelinase (nSMase) may be activated resulting in sphingomyelin (SM) hydrolysis and ceramide (CER) production, which would facilitate ETX oligomer formation. Oligomerization results in a heptameric pore that induces a rapid loss of iK+, entry of Cl− and Na+ (not shown), followed later by an increase of iCa2+. ETX-affected cells also lose important coenzymes required for energy production, including NAD+, NADH and CoA, contributing the formation of the mitochondrial permeability transition pore (MPTP). This would facilitate the translocation of the apoptosis-inducing factor (AIF), a caspase independent cell death factor, from mitochondria to the nucleus. Dashed red arrows represent what is concluded and/or suggested by the current authors. A dashed black arrow shows direction of movement.

Figure 4.
Intracellular pathways involved in C. perfringens iota-toxin (ITX) action. ITX is a binary toxin composed of an enzyme component (Ia) and a binding component (Ib). The lipolysis-stimulated lipoprotein receptor (LSR) has been reported a cellular receptor for Ib; after binding, Ib oligomerizes in a heptameric, functional pore that allows the movement of ions, and the translocation of Ia. Cytotoxicity involves features of necrosis, including decreased ATP, increased cell permeability and cell swelling. Bak and Bax activation also occurs leading to cytochrome C release from mitochondria. In addition, both Ia and Ib are internalize in endosomes. While a small proportion of Ib is recycled back to the plasma membrane, Ia is translocated into the cytoplasm from late endosomes. Once in the cytoplasm, Ia exerts its ADP-ribosylating activity, leading to depolymerization of actin which results in morphologic changes. Cytosolic stability of Ia would induce, likely via depolymerization of actin, a delayed caspase-3 activation and apoptosis. A dashed red arrow represents what is concluded and/or suggested by the current authors. A dashed black arrow shows direction of movement.
Figure 4.
Intracellular pathways involved in C. perfringens iota-toxin (ITX) action. ITX is a binary toxin composed of an enzyme component (Ia) and a binding component (Ib). The lipolysis-stimulated lipoprotein receptor (LSR) has been reported a cellular receptor for Ib; after binding, Ib oligomerizes in a heptameric, functional pore that allows the movement of ions, and the translocation of Ia. Cytotoxicity involves features of necrosis, including decreased ATP, increased cell permeability and cell swelling. Bak and Bax activation also occurs leading to cytochrome C release from mitochondria. In addition, both Ia and Ib are internalize in endosomes. While a small proportion of Ib is recycled back to the plasma membrane, Ia is translocated into the cytoplasm from late endosomes. Once in the cytoplasm, Ia exerts its ADP-ribosylating activity, leading to depolymerization of actin which results in morphologic changes. Cytosolic stability of Ia would induce, likely via depolymerization of actin, a delayed caspase-3 activation and apoptosis. A dashed red arrow represents what is concluded and/or suggested by the current authors. A dashed black arrow shows direction of movement.

Figure 5.
Intracellular pathways involved in C. perfringens enterotoxin (CPE) action. Claudins are cellular receptors for CPE. Initial CPE binding to claudins results in the formation of a small complex. Interaction of six small complexes lead to CPE oligomerization and formation of a pre-pore on the plasma membrane, a large complex named CH-1. Assembly of β-hairping loops from CPE into a β-barrel permits the insertion of a cation-permeating pore in the plasma membrane. An influx of Ca2+ occurs, which stimulates the activity of calpains, which in turn, leads to caspase-3 activation and apoptosis, or a mechanism of cell death with features of necrosis. A dashed red arrow represents what is concluded and/or suggested by the authors. A dashed black arrow shows direction of movement.
Figure 5.
Intracellular pathways involved in C. perfringens enterotoxin (CPE) action. Claudins are cellular receptors for CPE. Initial CPE binding to claudins results in the formation of a small complex. Interaction of six small complexes lead to CPE oligomerization and formation of a pre-pore on the plasma membrane, a large complex named CH-1. Assembly of β-hairping loops from CPE into a β-barrel permits the insertion of a cation-permeating pore in the plasma membrane. An influx of Ca2+ occurs, which stimulates the activity of calpains, which in turn, leads to caspase-3 activation and apoptosis, or a mechanism of cell death with features of necrosis. A dashed red arrow represents what is concluded and/or suggested by the authors. A dashed black arrow shows direction of movement.
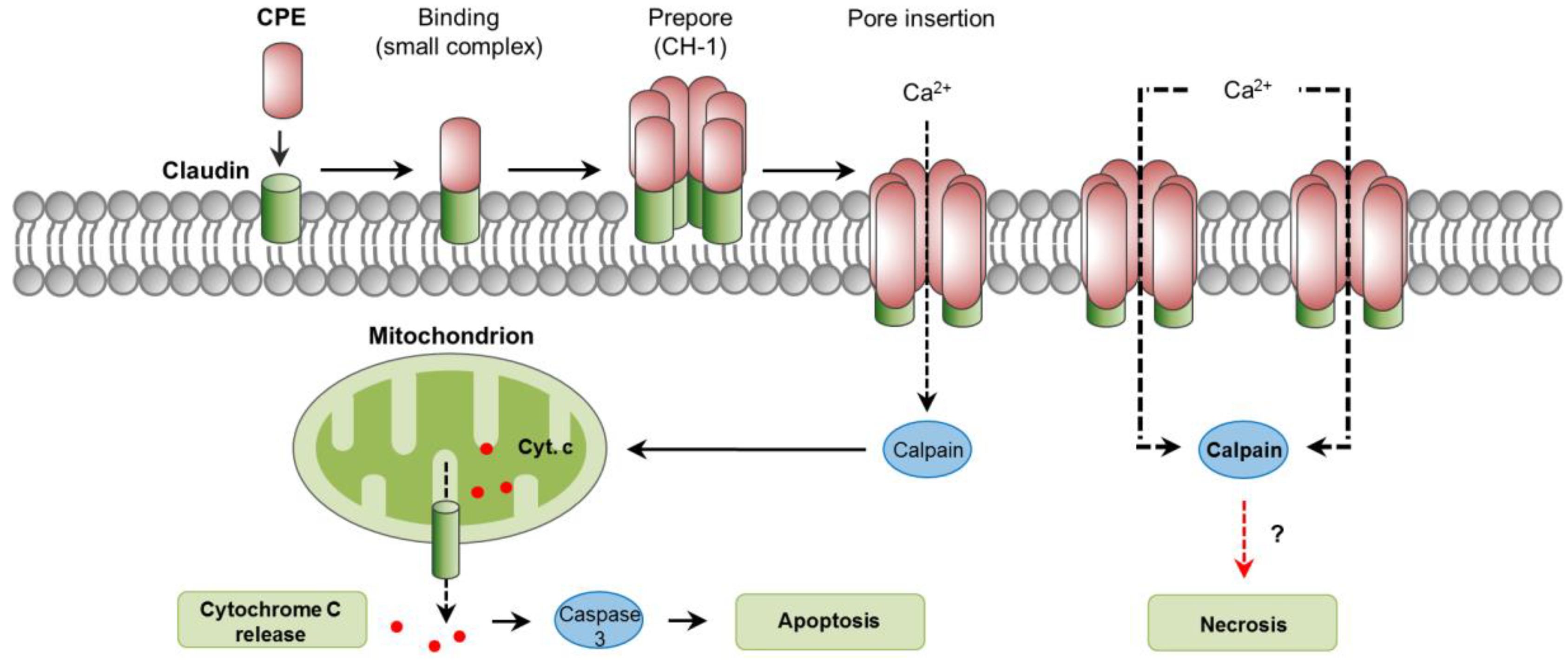

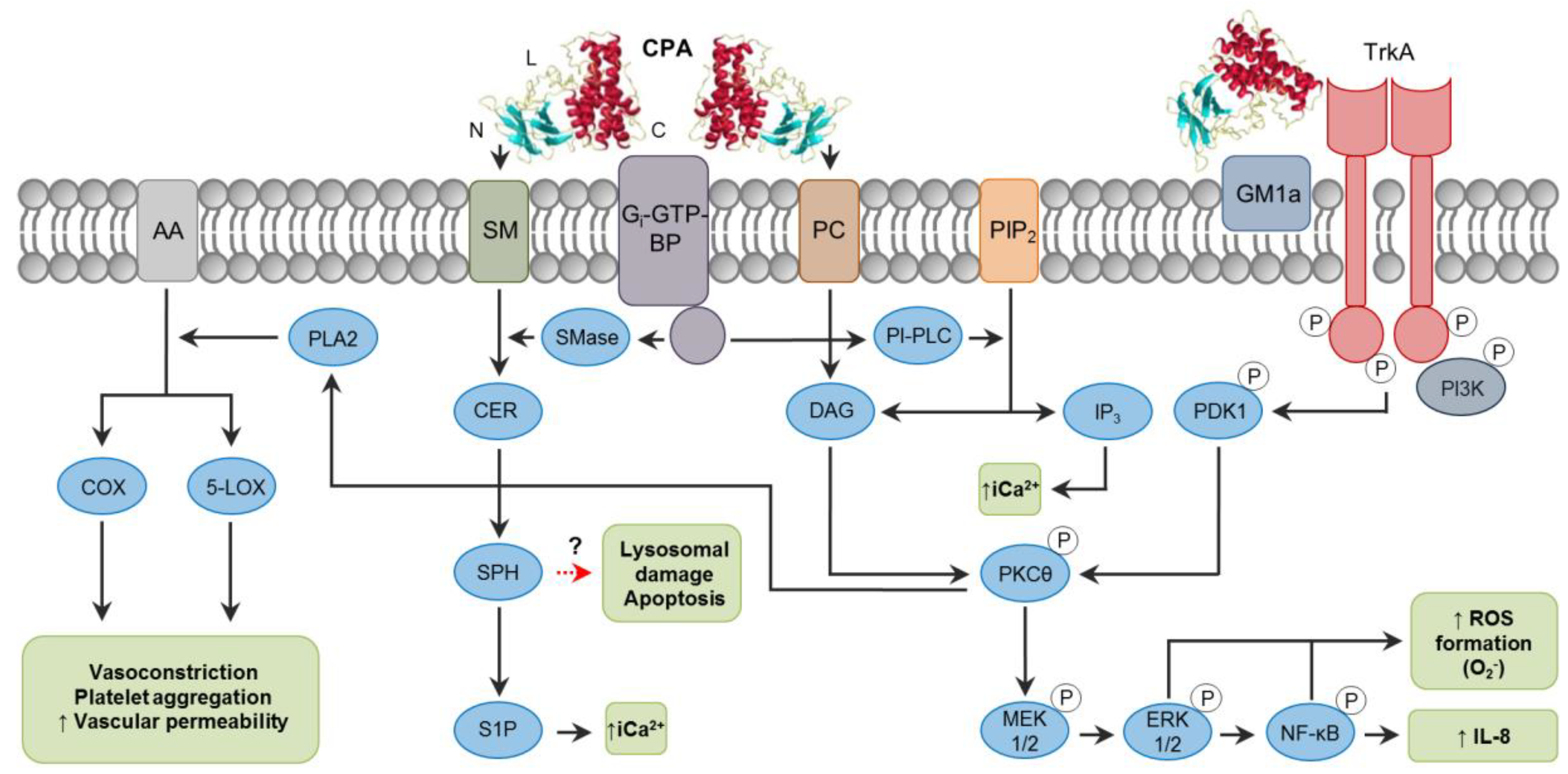{kind=link}
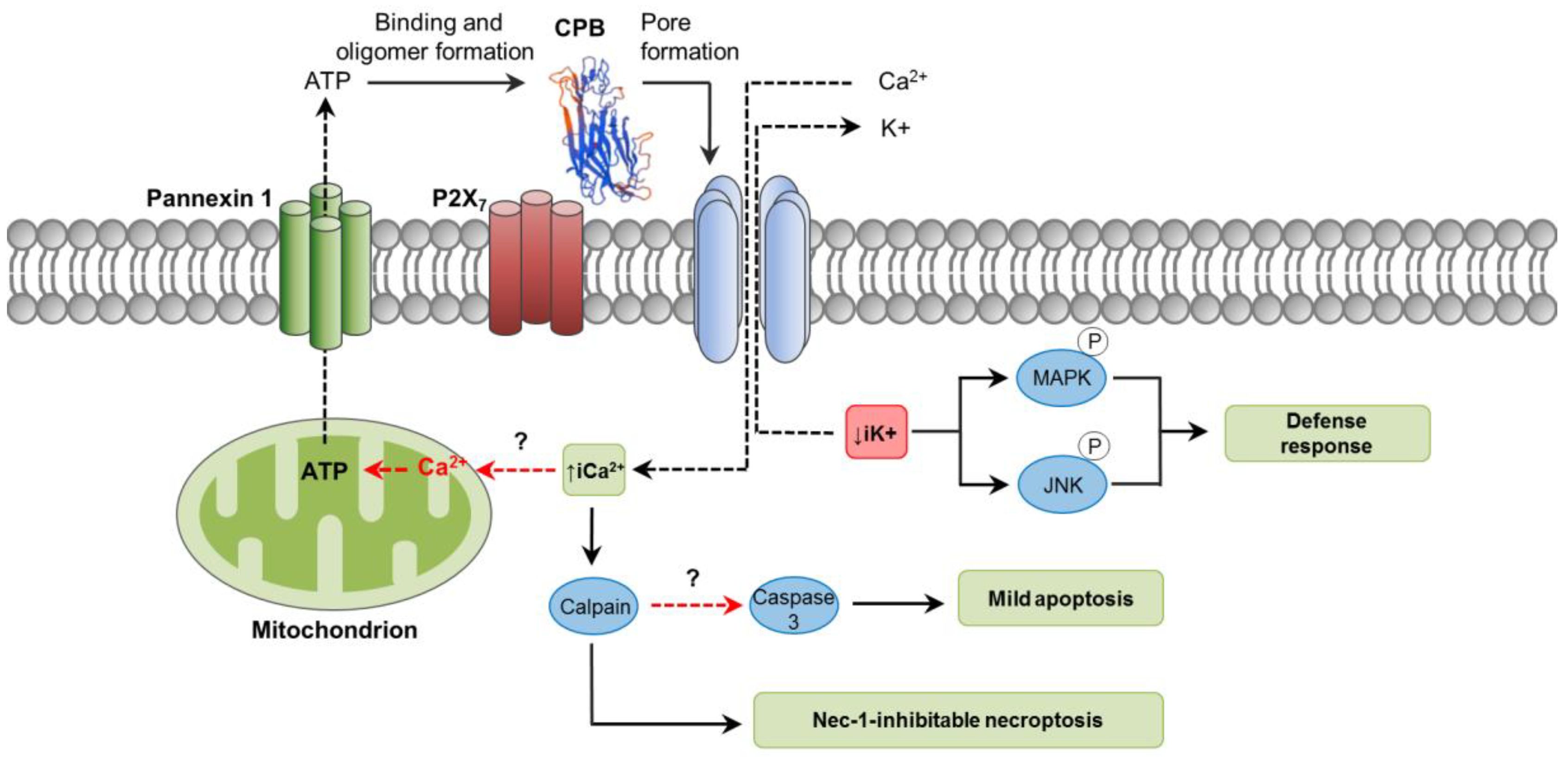{kind=link}
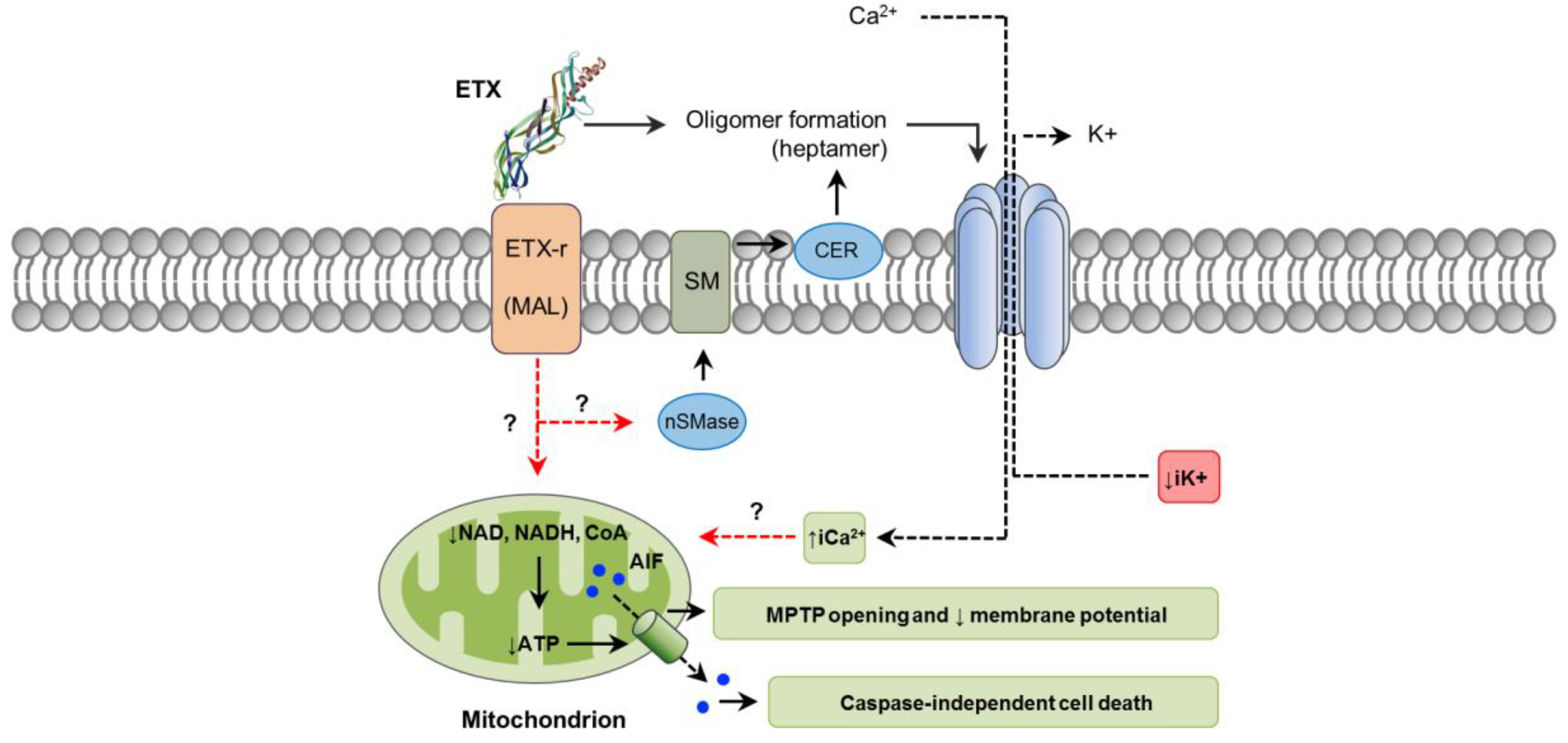{kind=link}
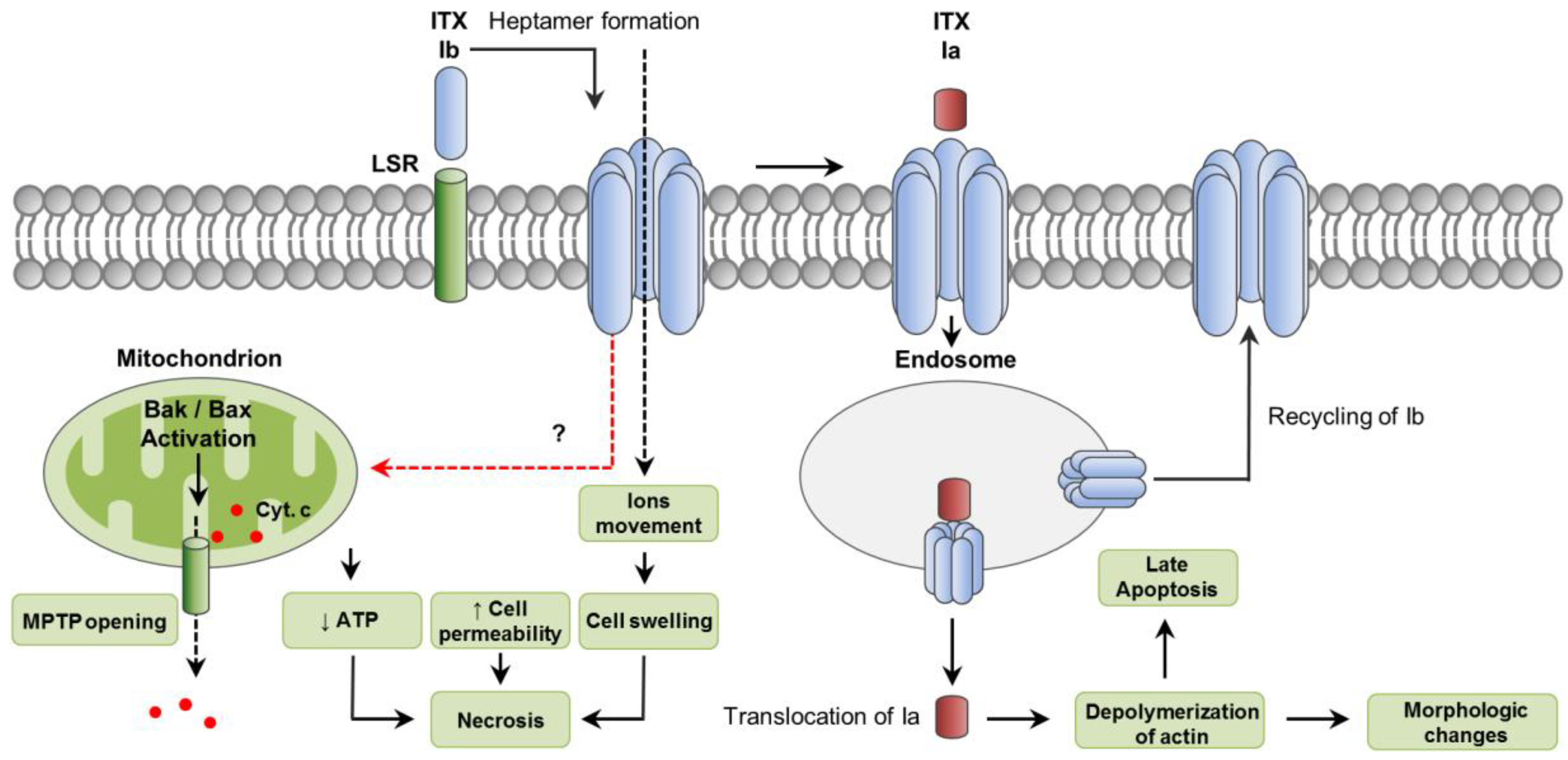{kind=link}
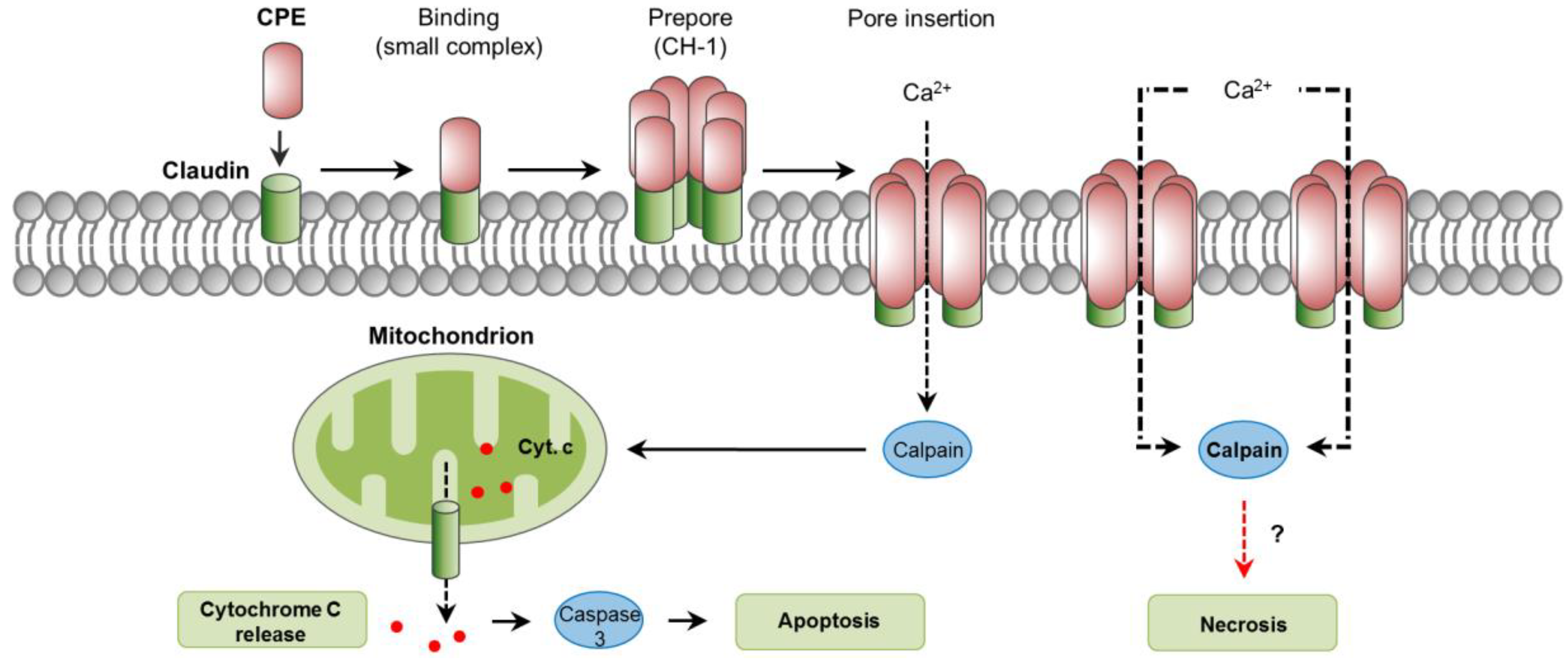{kind=link}
Table 1.
Revised classification system of Clostridium perfringens based on the production of six major toxins [3].
Table 1.
Revised classification system of Clostridium perfringens based on the production of six major toxins [3].
| Type | Toxin Produced | |||||
|---|---|---|---|---|---|---|
| α (CPA) | β (CPB) | ε (ETX) | ι (ITX) | CPE | NetB | |
| A | + | − | − | − | − | − |
| B | + | + | + | − | − | − |
| C | + | + | − | − | +/− | − |
| D | + | − | + | − | +/− | − |
| E | + | − | − | + | +/− | − |
| F | + | − | − | − | + | − |
| G | + | − | − | − | − | + |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Navarro, M.A.; McClane, B.A.; Uzal, F.A. Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins. Toxins 2018, 10, 212. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10050212
AMA Style
Navarro MA, McClane BA, Uzal FA. Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins. Toxins. 2018; 10(5):212. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10050212
Chicago/Turabian StyleNavarro, Mauricio A., Bruce A. McClane, and Francisco A. Uzal. 2018. "Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins" Toxins 10, no. 5: 212. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10050212
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.